Optical Methods to Study Protein-DNA Interactions in Vitro and in Living Cells at the Single-Molecule Level

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
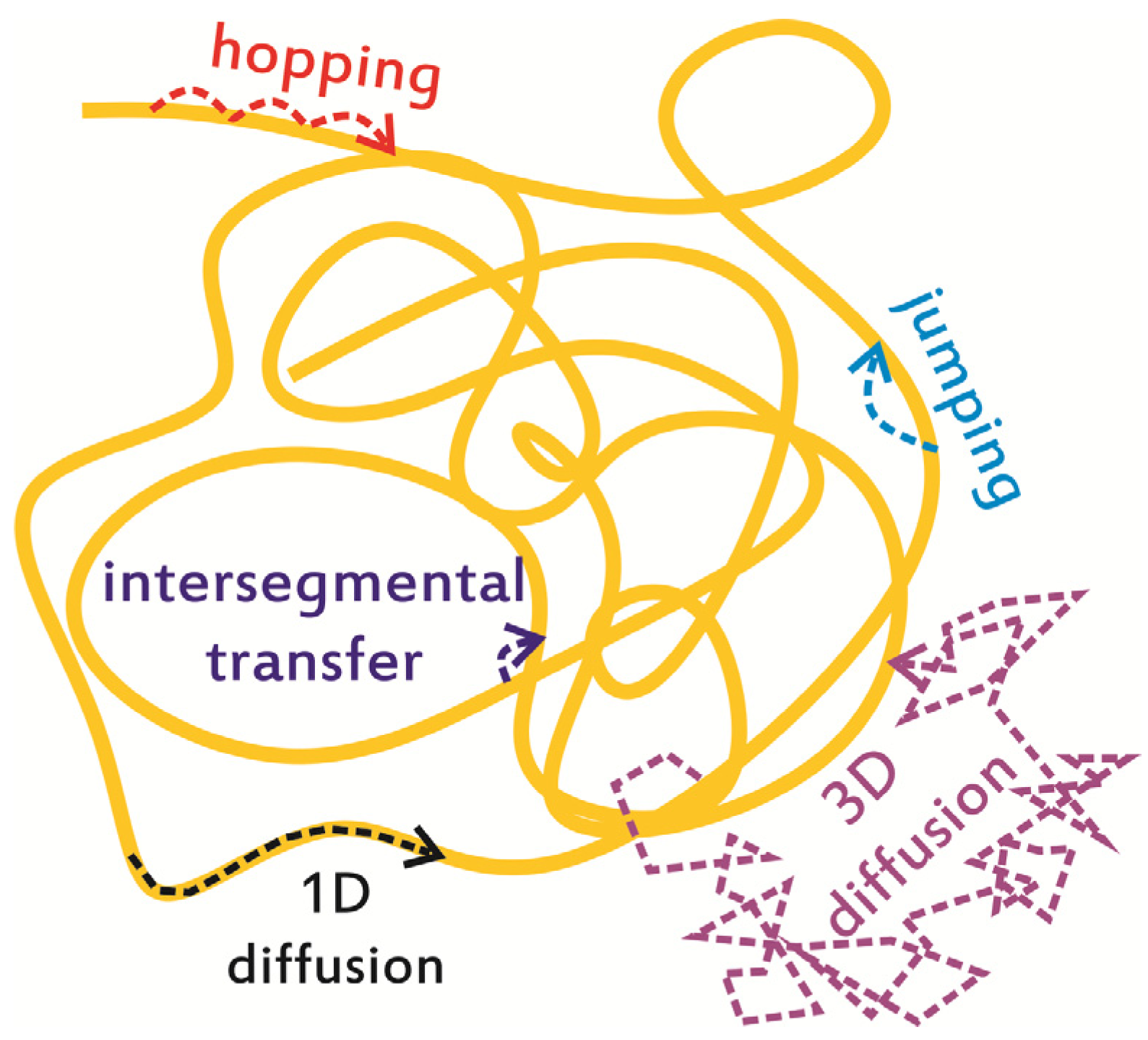
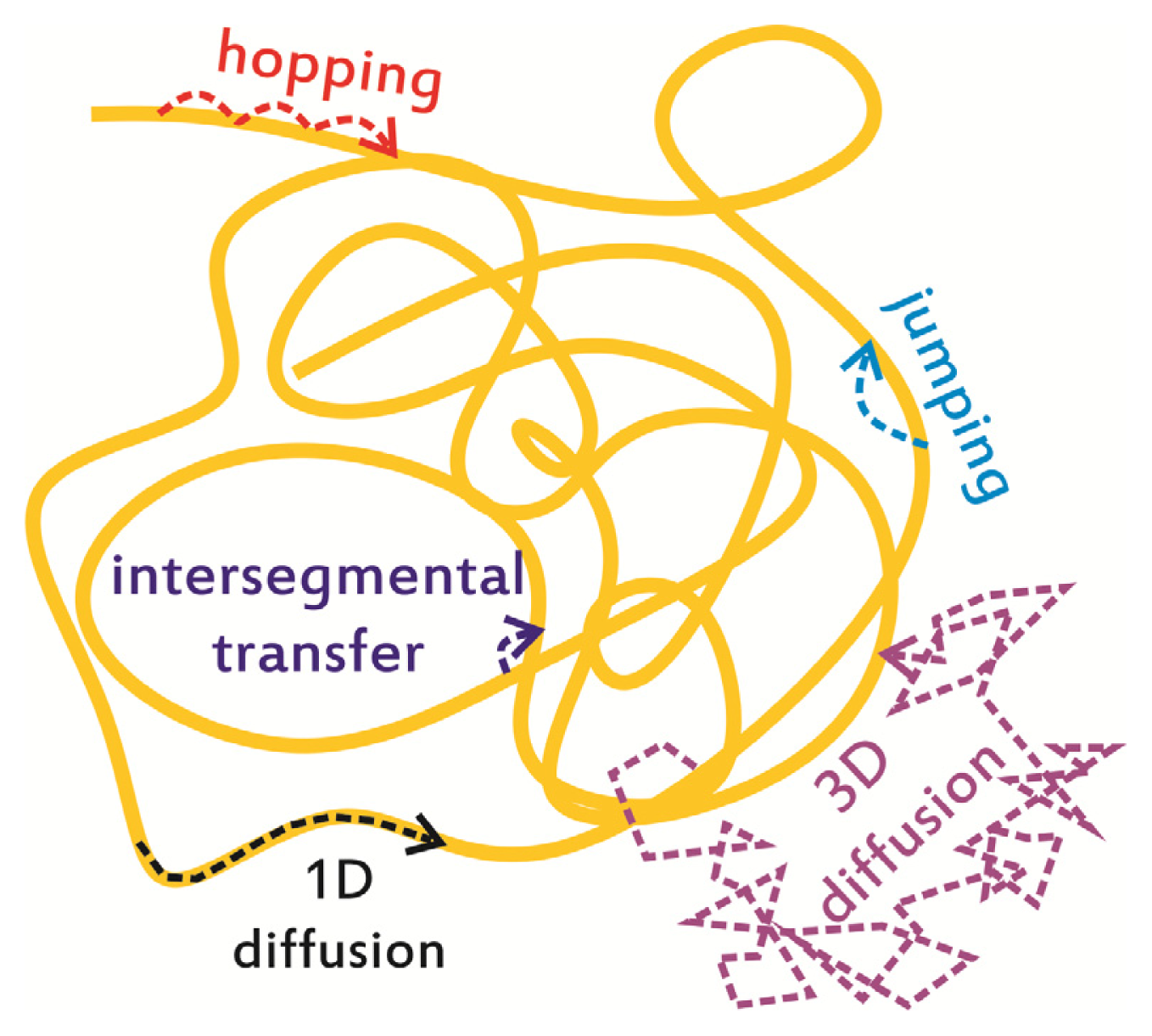
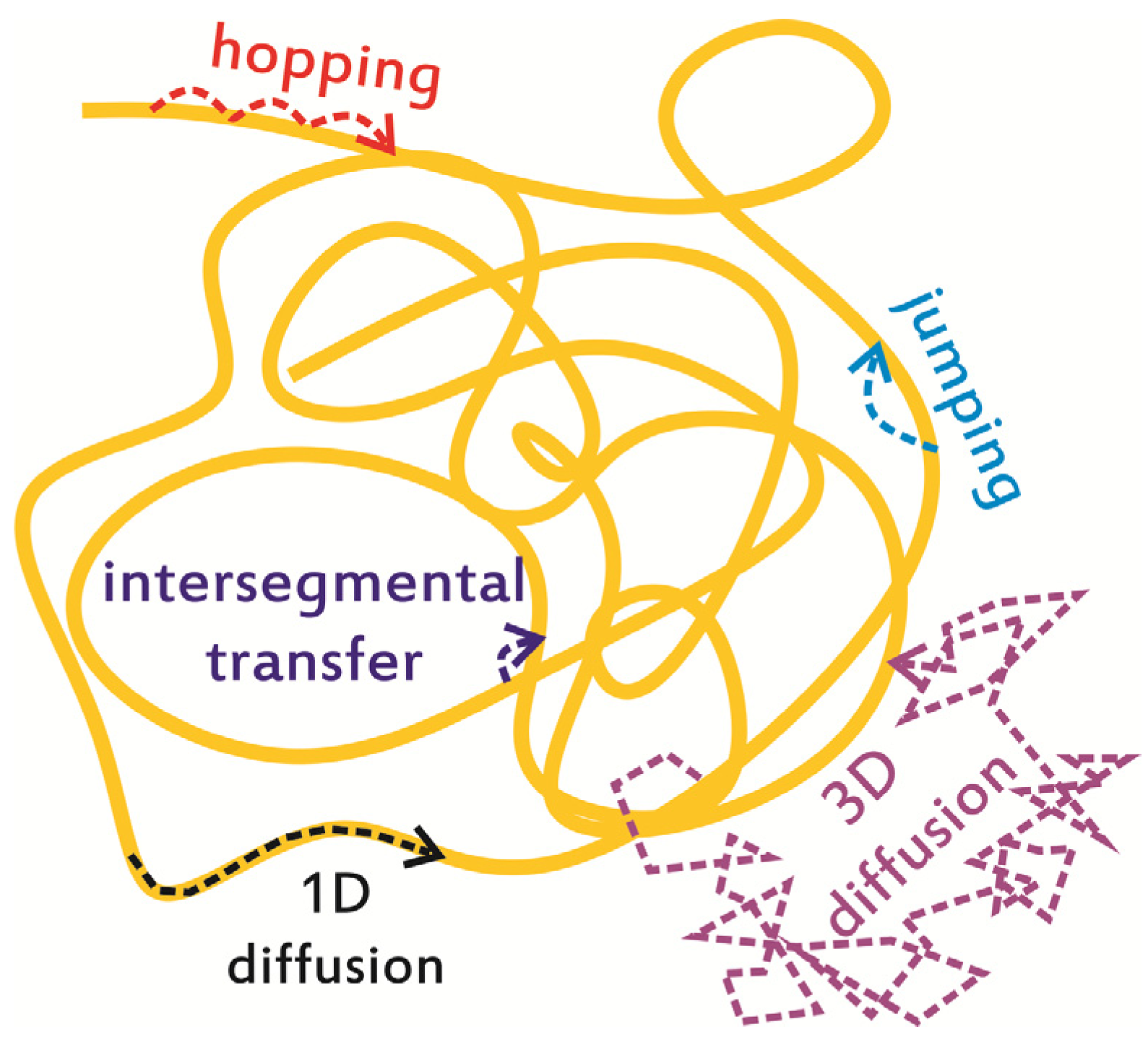
2. In Vitro Monitoring of Facilitated Diffusion and Protein-DNA Interactions at the Single-Molecule Level
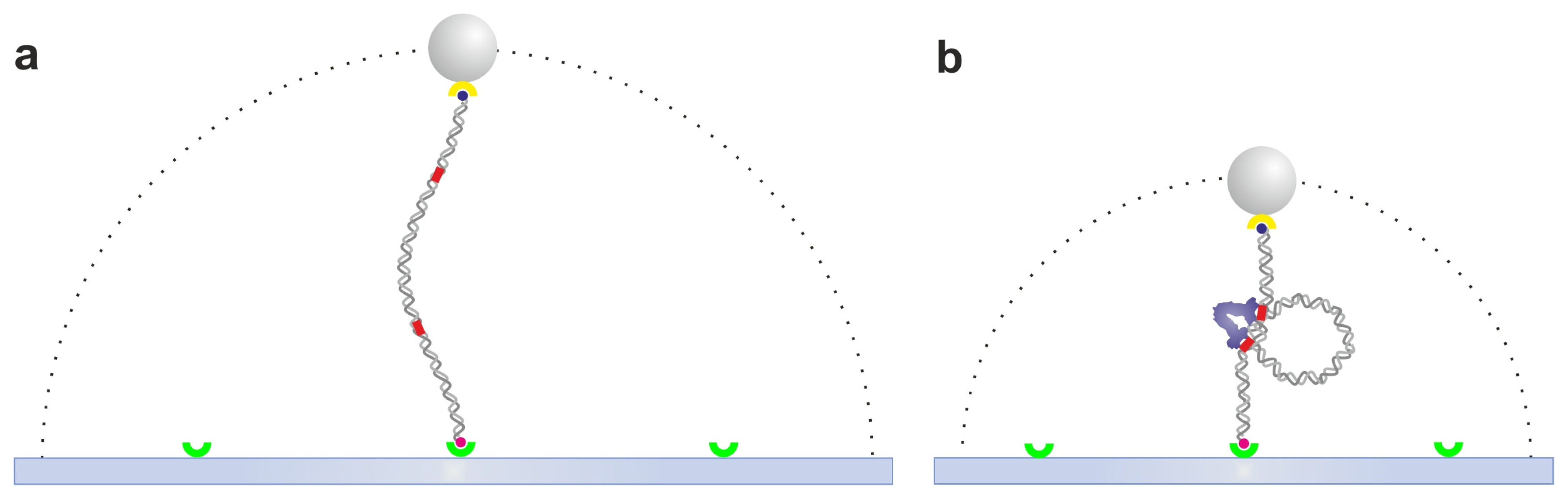
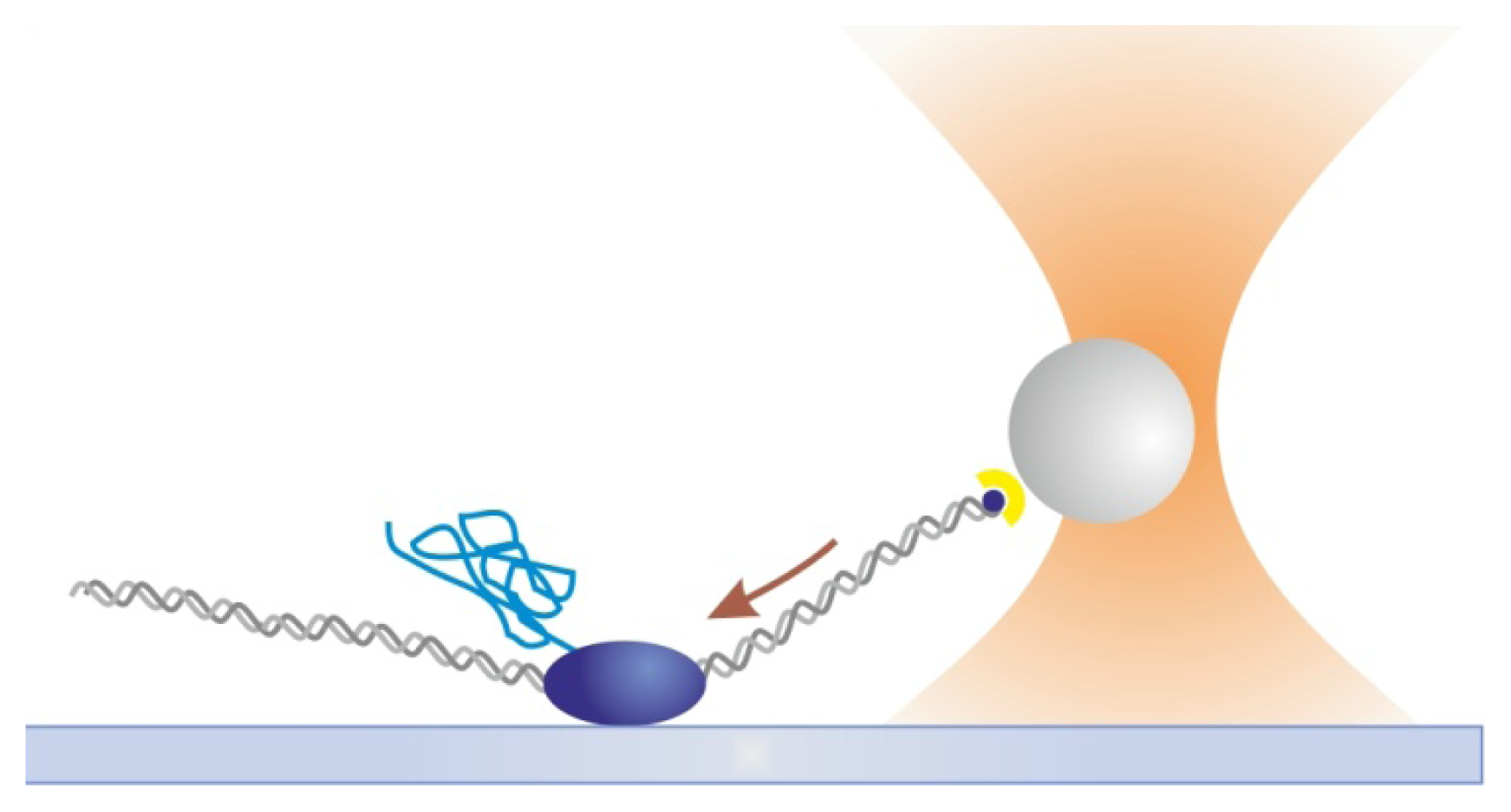
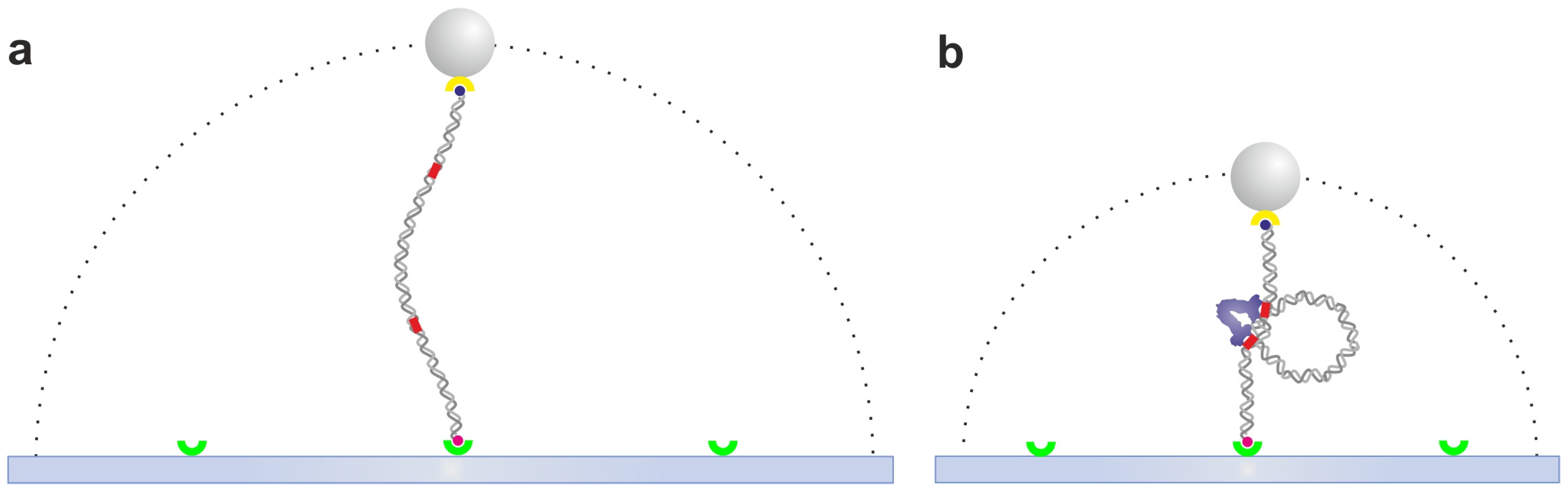
2.1. Tethered Particle Motion
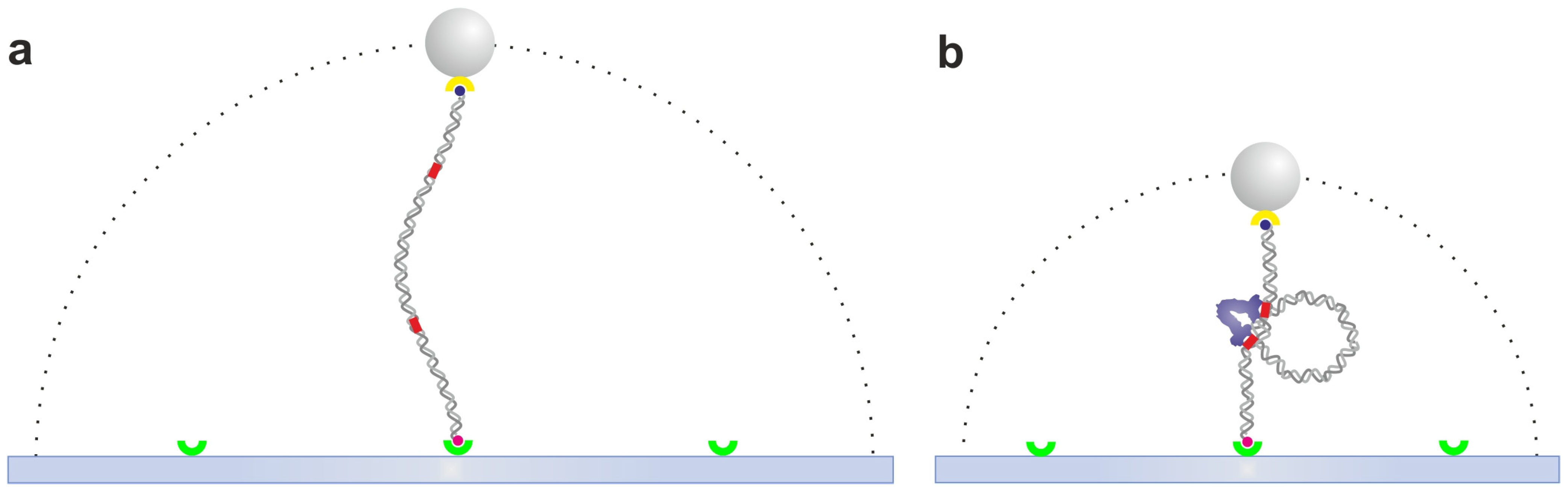
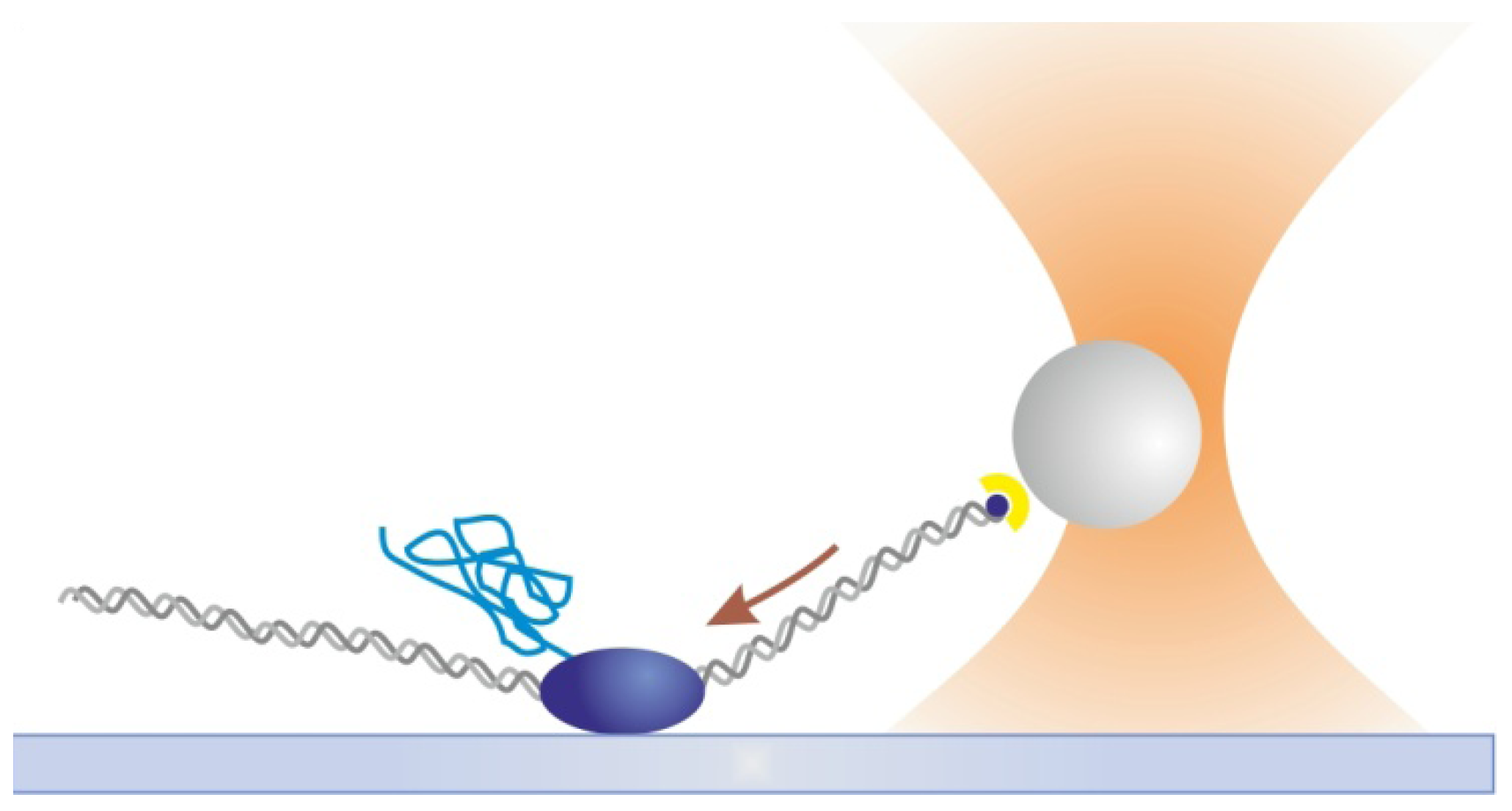
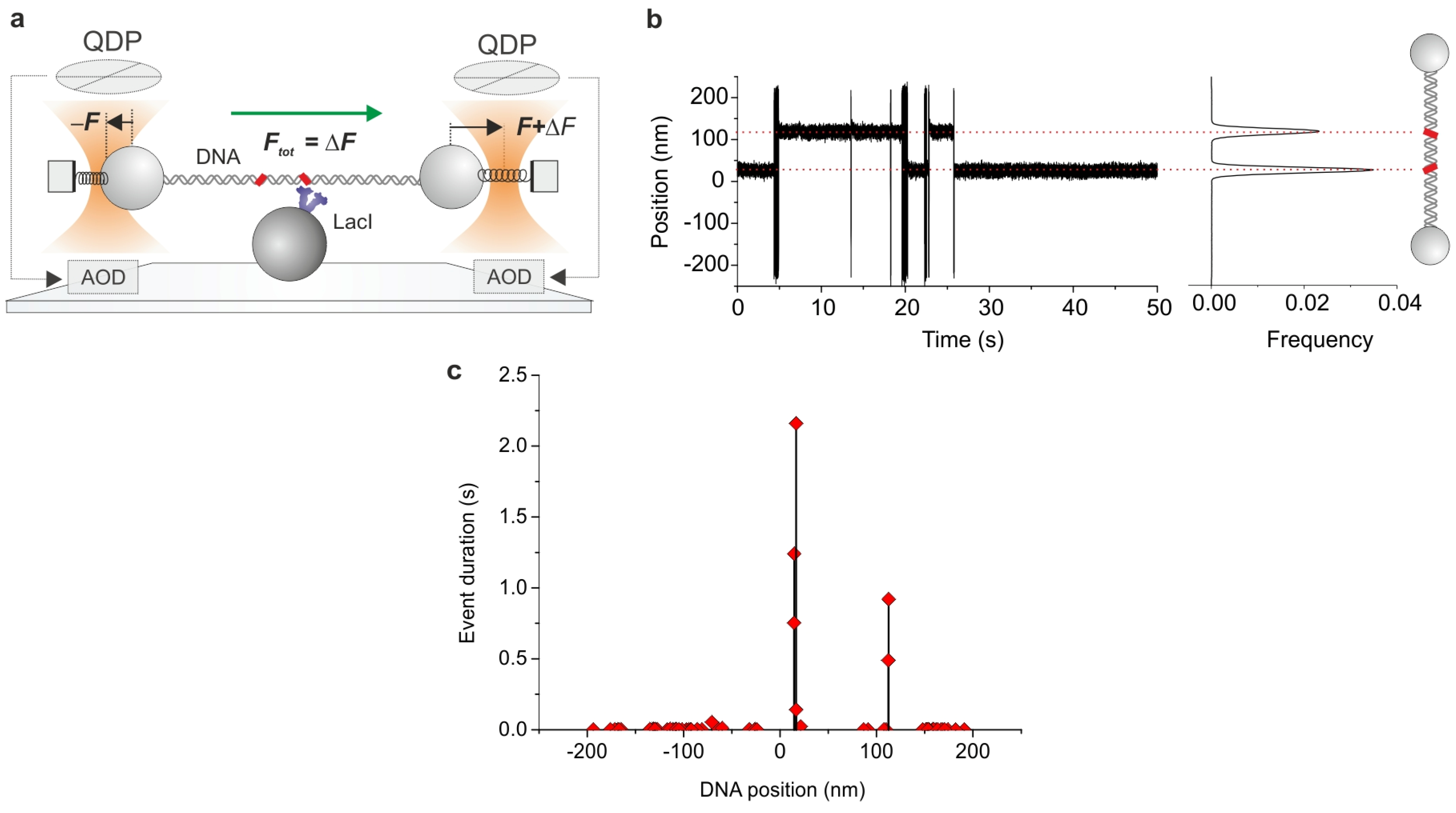
2.2. Optical Tweezers
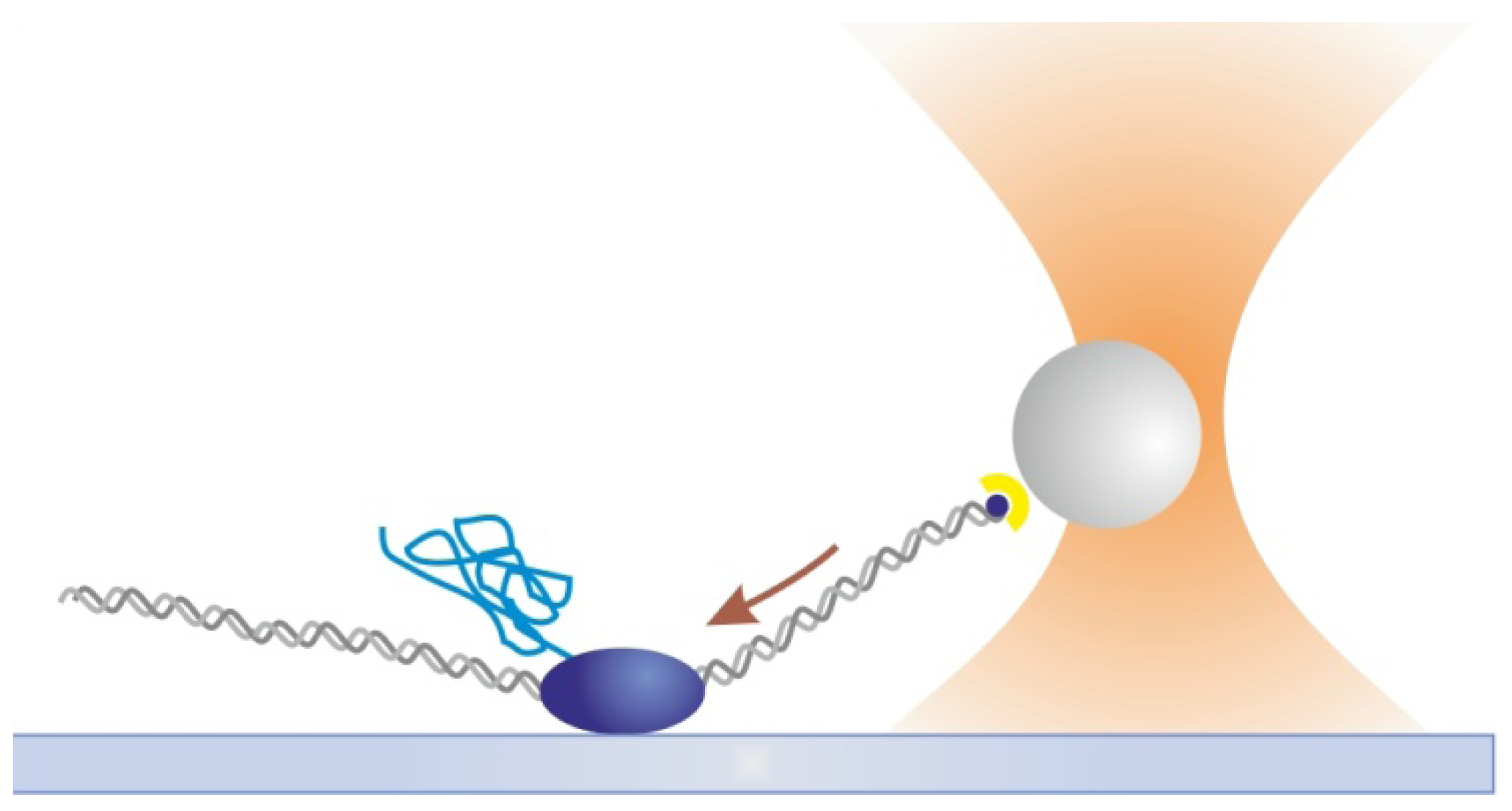
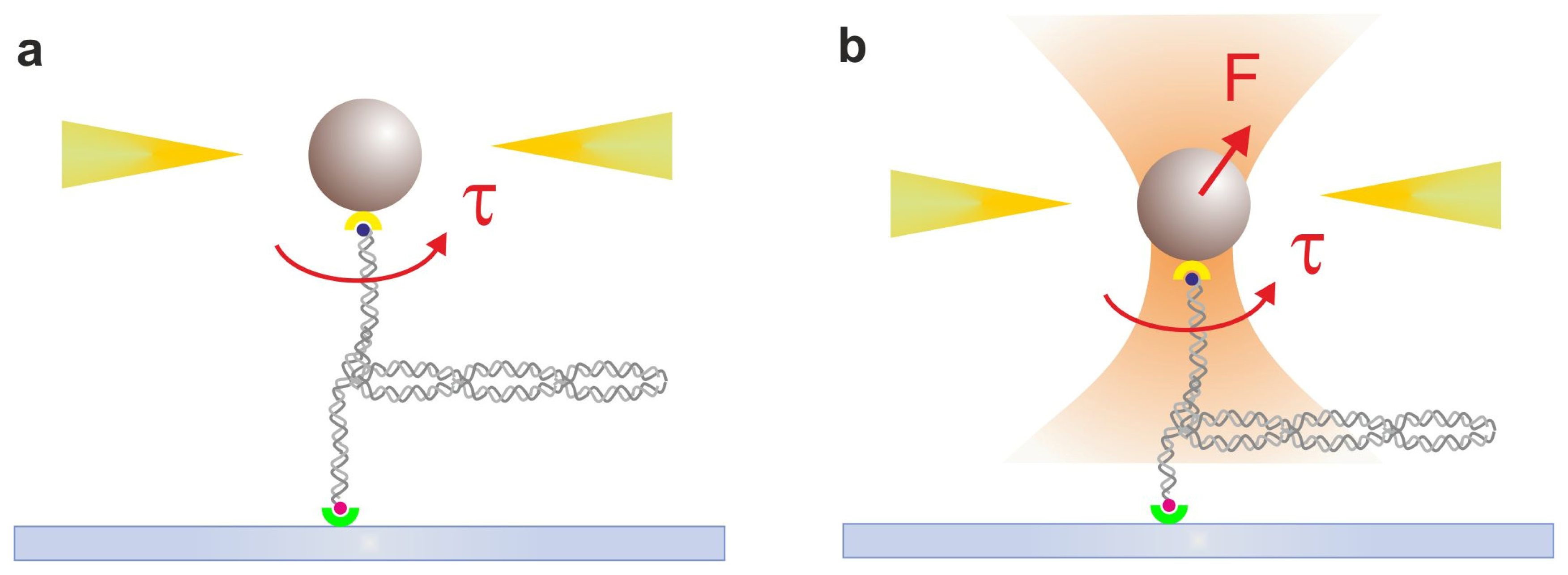
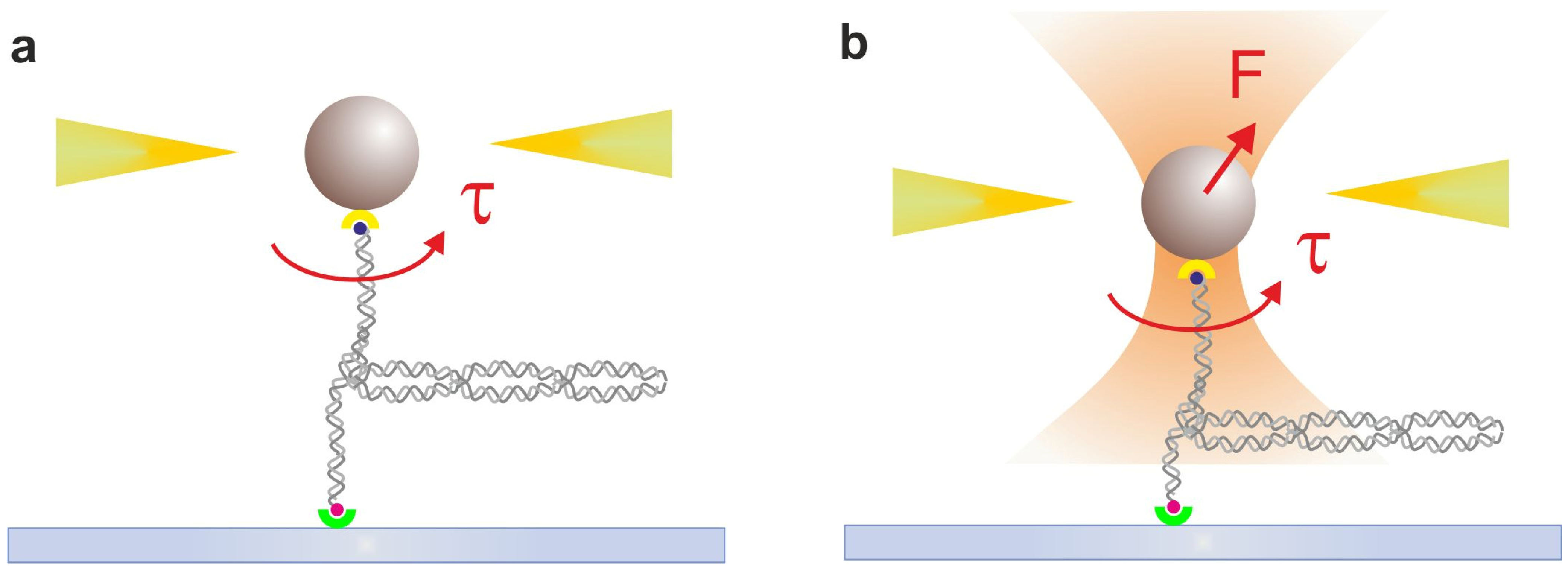
2.3. Magnetic Tweezers
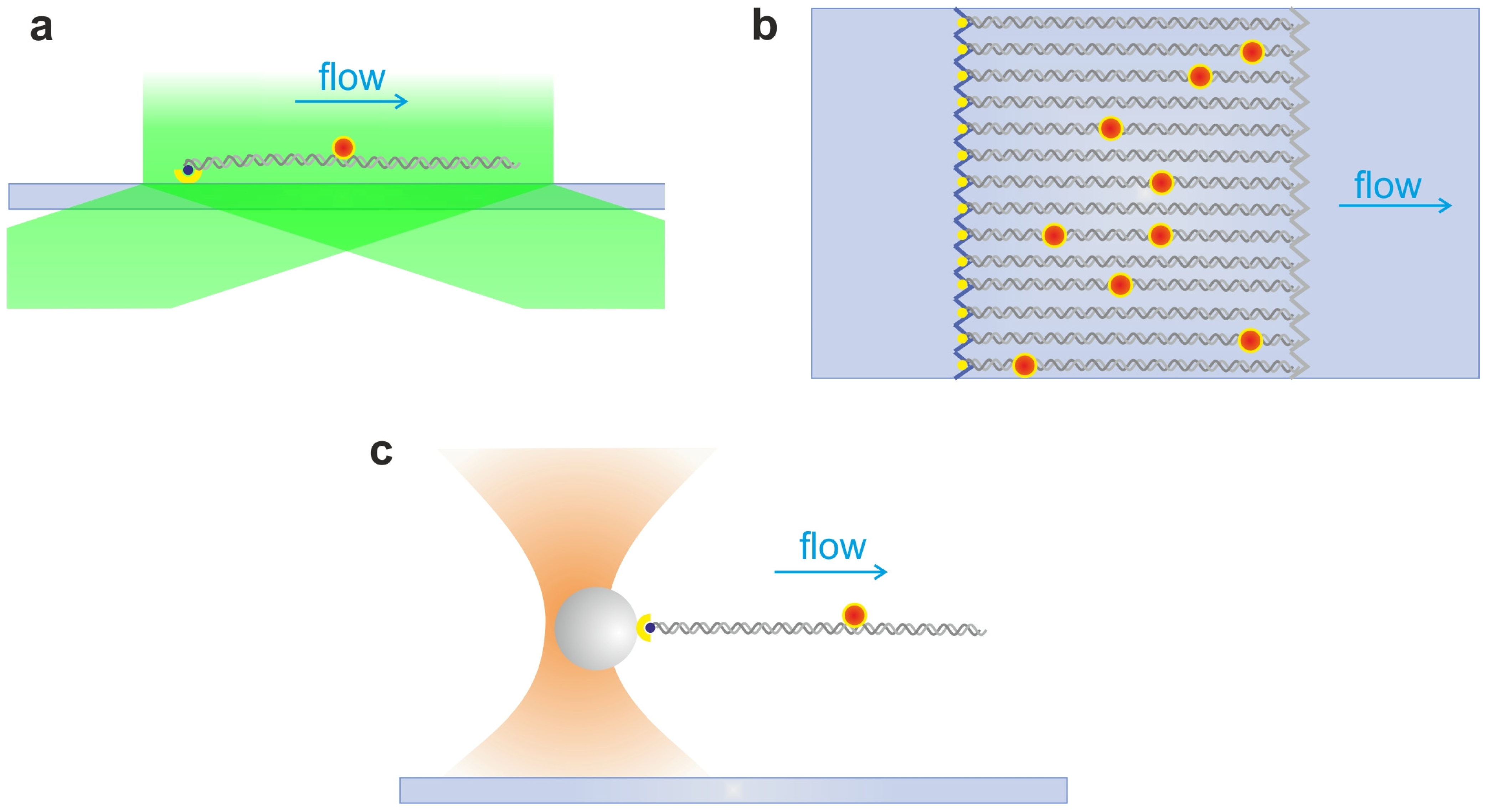
2.4. DNA Hydrodynamic Stretching and Curtains
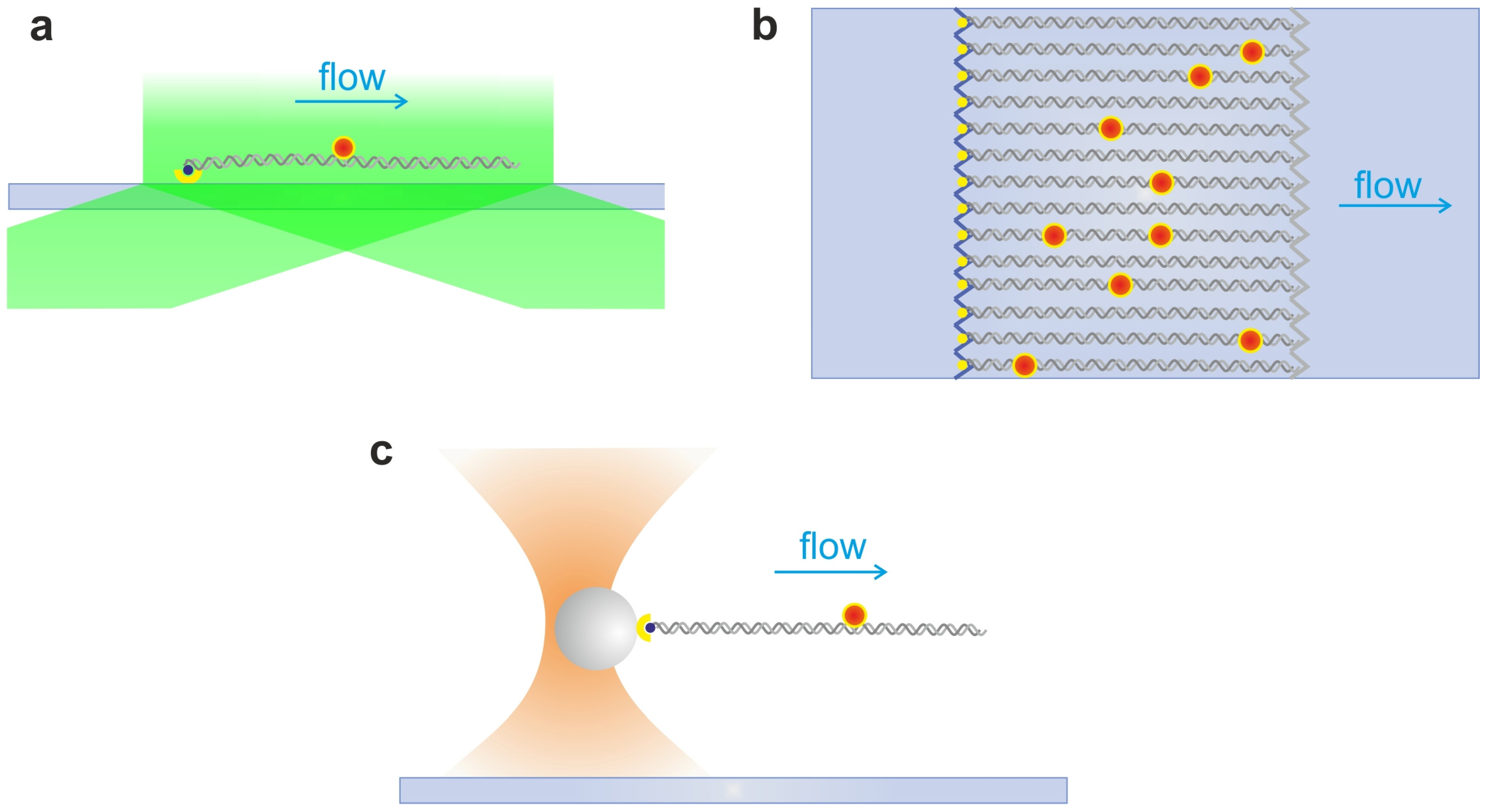
2.5. Single Optical Tweezers and Flow Extended DNA
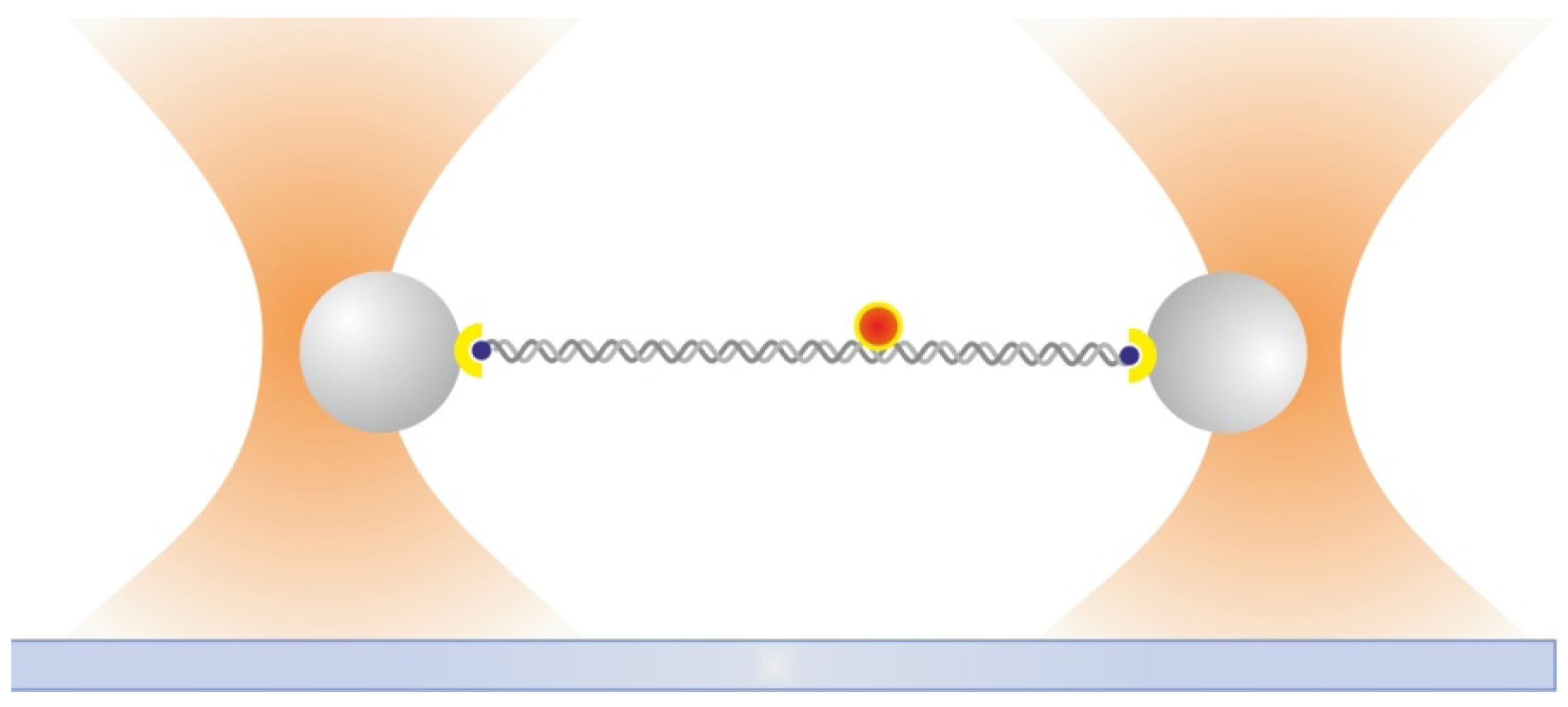
2.6. Dual Optical Tweezers Assays
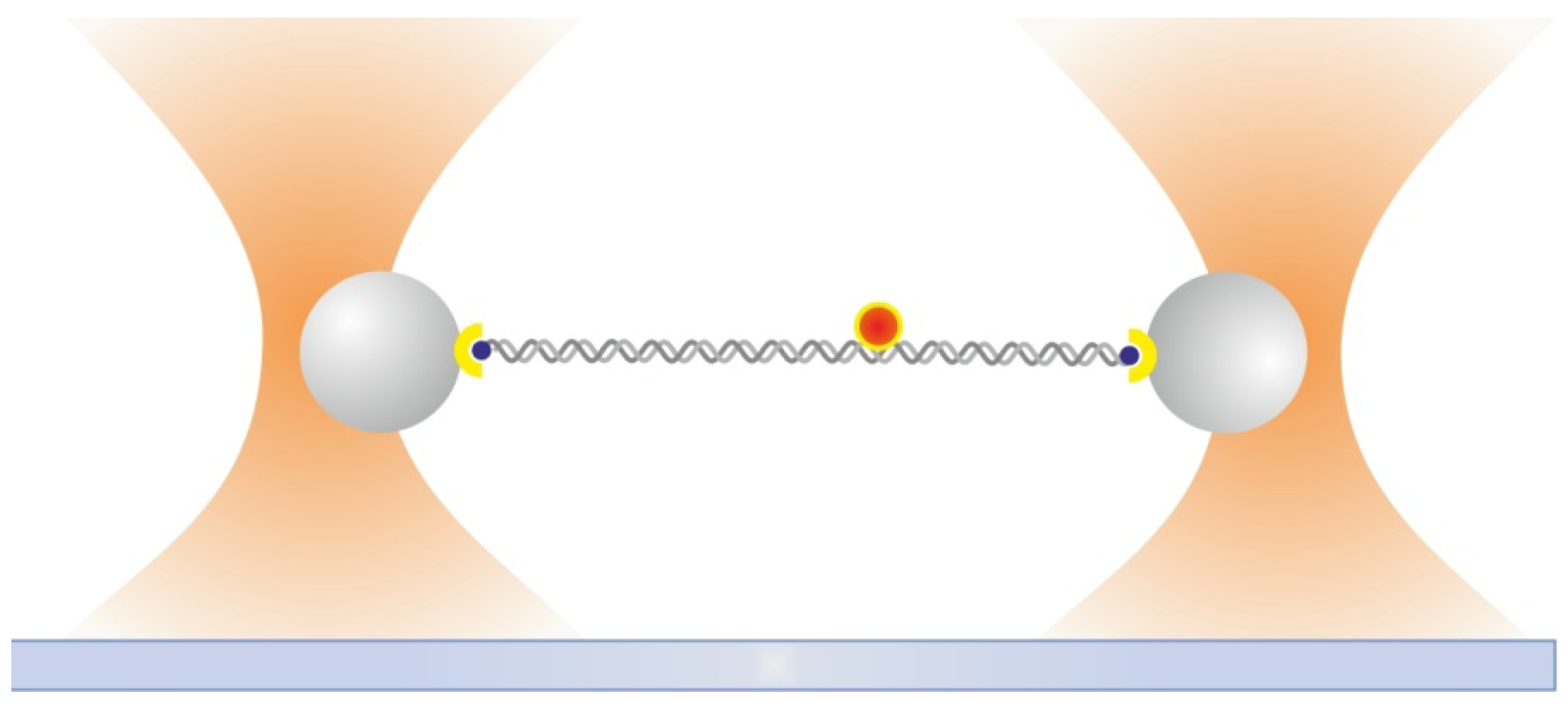
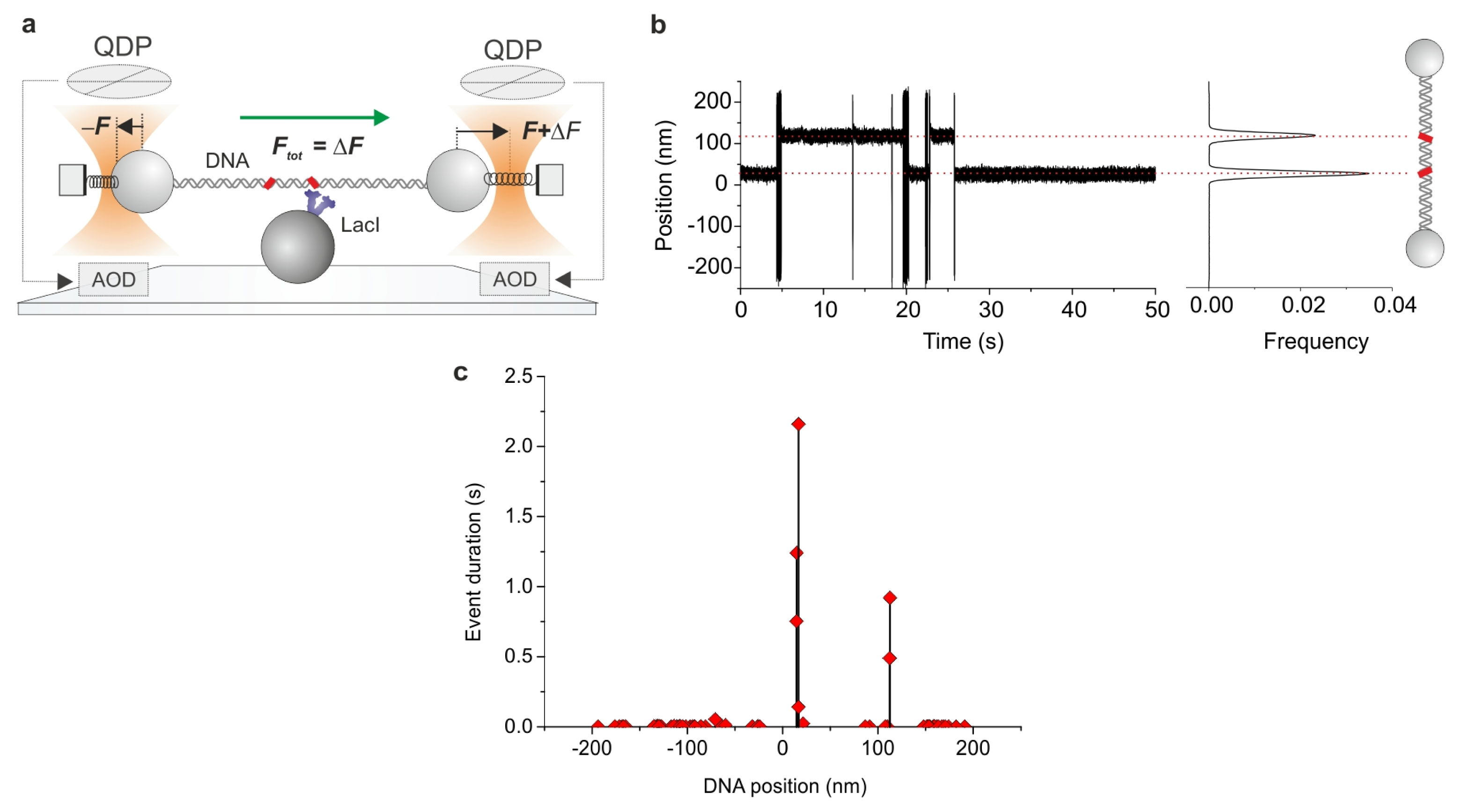
2.7. Combining Dual Optical Tweezers and Single-Molecule Fluorescence Microscopy
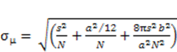
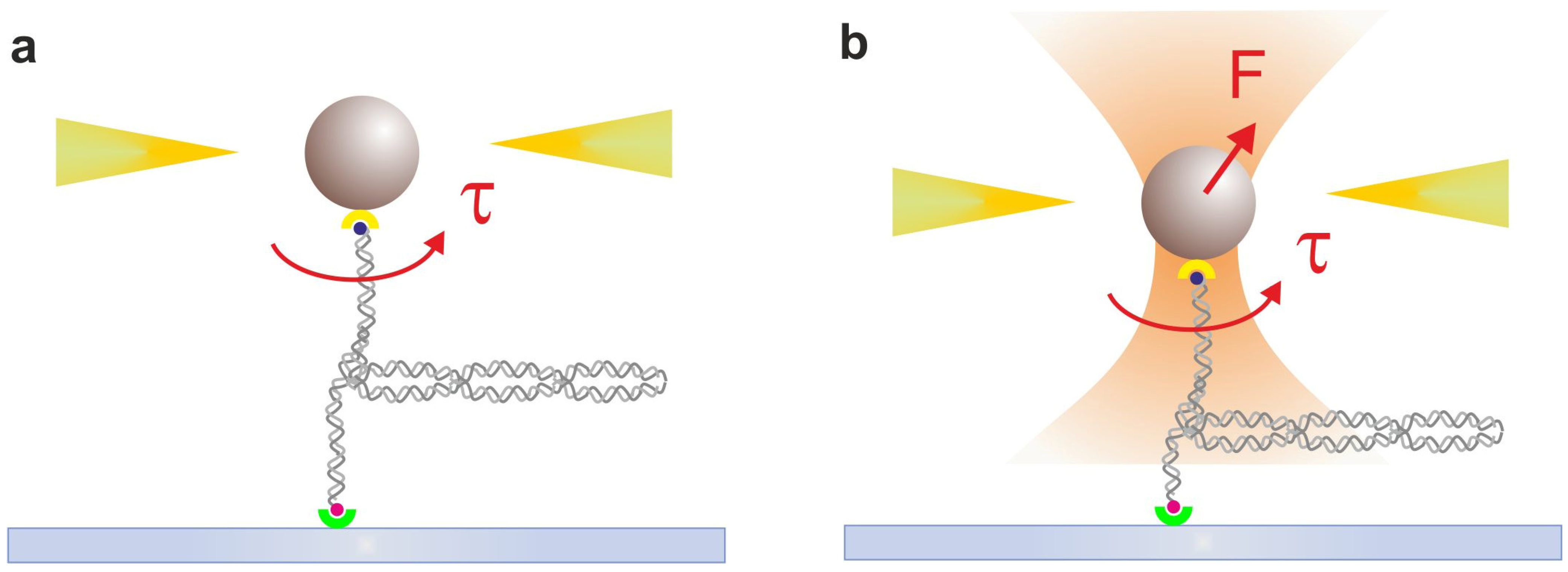
2.8. Ultrafast Force-Clamp Spectroscopy
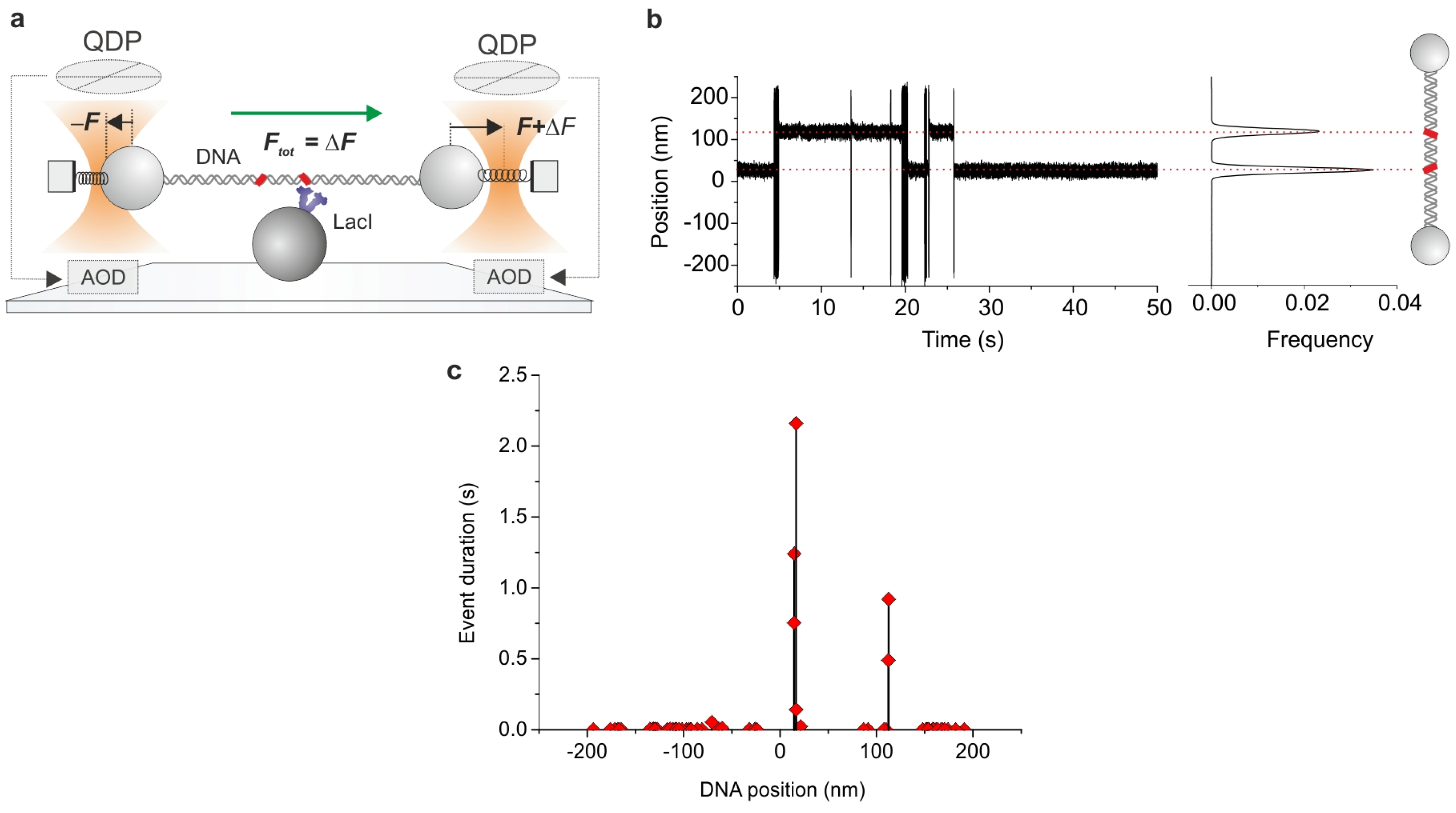
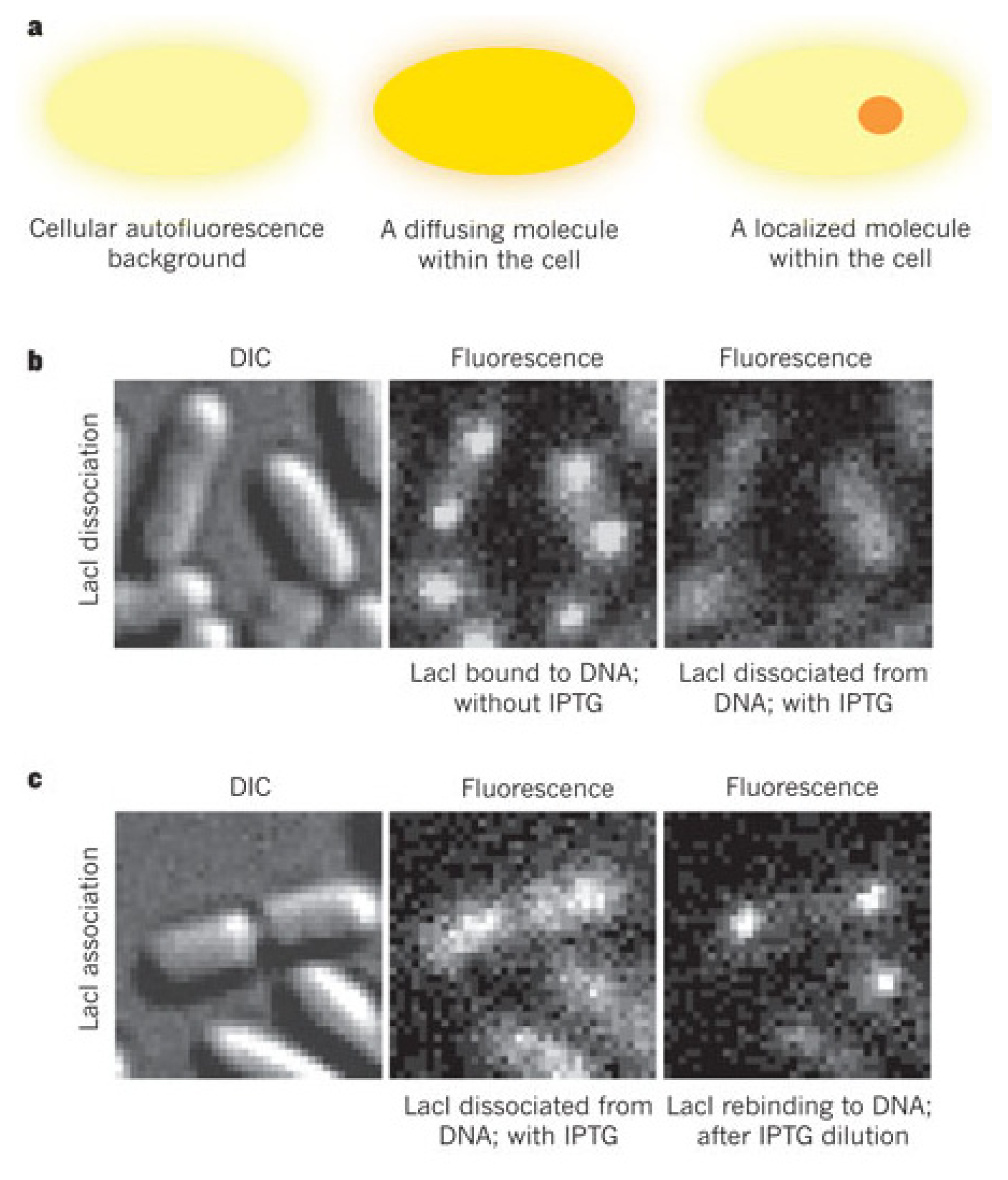
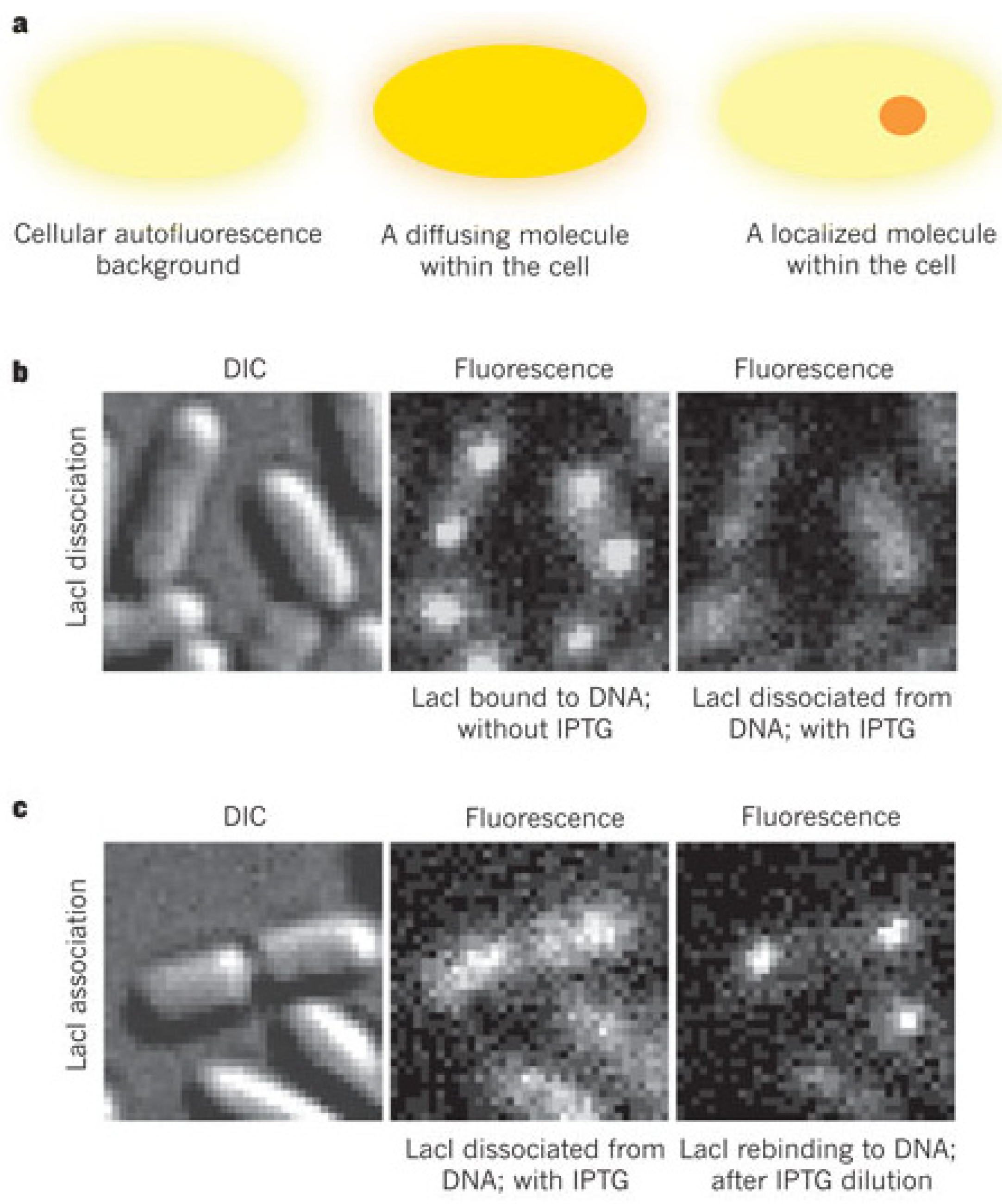
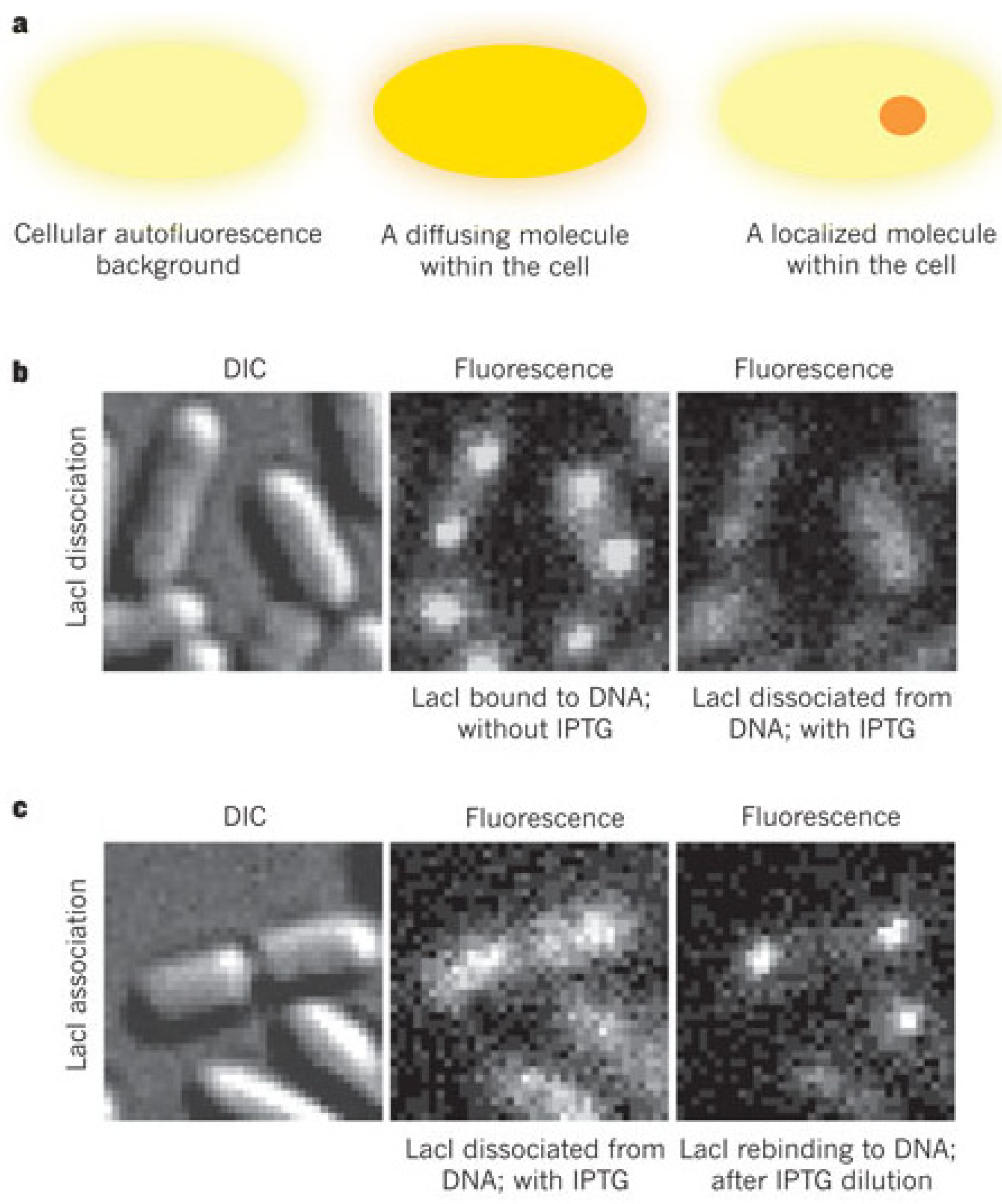
3. Probing Facilitated Diffusion and Protein-DNA Interaction Dynamics In Vivo
4. Conclusions
Acknowledgements
Conflict of Interest
References
- Furini, S.; Domene, C.; Cavalcanti, S. Insights into the sliding movement of the lac repressor nonspecifically bound to DNA. J. Phys. Chem. B 2010, 114, 2238–2245. [Google Scholar] [CrossRef]
- Roberts, R.J.; Vincze, T.; Posfai, J.; Macelis, D. REBASE—Enzymes and genes for DNA restriction and modification. Nucleic Acids Res. 2007, 35, D269–D270. [Google Scholar] [CrossRef]
- Von Hippel, P.H.; Berg, O.G. Facilitated target location in biological systems. J. Biol. Chem. 1989, 264, 675–678. [Google Scholar]
- Gilbert, W.; Muller-Hill, B. Isolation of the lac repressor. Proc. Natl. Acad. Sci. USA 1966, 56, 1891–1898. [Google Scholar] [CrossRef]
- Xie, X.S.; Choi, P.J.; Li, G.W.; Lee, N.K.; Lia, G. Single-molecule approach to molecular biology in living bacterial cells. Annu. Rev. Biophys. 2008, 37, 417–444. [Google Scholar] [CrossRef]
- Halford, S.E.; Marko, J.F. How do site-specific DNA-binding proteins find their targets? Nucleic Acids Res. 2004, 32, 3040–3052. [Google Scholar] [CrossRef]
- Droge, P.; Muller-Hill, B. High local protein concentrations at promoters: Strategies in prokaryotic and eukaryotic cells. Bioessays 2001, 23, 179–183. [Google Scholar] [CrossRef]
- Normanno, D.; Dahan, M.; Darzacq, X. Intra-nuclear mobility and target search mechanisms of transcription factors: A single-molecule perspective on gene expression. Biochim. Biophys. Acta 2012, 1819, 482–493. [Google Scholar] [CrossRef]
- Jacob, F.; Monod, J. Genetic regulatory mechanisms in the synthesis of proteins. J. Mol. Biol. 1961, 3, 318–356. [Google Scholar] [CrossRef]
- Riggs, A.D.; Bourgeois, S.; Cohn, M. The lac repressor-operator interaction. 3. Kinetic studies. J. Mol. Biol. 1970, 53, 401–417. [Google Scholar] [CrossRef]
- Schreiber, G. Kinetic studies of protein-protein interactions. Curr. Opin. Struct. Biol. 2002, 12, 41–47. [Google Scholar] [CrossRef]
- Richter, P.H.; Eigen, M. Diffusion controlled reaction rates in spheroidal geometry. Application to repressor-operator association and membrane bound enzymes. Biophys. Chem. 1974, 2, 255–263. [Google Scholar]
- Berg, O.G.; von Hippel, P.H. Diffusion-controlled macromolecular interactions. Annu. Rev. Biophys. Biophys. Chem. 1985, 14, 131–160. [Google Scholar] [CrossRef]
- Mirny, L.; Slutsky, M.; Wunderlich, Z.; Tafvizi, A.; Leith, J.; Kosmrlj, A. How a protein searches for its site on DNA: The mechanism of facilitated diffusion. J. Phys. A 2009, 42, 434013. [Google Scholar] [CrossRef]
- Halford, S.E. An end to 40 years of mistakes in DNA-protein association kinetics? Biochem. Soc. Trans. 2009, 37, 343–348. [Google Scholar] [CrossRef]
- Berg, O.G.; Ehrenberg, M. Association kinetics with coupled three- and one-dimensional diffusion. Chain-length dependence of the association rate of specific DNA sites. Biophys. Chem. 1982, 15, 41–51. [Google Scholar]
- Berg, O.G.; Winter, R.B.; von Hippel, P.H. Diffusion-driven mechanisms of protein translocation on nucleic acids. 1. Models and theory. Biochemistry 1981, 20, 6929–6948. [Google Scholar] [CrossRef]
- Winter, R.B.; Berg, O.G.; von Hippel, P.H. Diffusion-driven mechanisms of protein translocation on nucleic acids. 3. The Escherichia coli lac repressor-operator interaction: Kinetic measurements and conclusions. Biochemistry 1981, 20, 6961–6977. [Google Scholar]
- Berg, O.G.; Blomberg, C. Association kinetics with coupled diffusion III. Ionic-strength dependence of the lac repressor-operator association. Biophys. Chem. 1978, 8, 271–280. [Google Scholar]
- Berg, O.G.; Blomberg, C. Association kinetics with coupled diffusion. An extension to coiled-chain macromolecules applied to the lac repressor-operator system. Biophys. Chem. 1977, 7, 33–39. [Google Scholar]
- Berg, O.G.; Blomberg, C. Association kinetics with coupled diffusional flows. Special application to the lac repressor-operator system. Biophys. Chem. 1976, 4, 367–381. [Google Scholar]
- Hammar, P.; Leroy, P.; Mahmutovic, A.; Marklund, E.G.; Berg, O.G.; Elf, J. The lac repressor displays facilitated diffusion in living cells. Science 2012, 336, 1595–1598. [Google Scholar] [CrossRef]
- Slutsky, M.; Mirny, L.A. Kinetics of protein-DNA interaction: Facilitated target location in sequence-dependent potential. Biophys. J. 2004, 87, 4021–4035. [Google Scholar] [CrossRef]
- Wang, Y.M.; Austin, R.H.; Cox, E.C. Single molecule measurements of repressor protein 1D diffusion on DNA. Phys. Rev. Lett. 2006, 97, 048302. [Google Scholar] [CrossRef]
- Fickert, R.; Muller-Hill, B. How Lac repressor finds lac operator in vitro. J. Mol. Biol. 1992, 226, 59–68. [Google Scholar] [CrossRef]
- Halford, S.E.; Welsh, A.J.; Szczelkun, M.D. Enzyme-mediated DNA looping. Annu. Rev. Biophys. Biomol. Struct. 2004, 33, 1–24. [Google Scholar] [CrossRef]
- Gorman, J.; Greene, E.C. Visualizing one-dimensional diffusion of proteins along DNA. Nat. Struct. Mol. Biol. 2008, 15, 768–774. [Google Scholar] [CrossRef]
- Bustamante, C. In singulo biochemistry: When less is more. Annu. Rev. Biochem. 2008, 77, 45–50. [Google Scholar] [CrossRef]
- Tinoco, I., Jr.; Gonzalez, R.L., Jr. Biological mechanisms, one molecule at a time. Genes Dev. 2011, 25, 1205–1231. [Google Scholar] [CrossRef]
- Capitanio, M.; Vanzi, F.; Broggio, C.; Cicchi, R.; Normanno, D.; Romano, G.; Sacconi, L.; Pavone, F.S. Exploring molecular motors and switches at the single-molecule level. Microsc. Res. Tech. 2004, 65, 194–204. [Google Scholar] [CrossRef]
- Toprak, E.; Selvin, P.R. New fluorescent tools for watching nanometer-scale conformational changes of single molecules. Annu. Rev. Biophys. Biomol. Struct. 2007, 36, 349–369. [Google Scholar] [CrossRef]
- Schafer, D.A.; Gelles, J.; Sheetz, M.P.; Landick, R. Transcription by single molecules of RNA polymerase observed by light microscopy. Nature 1991, 352, 444–448. [Google Scholar] [CrossRef]
- Yin, H.; Landick, R.; Gelles, J. Tethered particle motion method for studying transcript elongation by a single RNA polymerase molecule. Biophys. J. 1994, 67, 2468–2478. [Google Scholar] [CrossRef]
- Matthews, K.S. DNA looping. Microbiol. Rev. 1992, 56, 123–136. [Google Scholar]
- Pouget, N.; Dennis, C.; Turlan, C.; Grigoriev, M.; Chandler, M.; Salome, L. Single-particle tracking for DNA tether length monitoring. Nucleic Acids Res. 2004, 32, e73. [Google Scholar] [CrossRef]
- Blumberg, S.; Gajraj, A.; Pennington, M.W.; Meiners, J.C. Three-dimensional characterization of tethered microspheres by total internal reflection fluorescence microscopy. Biophys. J. 2005, 89, 1272–1281. [Google Scholar] [CrossRef]
- Dietrich, H.R.C.; Rieger, B.; Wiertz, F.G.M.; de Groote, F.H.; Heering, H.A.; Young, I.T.; Garini, Y. Tethered particle motion mediated by scattering from gold nanoparticles and darkfield microscopy. J. Nanophotonics 2009, 3. [Google Scholar] [CrossRef]
- Vanzi, F.; Broggio, C.; Sacconi, L.; Pavone, F.S. Lac repressor hinge flexibility and DNA looping: Single molecule kinetics by tethered particle motion. Nucleic Acids Res. 2006, 34, 3409–3420. [Google Scholar] [CrossRef]
- Van den Broek, B.; Vanzi, F.; Normanno, D.; Pavone, F.S.; Wuite, G.J. Real-time observation of DNA looping dynamics of Type IIE restriction enzymes NaeI and NarI. Nucleic Acids Res. 2006, 34, 167–174. [Google Scholar] [CrossRef]
- Finzi, L.; Gelles, J. Measurement of lactose repressor-mediated loop formation and breakdown in single DNA molecules. Science 1995, 267, 378–380. [Google Scholar]
- Rutkauskas, D.; Zhan, H.L.; Matthews, K.S.; Pavone, F.S.; Vanzi, F. Tetramer opening in LacI-mediated DNA looping. Proc. Natl. Acad. Sci. USA 2009, 106, 16627–16632. [Google Scholar]
- Han, L.; Garcia, H.G.; Blumberg, S.; Towles, K.B.; Beausang, J.F.; Nelson, P.C.; Phillips, R. Concentration and length dependence of DNA looping in transcriptional regulation. PLoS One 2009, 4, e5621. [Google Scholar]
- Wong, O.K.; Guthold, M.; Erie, D.A.; Gelles, J. Interconvertible lac repressor-DNA loops revealed by single-molecule experiments. PLoS Biol. 2008, 6, e232. [Google Scholar] [CrossRef]
- Johnson, S.; Linden, M.; Phillips, R. Sequence dependence of transcription factor-mediated DNA looping. Nucleic Acids Res. 2012, 40, 7728–7738. [Google Scholar] [CrossRef]
- Chen, Y.F.; Milstein, J.N.; Meiners, J.C. Protein-mediated DNA loop formation and breakdown in a fluctuating environment. Phys. Rev. Lett. 2010, 104, 258103. [Google Scholar] [CrossRef]
- Chen, Y.F.; Milstein, J.N.; Meiners, J.C. Femtonewton entropic forces can control the formation of protein-mediated DNA loops. Phys. Rev. Lett. 2010, 104, 048301. [Google Scholar] [CrossRef]
- Pouget, N.; Turlan, C.; Destainville, N.; Salome, L.; Chandler, M. IS911 transpososome assembly as analysed by tethered particle motion. Nucleic Acids Res. 2006, 34, 4313–4323. [Google Scholar] [CrossRef]
- Zurla, C.; Manzo, C.; Dunlap, D.; Lewis, D.E.; Adhya, S.; Finzi, L. Direct demonstration and quantification of long-range DNA looping by the lambda bacteriophage repressor. Nucleic Acids Res. 2009, 37, 2789–2795. [Google Scholar] [CrossRef]
- Manzo, C.; Zurla, C.; Dunlap, D.D.; Finzi, L. The effect of nonspecific binding of lambda repressor on DNA looping dynamics. Biophys. J. 2012, 103, 1753–1761. [Google Scholar] [CrossRef]
- Laurens, N.; Rusling, D.A.; Pernstich, C.; Brouwer, I.; Halford, S.E.; Wuite, G.J. DNA looping by FokI: The impact of twisting and bending rigidity on protein-induced looping dynamics. Nucleic Acids Res. 2012, 40, 4988–4997. [Google Scholar] [CrossRef]
- Tolic-Norrelykke, S.F.; Rasmussen, M.B.; Pavone, F.S.; Berg-Sorensen, K.; Oddershede, L.B. Stepwise bending of DNA by a single TATA-box binding protein. Biophys. J. 2006, 90, 3694–3703. [Google Scholar] [CrossRef]
- Mumm, J.P.; Landy, A.; Gelles, J. Viewing single lambda site-specific recombination events from start to finish. EMBO J. 2006, 25, 4586–4595. [Google Scholar] [CrossRef]
- Fan, H.F. Real-time single-molecule tethered particle motion experiments reveal the kinetics and mechanisms of Cre-mediated site-specific recombination. Nucleic Acids Res. 2012, 40, 6208–6222. [Google Scholar] [CrossRef]
- Fan, H.F.; Li, H.W. Studying RecBCD helicase translocation along chi-DNA using tethered particle motion with a stretching force. Biophys. J. 2009, 96, 1875–1883. [Google Scholar] [CrossRef]
- Plenat, T.; Tardin, C.; Rousseau, P.; Salome, L. High-throughput single-molecule analysis of DNA-protein interactions by tethered particle motion. Nucleic Acids Res. 2012, 40, e89. [Google Scholar] [CrossRef]
- Manghi, M.; Tardin, C.; Baglio, J.; Rousseau, P.; Salome, L.; Destainville, N. Probing DNA conformational changes with high temporal resolution by tethered particle motion. Phys. Boil. 2010, 7, 046003. [Google Scholar] [CrossRef]
- Beausang, J.F.; Zurla, C.; Manzo, C.; Dunlap, D.; Finzi, L.; Nelson, P.C. DNA looping kinetics analyzed using diffusive hidden Markov model. Biophys. J. 2007, 92, L64–L66. [Google Scholar] [CrossRef]
- Manzo, C.; Finzi, L. Quantitative analysis of DNA-looping kinetics from tethered particle motion experiments. Methods Enzymol. 2010, 475, 199–220. [Google Scholar] [CrossRef]
- Vanzi, F.; Sacconi, L.; Pavone, F.S. Analysis of kinetics in noisy systems: Application to single molecule tethered particle motion. Biophys. J. 2007, 93, 21–36. [Google Scholar] [CrossRef]
- Segall, D.E.; Nelson, P.C.; Phillips, R. Volume-exclusion effects in tethered-particle experiments: Bead size matters. Phys. Rev. Lett. 2006, 96, 088306:1–088306:4. [Google Scholar]
- Milstein, J.N.; Chen, Y.F.; Meiners, J.C. Bead size effects on protein-mediated DNA looping in tethered-particle motion experiments. Biopolymers 2011, 95, 144–150. [Google Scholar] [CrossRef]
- Ashkin, A.; Dziedzic, J.M.; Bjorkholm, J.E.; Chu, S. Observation of a single-beam gradient force optical trap for dielectric particles. Opt. Lett. 1986, 11, 288–290. [Google Scholar] [CrossRef]
- Grier, D.G. A revolution in optical manipulation. Nature 2003, 424, 810–816. [Google Scholar] [CrossRef]
- Svoboda, K.; Block, S.M. Biological applications of optical forces. Annu. Rev. Biophys. Biomol. Struct. 1994, 23, 247–285. [Google Scholar] [CrossRef]
- Wallin, A.E.; Ojala, H.; Ziedaite, G.; Degerth, L.; Bamford, D.; Haeggström, E. High-resolution optical tweezers for investigating DNA-binding/translocating molecular motors. Proc. SPIE 2009, 740007. [Google Scholar] [CrossRef]
- Wang, M.D.; Schnitzer, M.J.; Yin, H.; Landick, R.; Gelles, J.; Block, S.M. Force and velocity measured for single molecules of RNA polymerase. Science 1998, 282, 902–907. [Google Scholar] [CrossRef]
- Dalal, R.V.; Larson, M.H.; Neuman, K.C.; Gelles, J.; Landick, R.; Block, S.M. Pulling on the nascent RNA during transcription does not alter kinetics of elongation or ubiquitous pausing. Mol. Cell 2006, 23, 231–239. [Google Scholar] [CrossRef]
- Sakata-Sogawa, K.; Shimamoto, N. RNA polymerase can track a DNA groove during promoter search. Proc. Natl. Acad. Sci. USA 2004, 101, 14731–14735. [Google Scholar] [CrossRef]
- Koch, S.J.; Wang, M.D. Dynamic force spectroscopy of protein-DNA interactions by unzipping DNA. Phys. Rev. Lett. 2003, 91, 028103. [Google Scholar] [CrossRef]
- Essevaz-Roulet, B.; Bockelmann, U.; Heslot, F. Mechanical separation of the complementary strands of DNA. Proc. Natl. Acad. Sci. USA 1997, 94, 11935–11940. [Google Scholar] [CrossRef]
- Bockelmann, U.; EssevazRoulet, B.; Heslot, F. Molecular stick-slip motion revealed by opening DNA with piconewton forces. Phys. Rev. Lett. 1997, 79, 4489–4492. [Google Scholar] [CrossRef]
- Koch, S.J.; Shundrovsky, A.; Jantzen, B.C.; Wang, M.D. Probing protein-DNA interactions by unzipping a single DNA double helix. Biophys. J. 2002, 83, 1098–1105. [Google Scholar] [CrossRef]
- Jiang, J.; Bai, L.; Surtees, J.A.; Gemici, Z.; Wang, M.D.; Alani, E. Detection of high-affinity and sliding clamp modes for MSH2-MSH6 by single-molecule unzipping force analysis. Mol. Cell 2005, 20, 771–781. [Google Scholar] [CrossRef]
- Hall, M.A.; Shundrovsky, A.; Bai, L.; Fulbright, R.M.; Lis, J.T.; Wang, M.D. High-resolution dynamic mapping of histone-DNA interactions in a nucleosome. Nat. Struct. Mol. Biol. 2009, 16, 124–129. [Google Scholar] [CrossRef]
- Johnson, D.S.; Bai, L.; Smith, B.Y.; Patel, S.S.; Wang, M.D. Single-molecule studies reveal dynamics of DNA unwinding by the ring-shaped T7 helicase. Cell 2007, 129, 1299–1309. [Google Scholar] [CrossRef]
- Sun, B.; Johnson, D.S.; Patel, G.; Smith, B.Y.; Pandey, M.; Patel, S.S.; Wang, M.D. ATP-induced helicase slippage reveals highly coordinated subunits. Nature 2011, 478, 132–135. [Google Scholar]
- Perkins, T.T.; Dalal, R.V.; Mitsis, P.G.; Block, S.M. Sequence-dependent pausing of single lambda exonuclease molecules. Science 2003, 301, 1914–1918. [Google Scholar] [CrossRef]
- Perkins, T.T.; Li, H.W.; Dalal, R.V.; Gelles, J.; Block, S.M. Forward and reverse motion of single RecBCD molecules on DNA. Biophys. J. 2004, 86, 1640–1648. [Google Scholar] [CrossRef]
- Brower-Toland, B.D.; Smith, C.L.; Yeh, R.C.; Lis, J.T.; Peterson, C.L.; Wang, M.D. Mechanical disruption of individual nucleosomes reveals a reversible multistage release of DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 1960–1965. [Google Scholar]
- Capitanio, M.; Cicchi, R.; Saverio Pavone, F. Continuous and time-shared multiple optical tweezers for the study of single motor proteins. Opt. Lasers Eng. 2007, 45, 450–457. [Google Scholar] [CrossRef]
- Visscher, K.; Gross, S.P.; Block, S.M. Construction of multiple-beam optical traps with nanometer-resolution position sensing. IEEE J. Sel. Top. Quantum Electron. 1996, 2, 1066–1076. [Google Scholar] [CrossRef]
- Capitanio, M.; Cicchi, R.; Pavone, F.S. Position control and optical manipulation for nanotechnology applications. Eur. Phys. J. B 2005, 46, 1–8. [Google Scholar] [CrossRef]
- Carter, A.R.; King, G.M.; Ulrich, T.A.; Halsey, W.; Alchenberger, D.; Perkins, T.T. Stabilization of an optical microscope to 0.1 nm in three dimensions. Appl. Opt. 2007, 46, 421–427. [Google Scholar]
- Capitanio, M.; Romano, G.; Ballerini, R.; Giuntini, M.; Pavone, F.S.; Dunlap, D.; Finzi, L. Calibration of optical tweezers with differential interference contrast signals. Rev. Sci. Instrum. 2002, 73, 1687–1696. [Google Scholar] [CrossRef]
- Tolic-Norrelykke, S.F.; Schaffer, E.; Howard, J.; Pavone, F.S.; Julicher, F.; Flyvbjerg, H. Calibration of optical tweezers with positional detection in the back focal plane. Rev. Sci. Instrum. 2006, 77, 103101–103111. [Google Scholar] [CrossRef]
- Moffitt, J.R.; Chemla, Y.R.; Smith, S.B.; Bustamante, C. Recent advances in optical tweezers. Annu. Rev. Biochem. 2008, 77, 205–228. [Google Scholar] [CrossRef]
- Neuman, K.C.; Block, S.M. Optical trapping. Rev. Sci. Instrum. 2004, 75, 2787–2809. [Google Scholar] [CrossRef]
- Bustamante, C.; Chemla, Y.R.; Moffitt, J.R. High-resolution dual-trap optical tweezers with differential detection: An introduction. Cold Spring Harb. Protoc. 2009, 2009. [Google Scholar] [CrossRef]
- Abbondanzieri, E.A.; Greenleaf, W.J.; Shaevitz, J.W.; Landick, R.; Block, S.M. Direct observation of base-pair stepping by RNA polymerase. Nature 2005, 438, 460–465. [Google Scholar] [CrossRef]
- Cheng, W.; Arunajadai, S.G.; Moffitt, J.R.; Tinoco, I., Jr.; Bustamante, C. Single-base pair unwinding and asynchronous RNA release by the hepatitis C virus NS3 helicase. Science 2011, 333, 1746–1749. [Google Scholar] [CrossRef]
- Capitanio, M.; Canepari, M.; Maffei, M.; Beneventi, D.; Monico, C.; Vanzi, F.; Bottinelli, R.; Pavone, F.S. Ultrafast force-clamp spectroscopy of single molecules reveals load dependence of myosin working stroke. Nat. Methods 2012, 9, 1013–1019. [Google Scholar] [CrossRef]
- Deufel, C.; Forth, S.; Simmons, C.R.; Dejgosha, S.; Wang, M.D. Nanofabricated quartz cylinders for angular trapping: DNA supercoiling torque detection. Nat. Methods 2007, 4, 223–225. [Google Scholar] [CrossRef]
- La Porta, A.; Wang, M.D. Optical torque wrench: Angular trapping, rotation, and torque detection of quartz microparticles. Phys. Rev. Lett. 2004, 92, 190801. [Google Scholar] [CrossRef]
- Pedaci, F.; Huang, Z.; van Oene, M.; Dekker, N.H. Calibration of the optical torque wrench. Opt. Express 2012, 20, 3787–3802. [Google Scholar]
- Bryant, Z.; Oberstrass, F.C.; Basu, A. Recent developments in single-molecule DNA mechanics. Curr. Opin. Struct. Biol. 2012, 22, 304–312. [Google Scholar] [CrossRef]
- Charvin, G.; Strick, T.R.; Bensimon, D.; Croquette, V. Tracking topoisomerase activity at the single-molecule level. Annu. Rev. Biophys. Biomol. Struct. 2005, 34, 201–219. [Google Scholar] [CrossRef]
- Koster, D.A.; Crut, A.; Shuman, S.; Bjornsti, M.A.; Dekker, N.H. Cellular strategies for regulating DNA supercoiling: A single-molecule perspective. Cell 2010, 142, 519–530. [Google Scholar] [CrossRef]
- Revyakin, A.; Ebright, R.H.; Strick, T.R. Promoter unwinding and promoter clearance by RNA polymerase: Detection by single-molecule DNA nanomanipulation. Proc. Natl. Acad. Sci. USA 2004, 101, 4776–4780. [Google Scholar] [CrossRef]
- Howan, K.; Smith, A.J.; Westblade, L.F.; Joly, N.; Grange, W.; Zorman, S.; Darst, S.A.; Savery, N.J.; Strick, T.R. Initiation of transcription-coupled repair characterized at single-molecule resolution. Nature 2012, 490, 431–434. [Google Scholar]
- Seidel, R.; van Noort, J.; van der Scheer, C.; Bloom, J.G.; Dekker, N.H.; Dutta, C.F.; Blundell, A.; Robinson, T.; Firman, K.; Dekker, C. Real-time observation of DNA translocation by the type I restriction modification enzyme EcoR124I. Nat. Struct. Mol. Biol. 2004, 11, 838–843. [Google Scholar] [CrossRef]
- Normanno, D.; Vanzi, F.; Pavone, F.S. Single-molecule manipulation reveals supercoiling-dependent modulation of lac repressor-mediated DNA looping. Nucleic Acids Res. 2008, 36, 2505–2513. [Google Scholar] [CrossRef]
- Aussel, L.; Barre, F.X.; Aroyo, M.; Stasiak, A.; Stasiak, A.Z.; Sherratt, D. FtsK is a DNA motor protein that activates chromosome dimer resolution by switching the catalytic state of the XerC and XerD recombinases. Cell 2002, 108, 195–205. [Google Scholar] [CrossRef]
- Saleh, O.A.; Bigot, S.; Barre, F.X.; Allemand, J.F. Analysis of DNA supercoil induction by FtsK indicates translocation without groove-tracking. Nat. Struct. Mol. Biol. 2005, 12, 436–440. [Google Scholar] [CrossRef]
- Liu, L.F.; Wang, J.C. Supercoiling of the DNA template during transcription. Proc. Natl. Acad. Sci. USA 1987, 84, 7024–7027. [Google Scholar] [CrossRef]
- Sacconi, L.; Romano, G.; Ballerini, R.; Capitanio, M.; De Pas, M.; Giuntini, M.; Dunlap, D.; Finzi, L.; Pavone, F.S. Three-dimensional magneto-optic trap for micro-object manipulation. Opt. Lett. 2001, 26, 1359–1361. [Google Scholar] [CrossRef]
- Romano, G.; Sacconi, L.; Capitanio, M.; Pavone, F.S. Force and torque measurements using magnetic micro beads for single molecule biophysics. Opt. Commun. 2003, 215, 323–331. [Google Scholar] [CrossRef]
- Normanno, D.; Capitanio, M.; Pavone, F.S. Spin absorption, windmill, and magneto-optic effects in optical angular momentum transfer. Phys. Rev. A 2004, 70, 053829. [Google Scholar] [CrossRef]
- Capitanio, M.; Normanno, D.; Pavone, F.S. High-precision measurements of light-induced torque on absorbing microspheres. Opt. Lett. 2004, 29, 2231–2233. [Google Scholar] [CrossRef]
- Lipfert, J.; Kerssemakers, J.W.; Jager, T.; Dekker, N.H. Magnetic torque tweezers: Measuring torsional stiffness in DNA and RecA-DNA filaments. Nat. Methods 2010, 7, 977–980. [Google Scholar] [CrossRef]
- Celedon, A.; Nodelman, I.M.; Wildt, B.; Dewan, R.; Searson, P.; Wirtz, D.; Bowman, G.D.; Sun, S.X. Magnetic tweezers measurement of single molecule torque. Nano Lett. 2009, 9, 1720–1725. [Google Scholar]
- Kruithof, M.; Chien, F.T.; Routh, A.; Logie, C.; Rhodes, D.; van Noort, J. Single-molecule force spectroscopy reveals a highly compliant helical folding for the 30-nm chromatin fiber. Nat. Struct. Mol. Biol. 2009, 16, 534–540. [Google Scholar] [CrossRef]
- De Vlaminck, I.; Dekker, C. Recent advances in magnetic tweezers. Annu. Rev. Biophys. 2012, 41, 453–472. [Google Scholar] [CrossRef]
- Gosse, C.; Croquette, V. Magnetic tweezers: Micromanipulation and force measurement at the molecular level. Biophys. J. 2002, 82, 3314–3329. [Google Scholar] [CrossRef]
- Seol, Y.; Neuman, K.C. Magnetic tweezers for single-molecule manipulation. Methods Mol. Biol. 2011, 783, 265–293. [Google Scholar] [CrossRef]
- Bensimon, A.; Simon, A.; Chiffaudel, A.; Croquette, V.; Heslot, F.; Bensimon, D. Alignment and sensitive detection of DNA by a moving interface. Science 1994, 265, 2096–2098. [Google Scholar]
- Kim, J.H.; Larson, R.G. Single-molecule analysis of 1D diffusion and transcription elongation of T7 RNA polymerase along individual stretched DNA molecules. Nucleic Acids Res. 2007, 35, 3848–3858. [Google Scholar] [CrossRef]
- Lebofsky, R.; Bensimon, A. Single DNA molecule analysis: Applications of molecular combing. Briefings Funct. Genomics Proteomics 2003, 1, 385–396. [Google Scholar] [CrossRef]
- Fazio, T.; Visnapuu, M.L.; Wind, S.; Greene, E.C. DNA curtains and nanoscale curtain rods: High-throughput tools for single molecule imaging. Langmuir 2008, 24, 10524–10531. [Google Scholar] [CrossRef]
- Wang, Y.M.; Tegenfeldt, J.O.; Reisner, W.; Riehn, R.; Guan, X.J.; Guo, L.; Golding, I.; Cox, E.C.; Sturm, J.; Austin, R.H. Single-molecule studies of repressor-DNA interactions show long-range interactions. Proc. Natl. Acad. Sci. USA 2005, 102, 9796–9801. [Google Scholar]
- Elf, J.; Li, G.W.; Xie, X.S. Probing transcription factor dynamics at the single-molecule level in a living cell. Science 2007, 316, 1191–1194. [Google Scholar] [CrossRef]
- Kabata, H.; Kurosawa, O.; Arai, I.; Washizu, M.; Margarson, S.A.; Glass, R.E.; Shimamoto, N. Visualization of single molecules of RNA polymerase sliding along DNA. Science 1993, 262, 1561–1563. [Google Scholar]
- Blainey, P.C.; van Oijen, A.M.; Banerjee, A.; Verdine, G.L.; Xie, X.S. A base-excision DNA-repair protein finds intrahelical lesion bases by fast sliding in contact with DNA. Proc. Natl. Acad. Sci. USA 2006, 103, 5752–5757. [Google Scholar]
- Gorman, J.; Chowdhury, A.; Surtees, J.A.; Shimada, J.; Reichman, D.R.; Alani, E.; Greene, E.C. Dynamic basis for one-dimensional DNA scanning by the mismatch repair complex Msh2-Msh6. Mol. Cell 2007, 28, 359–370. [Google Scholar] [CrossRef]
- Tafvizi, A.; Huang, F.; Leith, J.S.; Fersht, A.R.; Mirny, L.A.; van Oijen, A.M. Tumor suppressor p53 slides on DNA with low friction and high stability. Biophys. J. 2008, 95, L01–L03. [Google Scholar] [CrossRef]
- Tafvizi, A.; Huang, F.; Fersht, A.R.; Mirny, L.A.; van Oijen, A.M. A single-molecule characterization of p53 search on DNA. Proc. Natl. Acad. Sci. USA 2011, 108, 563–568. [Google Scholar]
- Graneli, A.; Yeykal, C.C.; Robertson, R.B.; Greene, E.C. Long-distance lateral diffusion of human Rad51 on double-stranded DNA. Proc. Natl. Acad. Sci. USA 2006, 103, 1221–1226. [Google Scholar] [CrossRef]
- Bonnet, I.; Biebricher, A.; Porte, P.L.; Loverdo, C.; Benichou, O.; Voituriez, R.; Escude, C.; Wende, W.; Pingoud, A.; Desbiolles, P. Sliding and jumping of single EcoRV restriction enzymes on non-cognate DNA. Nucleic Acids Res. 2008, 36, 4118–4127. [Google Scholar] [CrossRef]
- Schurr, J.M. The one-dimensional diffusion coefficient of proteins absorbed on DNA. Hydrodynamic considerations. Biophys. Chem. 1979, 9, 413–414. [Google Scholar] [CrossRef]
- Blainey, P.C.; Luo, G.; Kou, S.C.; Mangel, W.F.; Verdine, G.L.; Bagchi, B.; Xie, X.S. Nonspecifically bound proteins spin while diffusing along DNA. Nat. Struct. Mol. Biol. 2009, 16, 1224–1229. [Google Scholar] [CrossRef]
- DeSantis, M.C.; Li, J.L.; Wang, Y.M. Protein sliding and hopping kinetics on DNA. Phys. Rev. E 2011, 83, 021907. [Google Scholar] [CrossRef]
- Loverdo, C.; Benichou, O.; Voituriez, R.; Biebricher, A.; Bonnet, I.; Desbiolles, P. Quantifying hopping and jumping in facilitated diffusion of DNA-binding proteins. Phys. Rev. Lett. 2009, 102, 188101. [Google Scholar] [CrossRef]
- Dikic, J.; Menges, C.; Clarke, S.; Kokkinidis, M.; Pingoud, A.; Wende, W.; Desbiolles, P. The rotation-coupled sliding of EcoRV. Nucleic Acids Res. 2012, 40, 4064–4070. [Google Scholar] [CrossRef]
- Gorman, J.; Plys, A.J.; Visnapuu, M.L.; Alani, E.; Greene, E.C. Visualizing one-dimensional diffusion of eukaryotic DNA repair factors along a chromatin lattice. Nat. Struct. Mol. Biol. 2010, 17, 932–938. [Google Scholar] [CrossRef]
- Finkelstein, I.J.; Visnapuu, M.L.; Greene, E.C. Single-molecule imaging reveals mechanisms of protein disruption by a DNA translocase. Nature 2010, 468, 983–987. [Google Scholar] [CrossRef]
- Amitani, I.; Liu, B.A.; Dombrowski, C.C.; Baskin, R.J.; Kowalczykowski, S.C. Watching individual proteins acting on single molecules of DNA. Methods Enzymol. 2010, 472, 261–291. [Google Scholar] [CrossRef]
- Bianco, P.R.; Brewer, L.R.; Corzett, M.; Balhorn, R.; Yeh, Y.; Kowalczykowski, S.C.; Baskin, R.J. Processive translocation and DNA unwinding by individual RecBCD enzyme molecules. Nature 2001, 409, 374–378. [Google Scholar]
- Spies, M.; Bianco, P.R.; Dillingham, M.S.; Handa, N.; Baskin, R.J.; Kowalczykowski, S.C. A molecular throttle: The recombination hotspot chi controls DNA translocation by the RecBCD helicase. Cell 2003, 114, 647–654. [Google Scholar] [CrossRef]
- Spies, M.; Amitani, I.; Baskin, R.J.; Kowalczykowski, S.C. RecBCD enzyme switches lead motor subunits in response to chi recognition. Cell 2007, 131, 694–705. [Google Scholar] [CrossRef]
- Van Mameren, J.; Peterman, E.J.; Wuite, G.J. See me, feel me: Methods to concurrently visualize and manipulate single DNA molecules and associated proteins. Nucleic Acids Res. 2008, 36, 4381–4389. [Google Scholar] [CrossRef]
- Kim, J.H.; Dukkipati, V.R.; Pang, S.W.; Larson, R.G. Stretching and immobilization of DNA for studies of protein–DNA interactions at the single-molecule level. Nanoscale Res. Lett. 2007, 2, 185–201. [Google Scholar] [CrossRef]
- Moffitt, J.R.; Chemla, Y.R.; Izhaky, D.; Bustamante, C. Differential detection of dual traps improves the spatial resolution of optical tweezers. Proc. Natl. Acad. Sci. USA 2006, 103, 9006–9011. [Google Scholar] [CrossRef]
- Shaevitz, J.W.; Abbondanzieri, E.A.; Landick, R.; Block, S.M. Backtracking by single RNA polymerase molecules observed at near-base-pair resolution. Nature 2003, 426, 684–687. [Google Scholar] [CrossRef]
- Wuite, G.J.L.; Davenport, R.J.; Rappaport, A.; Bustamante, C. An integrated laser trap/flow control video microscope for the study of single biomolecules. Biophys. J. 2000, 79, 1155–1167. [Google Scholar] [CrossRef]
- Morin, J.A.; Cao, F.J.; Lazaro, J.M.; Arias-Gonzalez, J.R.; Valpuesta, J.M.; Carrascosa, J.L.; Salas, M.; Ibarra, B. Active DNA unwinding dynamics during processive DNA replication. Proc. Natl. Acad. Sci. USA 2012, 109, 8115–8120. [Google Scholar]
- Chemla, Y.R.; Aathavan, K.; Michaelis, J.; Grimes, S.; Jardine, P.J.; Anderson, D.L.; Bustamante, C. Mechanism of force generation of a viral DNA packaging motor. Cell 2005, 122, 683–692. [Google Scholar] [CrossRef]
- Moffitt, J.R.; Chemla, Y.R.; Aathavan, K.; Grimes, S.; Jardine, P.J.; Anderson, D.L.; Bustamante, C. Intersubunit coordination in a homomeric ring ATPase. Nature 2009, 457, 446–450. [Google Scholar] [CrossRef]
- Smith, D.E.; Tans, S.J.; Smith, S.B.; Grimes, S.; Anderson, D.L.; Bustamante, C. The bacteriophage straight phi29 portal motor can package DNA against a large internal force. Nature 2001, 413, 748–752. [Google Scholar] [CrossRef]
- Yu, J.; Moffitt, J.; Hetherington, C.L.; Bustamante, C.; Oster, G. Mechanochemistry of a viral DNA packaging motor. J. Mol. Biol. 2010, 400, 186–203. [Google Scholar] [CrossRef]
- Bennink, M.L.; Leuba, S.H.; Leno, G.H.; Zlatanova, J.; de Grooth, B.G.; Greve, J. Unfolding individual nucleosomes by stretching single chromatin fibers with optical tweezers. Nat. Struct. Biol. 2001, 8, 606–610. [Google Scholar] [CrossRef]
- Cui, Y.; Bustamante, C. Pulling a single chromatin fiber reveals the forces that maintain its higher-order structure. Proc. Natl. Acad. Sci. USA 2000, 97, 127–132. [Google Scholar] [CrossRef]
- Hodges, C.; Bintu, L.; Lubkowska, L.; Kashlev, M.; Bustamante, C. Nucleosomal fluctuations govern the transcription dynamics of RNA polymerase II. Science 2009, 325, 626–628. [Google Scholar] [CrossRef]
- Van den Broek, B.; Noom, M.C.; Wuite, G.J.L. DNA-tension dependence of restriction enzyme activity reveals mechanochemical properties of the reaction pathway. Nucleic Acids Res. 2005, 33, 2676–2684. [Google Scholar] [CrossRef]
- Van den Broek, B.; Lomholt, M.A.; Kalisch, S.M.J.; Metzler, R.; Wuite, G.J.L. How DNA coiling enhances target localization by proteins. Proc. Natl. Acad. Sci. USA 2008, 105, 15738–15742. [Google Scholar]
- Candelli, A.; Wuite, G.J.; Peterman, E.J. Combining optical trapping, fluorescence microscopy and micro-fluidics for single molecule studies of DNA-protein interactions. Phys. Chem. Chem. Phys. 2011, 13, 7263–7272. [Google Scholar] [CrossRef]
- Capitanio, M.; Maggi, D.; Vanzi, F.; Pavone, F.S. FIONA in the trap: The advantages of combining optical tweezers and fluorescence. J. Opt. A 2007, 9, S157–S163. [Google Scholar] [CrossRef]
- Harada, Y.; Funatsu, T.; Murakami, K.; Nonoyama, Y.; Ishihama, A.; Yanagida, T. Single-molecule imaging of RNA polymerase-DNA interactions in real time. Biophys. J. 1999, 76, 709–715. [Google Scholar] [CrossRef]
- Van Mameren, J.; Modesti, M.; Kanaar, R.; Wyman, C.; Peterman, E.J.G.; Wuite, G.J.L. Counting RAD51 proteins disassembling from nucleoprotein filaments under tension. Nature 2009, 457, 745–748. [Google Scholar] [CrossRef]
- Biebricher, A.; Wende, W.; Escude, C.; Pingoud, A.; Desbiolles, P. Tracking of single quantum dot labeled EcoRV sliding along DNA manipulated by double optical tweezers. Biophys. J. 2009, 96, L50–L52. [Google Scholar]
- Resch-Genger, U.; Grabolle, M.; Cavaliere-Jaricot, S.; Nitschke, R.; Nann, T. Quantum dots versus organic dyes as fluorescent labels. Nat. Methods 2008, 5, 763–775. [Google Scholar] [CrossRef]
- Dijk, M.A.; Kapitein, L.C.; Mameren, J.; Schmidt, C.F.; Peterman, E.J. Combining optical trapping and single-molecule fluorescence spectroscopy: Enhanced photobleaching of fluorophores. J. Phys. Chem. B 2004, 108, 6479–6484. [Google Scholar]
- Gross, P.; Farge, G.; Peterman, E.J.; Wuite, G.J. Combining optical tweezers, single-molecule fluorescence microscopy, and microfluidics for studies of DNA-protein interactions. Methods Enzymol. 2010, 475, 427–453. [Google Scholar] [CrossRef]
- Brau, R.R.; Tarsa, P.B.; Ferrer, J.M.; Lee, P.; Lang, M.J. Interlaced optical force-fluorescence measurements for single molecule biophysics. Biophys. J. 2006, 91, 1069–1077. [Google Scholar] [CrossRef]
- Comstock, M.J.; Ha, T.; Chemla, Y.R. Ultrahigh-resolution optical trap with single-fluorophore sensitivity. Nat. Methods 2011, 8, 335–340. [Google Scholar] [CrossRef]
- Toprak, E.; Kural, C.; Selvin, P.R. Super-accuracy and super-resolution getting around the diffraction limit. Methods Enzymol. 2010, 475, 1–26. [Google Scholar] [CrossRef]
- Hoffman, M.T.; Sheung, J.; Selvin, P.R. Fluorescence imaging with one nanometer accuracy: In vitro and in vivo studies of molecular motors. Methods Mol. Biol. 2011, 778, 33–56. [Google Scholar] [CrossRef]
- Walter, N.G.; Huang, C.Y.; Manzo, A.J.; Sobhy, M.A. Do-it-yourself guide: How to use the modern single-molecule toolkit. Nat. Methods 2008, 5, 475–489. [Google Scholar] [CrossRef]
- Van den Wildenberg, S.M.; Prevo, B.; Peterman, E.J. A brief introduction to single-molecule fluorescence methods. Methods Mol. Biol. 2011, 783, 81–99. [Google Scholar] [CrossRef]
- Thompson, R.E.; Larson, D.R.; Webb, W.W. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J. 2002, 82, 2775–2783. [Google Scholar] [CrossRef]
- Yildiz, A.; Forkey, J.N.; McKinney, S.A.; Ha, T.; Goldman, Y.E.; Selvin, P.R. Myosin V walks hand-over-hand: Single fluorophore imaging with 1.5-nm localization. Science 2003, 300, 2061–2065. [Google Scholar]
- Graham, J.S.; Johnson, R.C.; Marko, J.F. Concentration-dependent exchange accelerates turnover of proteins bound to double-stranded DNA. Nucleic Acids Res. 2011, 39, 2249–2259. [Google Scholar] [CrossRef]
- Li, G.W.; Berg, O.G.; Elf, J. Effects of macromolecular crowding and DNA looping on gene regulation kinetics. Nat. Phys. 2009, 5, 294–297. [Google Scholar] [CrossRef]
- Axelrod, D.; Koppel, D.E.; Schlessinger, J.; Elson, E.; Webb, W.W. Mobility measurement by analysis of fluorescence photobleaching recovery kinetics. Biophys. J. 1976, 16, 1055–1069. [Google Scholar] [CrossRef]
- Beaudouin, J.; Mora-Bermudez, F.; Klee, T.; Daigle, N.; Ellenberg, J. Dissecting the contribution of diffusion and interactions to the mobility of nuclear proteins. Biophys. J. 2006, 90, 1878–1894. [Google Scholar] [CrossRef]
- Sprague, B.L.; Muller, F.; Pego, R.L.; Bungay, P.M.; Stavreva, D.A.; McNally, J.G. Analysis of binding at a single spatially localized cluster of binding sites by fluorescence recovery after photobleaching. Biophys. J. 2006, 91, 1169–1191. [Google Scholar] [CrossRef]
- Phair, R.D.; Scaffidi, P.; Elbi, C.; Vecerova, J.; Dey, A.; Ozato, K.; Brown, D.T.; Hager, G.; Bustin, M.; Misteli, T. Global nature of dynamic protein-chromatin interactions in vivo: Three-dimensional genome scanning and dynamic interaction networks of chromatin proteins. Mol. Cell. Biol. 2004, 24, 6393–6402. [Google Scholar]
- Sprague, B.L.; McNally, J.G. FRAP analysis of binding: Proper and fitting. Trends Cell Biol. 2005, 15, 84–91. [Google Scholar] [CrossRef]
- Mueller, F.; Wach, P.; McNally, J.G. Evidence for a common mode of transcription factor interaction with chromatin as revealed by improved quantitative fluorescence recovery after photobleaching. Biophys. J. 2008, 94, 3323–3339. [Google Scholar] [CrossRef]
- Dundr, M.; Hoffmann-Rohrer, U.; Hu, Q.; Grummt, I.; Rothblum, L.I.; Phair, R.D.; Misteli, T. A kinetic framework for a mammalian RNA polymerase in vivo. Science 2002, 298, 1623–1626. [Google Scholar] [CrossRef]
- Kimura, H.; Sugaya, K.; Cook, P.R. The transcription cycle of RNA polymerase II in living cells. J. Cell Biol. 2002, 159, 777–782. [Google Scholar] [CrossRef]
- Darzacq, X.; Shav-Tal, Y.; de Turris, V.; Brody, Y.; Shenoy, S.M.; Phair, R.D.; Singer, R.H. In vivo dynamics of RNA polymerase II transcription. Nat. Struct. Mol. Biol. 2007, 14, 796–806. [Google Scholar] [CrossRef]
- Van Royen, M.E.; Farla, P.; Mattern, K.A.; Geverts, B.; Trapman, J.; Houtsmuller, A.B. Fluorescence recovery after photobleaching (FRAP) to study nuclear protein dynamics in living cells. Methods Mol. Biol. 2009, 464, 363–385. [Google Scholar]
- Schwille, P.; Haupts, U.; Maiti, S.; Webb, W.W. Molecular dynamics in living cells observed by fluorescence correlation spectroscopy with one- and two-photon excitation. Biophys. J. 1999, 77, 2251–2265. [Google Scholar] [CrossRef]
- Elson, E.L. Fluorescence correlation spectroscopy measures molecular transport in cells. Traffic 2001, 2, 789–796. [Google Scholar] [CrossRef]
- Bacia, K.; Kim, S.A.; Schwille, P. Fluorescence cross-correlation spectroscopy in living cells. Nat. Methods 2006, 3, 83–89. [Google Scholar] [CrossRef]
- Stasevich, T.J.; Mueller, F.; Michelman-Ribeiro, A.; Rosales, T.; Knutson, J.R.; McNally, J.G. Cross-validating FRAP and FCS to quantify the impact of photobleaching on in vivo binding estimates. Biophys. J. 2010, 99, 3093–3101. [Google Scholar] [CrossRef]
- Michelman-Ribeiro, A.; Mazza, D.; Rosales, T.; Stasevich, T.J.; Boukari, H.; Rishi, V.; Vinson, C.; Knutson, J.R.; McNally, J.G. Direct measurement of association and dissociation rates of DNA binding in live cells by fluorescence correlation spectroscopy. Biophys. J. 2009, 97, 337–346. [Google Scholar] [CrossRef]
- Larson, D.R.; Zenklusen, D.; Wu, B.; Chao, J.A.; Singer, R.H. Real-time observation of transcription initiation and elongation on an endogenous yeast gene. Science 2011, 332, 475–478. [Google Scholar] [CrossRef]
- Li, G.W.; Xie, X.S. Central dogma at the single-molecule level in living cells. Nature 2011, 475, 308–315. [Google Scholar] [CrossRef]
- Yu, J.; Xiao, J.; Ren, X.J.; Lao, K.Q.; Xie, X.S. Probing gene expression in live cells, one protein molecule at a time. Science 2006, 311, 1600–1603. [Google Scholar] [CrossRef]
- Ruusala, T.; Crothers, D.M. Sliding and intermolecular transfer of the lac repressor: Kinetic perturbation of a reaction intermediate by a distant DNA sequence. Proc. Natl. Acad. Sci. USA 1992, 89, 4903–4907. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Monico, C.; Capitanio, M.; Belcastro, G.; Vanzi, F.; Pavone, F.S. Optical Methods to Study Protein-DNA Interactions in Vitro and in Living Cells at the Single-Molecule Level. Int. J. Mol. Sci. 2013, 14, 3961-3992. https://doi.org/10.3390/ijms14023961
Monico C, Capitanio M, Belcastro G, Vanzi F, Pavone FS. Optical Methods to Study Protein-DNA Interactions in Vitro and in Living Cells at the Single-Molecule Level. International Journal of Molecular Sciences. 2013; 14(2):3961-3992. https://doi.org/10.3390/ijms14023961
Chicago/Turabian StyleMonico, Carina, Marco Capitanio, Gionata Belcastro, Francesco Vanzi, and Francesco S. Pavone. 2013. "Optical Methods to Study Protein-DNA Interactions in Vitro and in Living Cells at the Single-Molecule Level" International Journal of Molecular Sciences 14, no. 2: 3961-3992. https://doi.org/10.3390/ijms14023961
APA StyleMonico, C., Capitanio, M., Belcastro, G., Vanzi, F., & Pavone, F. S. (2013). Optical Methods to Study Protein-DNA Interactions in Vitro and in Living Cells at the Single-Molecule Level. International Journal of Molecular Sciences, 14(2), 3961-3992. https://doi.org/10.3390/ijms14023961