Multidrug Resistance and Cancer Stem Cells in Neuroblastoma and Hepatoblastoma




{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. MDR in Neuroblastoma and Hepatoblastoma
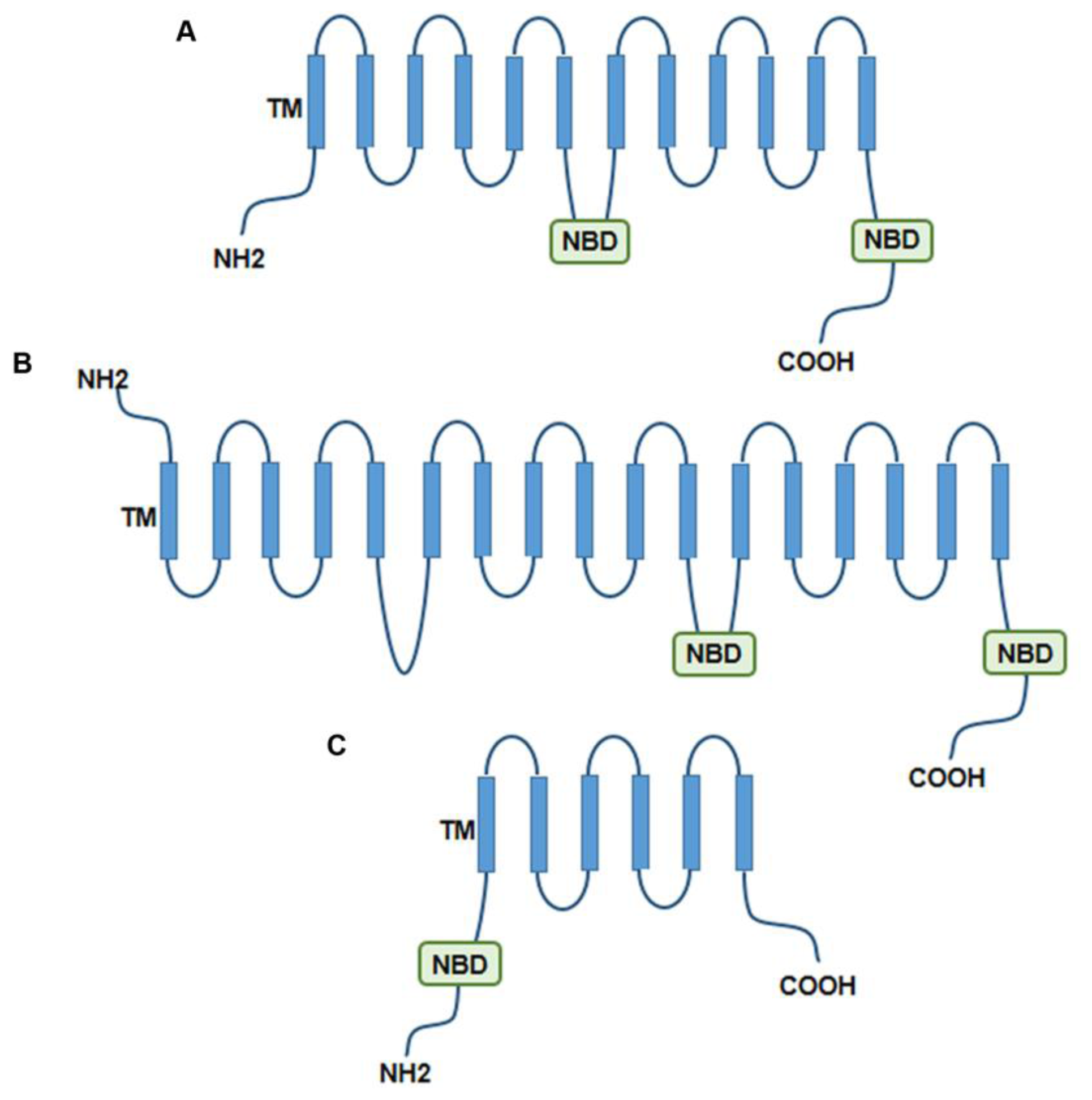
2.1. MDR Concepts and Proteins
2.2. Neuroblastoma and MDR
2.2.1. Epidemiology and Characteristics of Neuroblastoma
2.2.2. Clinical/Experimental Evidence of MDR in NB
2.3. Hepatoblastoma and MDR
2.3.1. Epidemiology and Characteristics of Hepatoblastoma
2.3.2. Clinical/Experimental Evidence of MDR in HB
3. CSCs in NB and HB
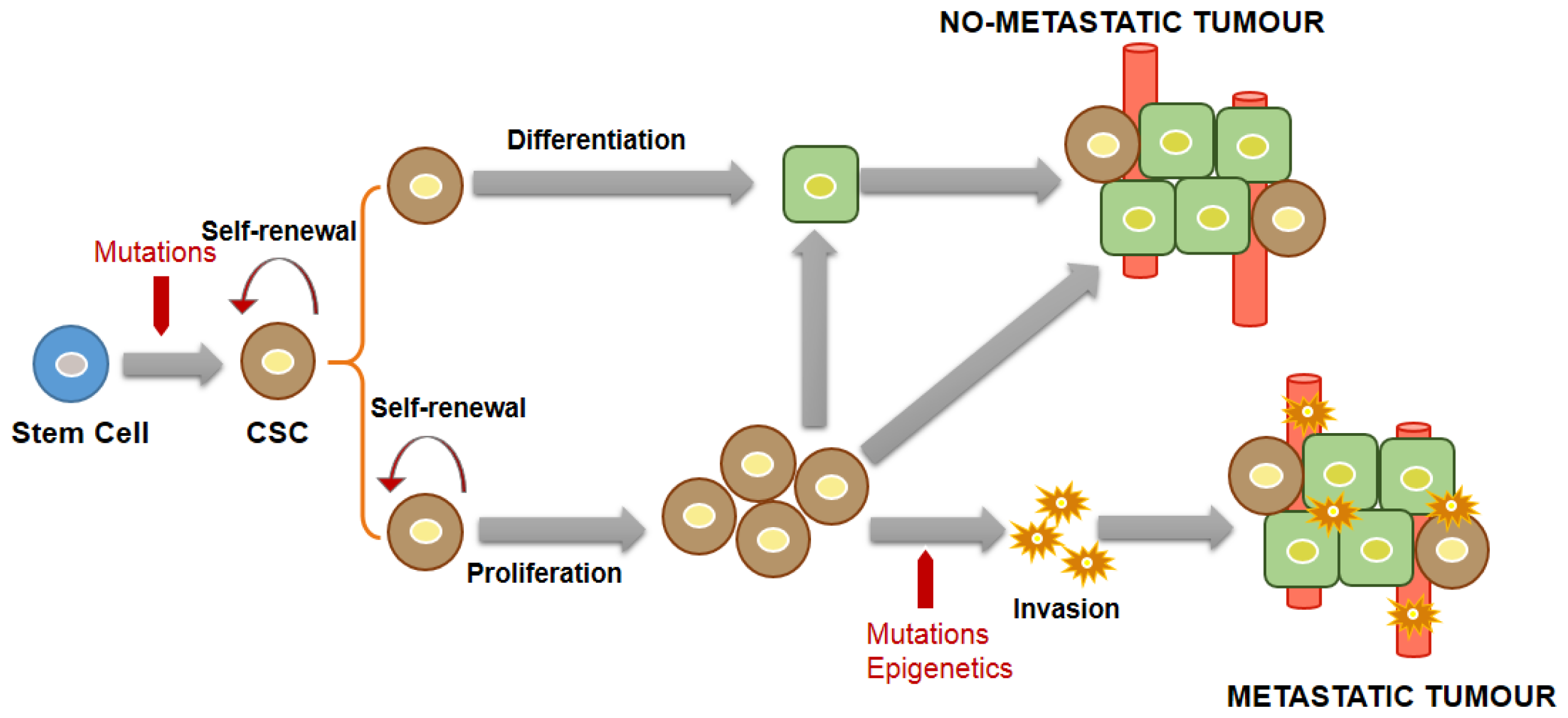
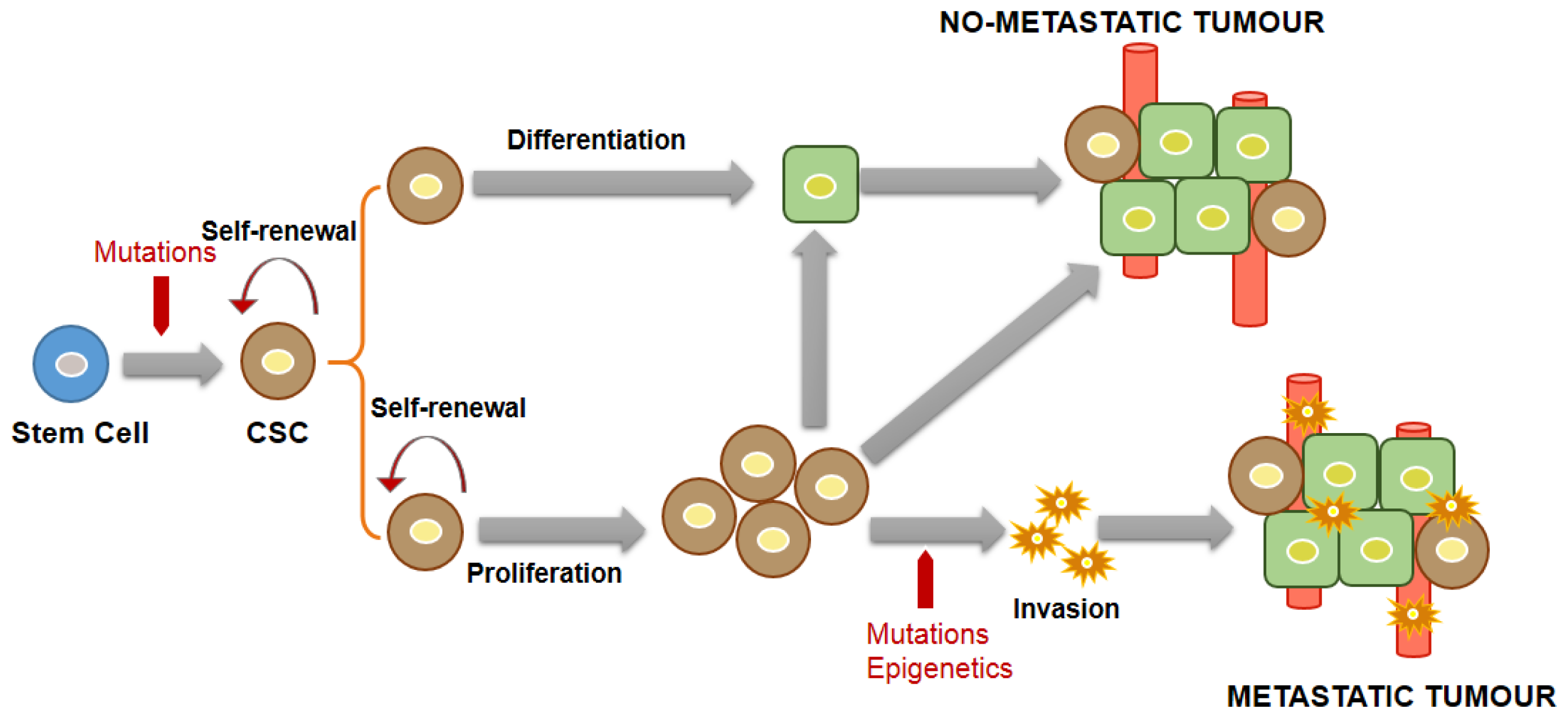
3.1. General Concepts and Characteristics of CSCs
3.2. CSC Markers in NB
3.3. CSC Markers in HB
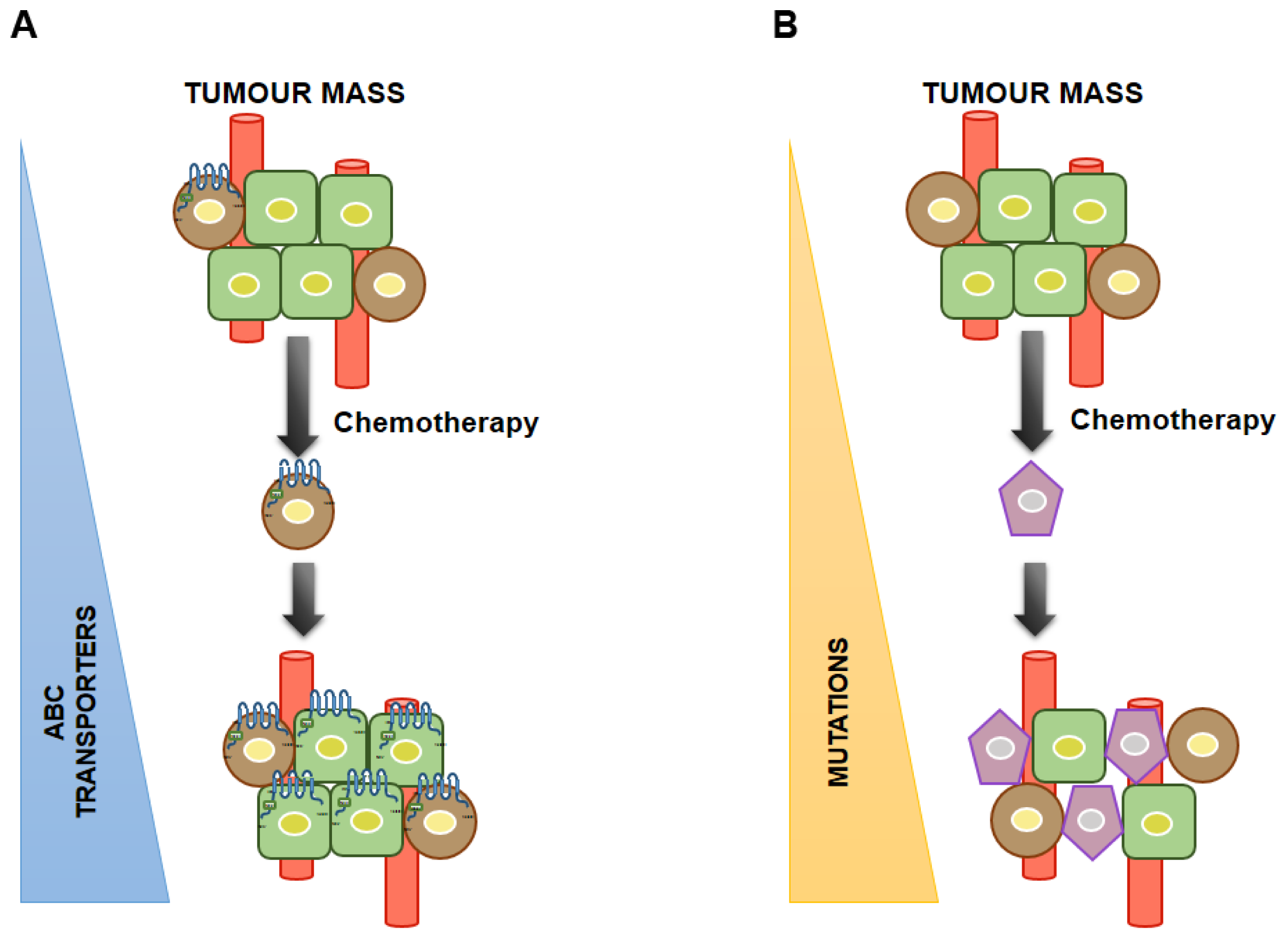
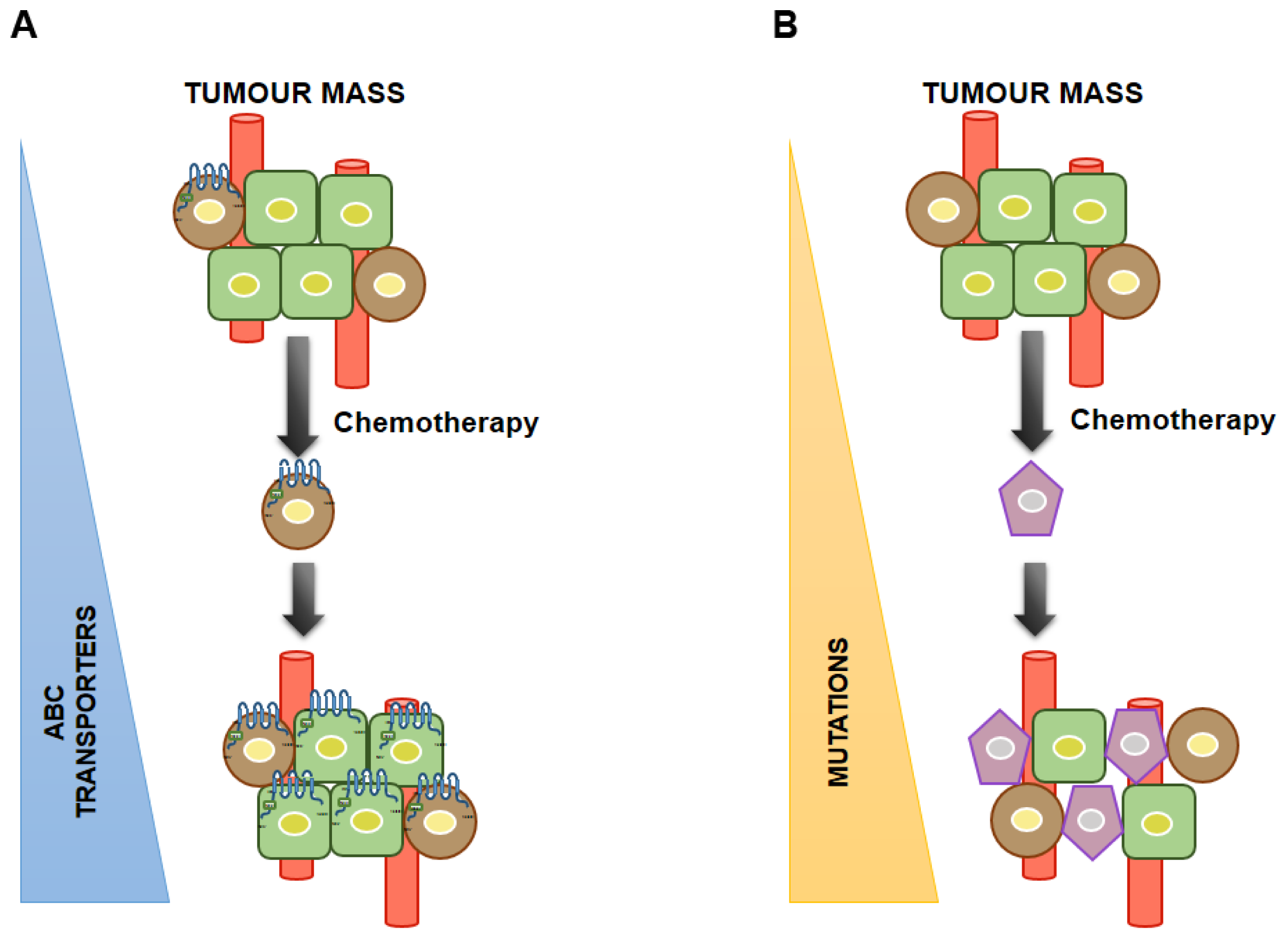
4. Association between CSCs and MDR and Its Diagnostic Significance
5. Future Perspective and Conclusions
Acknowledgments
Conflicts of Interest
References
- Gottesman, M.M. Mechanisms of cancer drug resistance. Annu. Rev. Med 2002, 53, 615–627. [Google Scholar]
- Gatti, L.; Zunino, F. Overview of tumor cell chemoresistance mechanisms. Methods Mol. Med 2005, 111, 127–148. [Google Scholar]
- Wilson, T.R.; Longley, D.B.; Johnston, P.G. Chemoresistance in solid tumours. Ann. Oncol 2006, 17, x315–x324. [Google Scholar]
- Ullah, M.F. Cancer multidrug resistance (MDR): A major impediment to effective chemotherapy. Asian Pac. J. Cancer Prev 2008, 9, 1–6. [Google Scholar]
- Fletcher, J.I.; Haber, M.; Henderson, M.J.; Norris, M.D. ABC transporters in cancer: More than just drug efflux pumps. Nat. Rev. Cancer 2010, 10, 147–156. [Google Scholar]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar]
- Greaves, M.; Maley, C.C.; Visvader, J.E. Cells of origin in cancer. Nature 2011, 469, 314–322. [Google Scholar]
- Magee, J.A.; Piskounova, E.; Morrison, S.J. Cancer stem cells: Impact, heterogeneity, and uncertainty. Cancer Cell 2012, 21, 283–296. [Google Scholar]
- Baiocchi, M.; Biffoni, M.; Ricci-Vitiani, L.; Pilozzi, E.; De Maria, R. New models for cancer research: Human cancer stem cell xenografts. Curr. Opin. Pharmacol 2010, 10, 380–384. [Google Scholar]
- Abdullah, L.N.; Chow, E.K. Mechanisms of chemoresistance in cancer stem cells. Clin. Transl. Med 2013, 2, 3. [Google Scholar]
- Moitra, K.; Lou, H.; Dean, M. Multidrug efflux pumps and cancer stem cells: Insights into multidrug resistance and therapeutic development. Clin. Pharmacol. Ther 2011, 89, 491–502. [Google Scholar]
- Vermeulen, L.; de Sousa e Melo, F.; Richel, D.J.; Medema, J.P. The developing cancer stem-cell model: Clinical challenges and opportunities. Lancet Oncol 2012, 13, e83–e89. [Google Scholar]
- Clevers, H. The cancer stem cell: Premises, promises and challenges. Nat. Med 2011, 17, 313–339. [Google Scholar]
- O’Brien, C.A.; Pollett, A.; Gallinger, S.; Dick, J.E. A human colon cancer cell capable of initiating tumour growth in immunodeficient mice. Nature 2007, 445, 106–110. [Google Scholar]
- Reddy, E.P. Liver stem cells and hepatocellular carcinoma. Hepatology 2009, 49, 318–329. [Google Scholar]
- Jiang, W.; Peng, J.; Zhang, Y.; Cho, W.C.; Jin, K. The implications of cancer stem cells for cancer therapy. Int. J. Mol. Sci 2012, 13, 16636–16657. [Google Scholar]
- Friedman, G.K.; Gillespie, G.Y. Cancer Stem Cells and Pediatric Solid Tumors. Cancers 2011, 3, 298–318. [Google Scholar]
- Dean, M.; Rzhetsky, A.; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res 2001, 11, 1156–1166. [Google Scholar]
- Nutrition, Metabolism & Genomics Group. Available online: http://nutrigene.4t.com/humanabc.htm (accessed on 10 October 2013).
- Albrecht, C.; McVey, J.H.; Elliott, J.I.; Sardini, A.; Kasza, I.; Mumford, A.D.; Naoumova, R.P.; Tuddenham, E.G.; Szabo, K.; Higgins, C.F. A novel missense mutation in ABCA1 results in altered protein trafficking and reduced phosphatidylserine translocation in a patient with Scott syndrome. Blood 2005, 106, 542–549. [Google Scholar]
- Martínez-Mir, A.; Paloma, E.; Allikmets, R.; Ayuso, C.; del Rio, T.; Dean, M.; Vilageliu, L.; Gonzàlez-Duarte, R.; Balcells, S. Retinitis pigmentosa caused by a homozygous mutation in the Stargardt disease gene ABCR. Nat. Genet 1998, 18, 11–12. [Google Scholar]
- Jacquemin, E. Progressive familial intrahepatic cholestasis. Genetic basis and treatment. Clin. Liver Dis 2000, 4, 753–763. [Google Scholar]
- Dean, M.; Allikmets, R. Complete characterization of the human ABC gene family. J. Bioenerg. Biomembr 2001, 33, 475–479. [Google Scholar]
- Pendse, S.S.; Briscoe, D.M.; Frank, M.H. P-glycoprotein and alloimmune T-cell activation. Clin. Appl. Immunol. Rev 2003, 4, 3–14. [Google Scholar]
- Hipfner, D.R.; Deeley, R.G.; Cole, S.P. Structural, mechanistic and clinical aspects of MRP1. Biochim. Biophys. Acta 1999, 1461, 359–376. [Google Scholar]
- Kartenbeck, J.; Leuschner, U.; Mayer, R.; Keppler, D. Absence of the canalicular isoform of the MRP gene-encoded conjugate export pump from the hepatocytes in Dubin-Johnson syndrome. Hepatology 1996, 23, 1061–1066. [Google Scholar]
- Nooter, K.; Brutel de la Riviere, G.; Look, M.P.; van Wingerden, K.E.; Henzen-Logmans, S.C.; Scheper, R.J.; Flens, M.J.; Klijn, J.G.; Stoter, G.; Foekens, J.A. The prognostic significance of expression of the multidrug resistance-associated protein (MRP) in primary breast cancer. Br. J. Cancer 1997, 76, 486–493. [Google Scholar]
- Nooter, K.; de la Riviere, G.B.; Klijn, J.; Stoter, G.; Foekens, J. Multidrug resistance protein in recurrent breast cancer. Lancet 1997, 349, 1885–1886. [Google Scholar]
- Rudas, M.; Filipits, M.; Taucher, S.; Stranzl, T.; Steger, G.G.; Jakesz, R.; Pirker, R.; Pohl, G. Expression of MRP1, LRP and Pgp in breast carcinoma patients treated with preoperative chemotherapy. Breast Cancer Res. Treat 2003, 81, 149–157. [Google Scholar]
- Berger, W.; Setinek, U.; Hollaus, P.; Zidek, T.; Steiner, E.; Elbling, L.; Cantonati, H.; Attems, J.; Gsur, A.; Micksche, M. Multidrug resistance markers P-glycoprotein, multidrug resistance protein 1, and lung resistance protein in non-small cell lung cancer: Prognostic implications. J. Cancer Res. Clin. Oncol 2005, 131, 355–363. [Google Scholar]
- Ota, E.; Abe, Y.; Oshika, Y.; Ozeki, Y.; Iwasaki, M.; Inoue, H.; Yamazaki, H.; Ueyama, Y.; Takagi, K.; Ogata, T.; et al. Expression of the multidrug resistance-associated protein (MRP) gene in non-small-cell lung cancer. Br. J. Cancer 1995, 72, 550–554. [Google Scholar]
- Hsia, T.C.; Lin, C.C.; Wang, J.J.; Ho, S.T.; Kao, A. Relationship between chemotherapy response of small cell lung cancer and P-glycoprotein or multidrug resistance-related protein expression. Lung 2002, 180, 173–179. [Google Scholar]
- Oshika, Y.; Nakamura, M.; Tokunaga, T.; Fukushima, Y.; Abe, Y.; Ozeki, Y.; Yamazaki, H.; Tamaoki, N.; Ueyama, Y. Multidrug resistance-associated protein and mutant p53 protein expression in non-small cell lung cancer. Mod. Pathol 1998, 11, 1059–1063. [Google Scholar]
- Kuo, T.H.; Liu, F.Y.; Chuang, C.Y.; Wu, H.S.; Wang, J.J.; Kao, A. To predict response chemotherapy using technetium-99m tetrofosmin chest images in patients with untreated small cell lung cancer and compare with p-glycoprotein, multidrug resistance related protein-1, and lung resistance-related protein expression. Nucl. Med. Biol 2003, 30, 627–632. [Google Scholar]
- Deeley, R.G.; Cole, S.P. Substrate recognition and transport by multidrug resistance protein 1 (ABCC1). FEBS Lett 2006, 580, 1103–1111. [Google Scholar]
- Norris, M.D.; Bordow, S.B.; Marshall, G.M.; Haber, M. Expression of the gene for multidrug-resistance–associated protein and outcome in patients with neuroblastoma. N. Engl. J. Med 1996, 334, 231–238. [Google Scholar]
- Warmann, S.; Hunger, M.; Teichmann, B.; Flemming, P.; Gratz, K.F.; Fuchs, J. The role of the MDR1 gene in the development of multidrug resistance in human hepatoblastoma: Clinical course and in vivo model. Cancer 2002, 95, 1795–1801. [Google Scholar]
- Oue, T.; Yoneda, A.; Uehara, S.; Yamanaka, H.; Fukuzawa, M. Increased expression of multidrug resistance-associated genes after chemotherapy in pediatric solid malignancies. J. Pediatr. Surg 2009, 44, 377–380. [Google Scholar]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med 2010, 362, 2202–2211. [Google Scholar]
- Cheung, N.K.; Dyer, M.A. Neuroblastoma: Developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 2013, 13, 397–411. [Google Scholar]
- Brodeur, G.M.; Seeger, R.C.; Schwab, M.; Varmus, H.E.; Bishop, J.M. Amplification of N-myc in untreated human neuroblastomas correlates with advanced disease stage. Science 1984, 224, 1121–1124. [Google Scholar]
- Seeger, R.C.; Brodeur, G.M.; Sather, H.; Dalton, A.; Siegel, S.E.; Wong, K.Y.; Hammond, D. Association of multiple copies of the N-myc oncogene with rapid progression of neuroblastomas. N. Engl. J. Med 1985, 313, 1111–1116. [Google Scholar]
- Bordow, S.B.; Haber, M.; Madafiglio, J.; Cheung, B.; Marshall, G.M.; Norris, M.D. Expression of the multidrug resistance-associated protein (MRP) gene correlates with amplification and overexpression of the N-myc oncogene in childhood neuroblastoma. Cancer Res 1994, 54, 5036–5040. [Google Scholar]
- Norris, M.D.; Bordow, S.B.; Haber, P.S.; Marshall, G.M.; Kavallaris, M.; Madafiglio, J.; Cohn, S.L.; Salwen, H.; Schmidt, M.L.; Hipfner, D.R.; et al. Evidence that the MYCN oncogene regulates MRP gene expression in neuroblastoma. Eur. J. Cancer 1997, 33, 1911–1916. [Google Scholar]
- Haber, M.; Bordow, S.B.; Gilbert, J.; Madafiglio, J.; Kavallaris, M.; Marshall, G.M.; Mechetner, E.B.; Fruehauf, J.P.; Tee, L.; Cohn, S.L.; et al. Altered expression of the MYCN oncogene modulates MRP gene expression and response to cytotoxic drugs in neuroblastoma cells. Oncogene 1999, 18, 2777–2782. [Google Scholar]
- Bader, P.; Schilling, F.; Schlaud, M.; Girgert, R.; Handgretinger, R.; Klingebiel, T.; Treuner, J.; Liu, C.; Niethammer, D.; Beck, J.F. Expression analysis of multidrug resistance associated genes in neuroblastomas. Oncol. Rep 1999, 6, 1143–1146. [Google Scholar]
- Burkhart, C.A.; Cheng, A.J.; Madafiglio, J.; Kavallaris, M.; Mili, M.; Marshall, G.M.; Weiss, W.A.; Khachigian, L.M.; Norris, M.D.; Haber, M. Effects of MYCN antisense oligonucleotide administration on tumorigenesis in a murine model of neuroblastoma. J. Natl. Cancer Inst 2003, 95, 1394–1403. [Google Scholar]
- Pajic, M.; Norris, M.D.; Cohn, S.L.; Haber, M. The role of the multidrug resistance-associated protein 1 gene in neuroblastoma biology and clinical outcome. Cancer Lett 2005, 228, 241–246. [Google Scholar]
- Matsunaga, T.; Shirasawa, H.; Hishiki, T.; Enomoto, H.; Kouchi, K.; Ohtsuka, Y.; Iwai, J.; Yoshida, H.; Tanabe, M.; Kobayashi, S.; et al. Expression of MRP and cMOAT in childhood neuroblastomas and malignant liver tumors and its relevance to clinical behavior. Jpn. J. Cancer Res 1998, 89, 1276–1283. [Google Scholar]
- Lu, Q.J.; Dong, F.; Zhang, J.H.; Li, X.H.; Ma, Y.; Jiang, W.G. Expression of multidrug resistance-related markers in primary neuroblastoma. Chin. Med. J. (Engl.) 2004, 117, 1358–1363. [Google Scholar]
- Haber, M.; Bordow, S.B.; Haber, P.S.; Marshall, G.M.; Stewart, B.W.; Norris, M.D. The prognostic value of MDR1 gene expression in primary untreated neuroblastoma. Eur. J. Cancer 1997, 33, 2031–2036. [Google Scholar]
- Bourhis, J.; Bénard, J.; Hartmann, O.; Boccon-Gibod, L.; Lemerle, J.; Riou, G. Correlation of MDR1 gene expression with chemotherapy in neuroblastoma. J. Natl. Cancer Inst 1989, 81, 1401–1405. [Google Scholar]
- Nakagawara, A.; Kadomatsu, K.; Sato, S.; Kohno, K.; Takano, H.; Akazawa, K.; Nose, Y.; Kuwano, M. Inverse correlation between expression of multidrug resistance gene and N-myc oncogene in human neuroblastomas. Cancer Res 1990, 50, 3043–3047. [Google Scholar]
- Favrot, M.; Combaret, V.; Goillot, E.; Wagner, J.P.; Bouffet, E.; Mazingue, F.; Thyss, A.; Bordigoni, P.; Delsol, G.; Bailly, C.; et al. Expression of P-glycoprotein restricted to normal cells in neuroblastoma biopsies. Br. J. Cancer 1991, 64, 233–238. [Google Scholar]
- Chan, H.S.; Haddad, G.; Thorner, P.S.; DeBoer, G.; Lin, Y.P.; Ondrusek, N.; Yeger, H.; Ling, V. P-glycoprotein expression as a predictor of the outcome of therapy for neuroblastoma. N. Engl. J. Med 1991, 325, 1608–1614. [Google Scholar]
- Manohar, C.F.; Bray, J.A.; Salwen, H.R.; Madafiglio, J.; Cheng, A.; Flemming, C.; Marshall, G.M.; Norris, M.D.; Haber, M.; Cohn, S.L. MYCN-mediated regulation of the MRP1 promoter in human neuroblastoma. Oncogene 2004, 23, 753–762. [Google Scholar]
- Henderson, M.J.; Haber, M.; Porro, A.; Munoz, M.A.; Iraci, N.; Xue, C.; Murray, J.; Flemming, C.L.; Smith, J.; Fletcher, J.I.; et al. ABCC multidrug transporters in childhood neuroblastoma: Clinical and biological effects independent of cytotoxic drug efflux. J. Natl. Cancer Inst 2011, 103, 1236–1251. [Google Scholar]
- Norris, M.D.; Smith, J.; Tanabe, K.; Tobin, P.; Flemming, C.; Scheffer, G.L.; Wielinga, P.; Cohn, S.L.; London, W.B.; Marshall, G.M.; et al. Expression of multidrug transporter MRP4/ABCC4 is a marker of poor prognosis in neuroblastoma and confers resistance to irinotecan in vitro. Mol. Cancer Ther 2005, 4, 547–553. [Google Scholar]
- Porro, A.; Haber, M.; Diolaiti, D.; Iraci, N.; Henderson, M.; Gherardi, S.; Valli, E.; Munoz, M.A.; Xue, C.; Flemming, C.; et al. Direct and coordinate regulation of ATP-binding cassette transporter genes by Myc factors generates specific transcription signatures that significantly affect the chemoresistance phenotype of cancer cells. J. Biol. Chem 2010, 285, 19532–19543. [Google Scholar]
- Spector, L.G.; Birch, J. The epidemiology of hepatoblastoma. Pediatr. Blood Cancer 2012, 59, 776–779. [Google Scholar]
- López-Terrada, D.; Alaggio, R.; de Dávila, M.T.; Czauderna, P.; Hiyama, E.; Katzenstein, H.; Leuschner, I.; Malogolowkin, M.; Meyers, R.; Ranganathan, S.; et al. Towards an international pediatric liver tumor consensus classification: Proceedings of the Los Angeles COG liver tumors symposium. Mod. Pathol 2013. [Google Scholar] [CrossRef]
- Czauderna, P.; Otte, J.B.; Aronson, D.C.; Gauthier, F.; Mackinlay, G.; Roebuck, D.; Plaschkes, J.; Perilongo, G. Childhood Liver Tumour Strategy Group of the International Society of Paediatric Oncology (SIOPEL). Guidelines for surgical treatment of hepatoblastoma in the modern era: Recommendations from the childhood liver tumour strategy group of the international society of paediatric oncology (SIOPEL). Eur. J. Cancer 2005, 41, 1031–1036. [Google Scholar]
- National Cancer Institute at the National Institutes of Health. Available online: http://www.cancer.gov/clinicaltrials/search/view?version=healthprofessional&cdrid=654889 (accessed on 8 October 2013).
- Wiederkehr, J.C.; Coelho, I.M.; Avilla, S.G.; Wiederkehr, B.A.; Wiederkehr, H.A. Liver Tumors in Infancy. In Hepatic Surgery; InTech: Rijeka, Croatia, 2013; Volume Chapter 18. [Google Scholar] [CrossRef]
- Minemura, M.; Tanimura, H.; Tabor, E. Overexpression of multidrug resistance genes MDR1 and cMOAT in human hepatocellular carcinoma and hepatoblastoma cell lines. Int. J. Oncol 1999, 15, 559–563. [Google Scholar]
- Bader, P.; Fuchs, J.; Wenderoth, M.; von Schweinitz, D.; Niethammer, D.; Beck, J.F. Altered expression of resistance associated genes in hepatoblastoma xenografts incorporated in mice following treatment with doxorubicin or cisplatin. Anticancer Res 1998, 18, 3127–3132. [Google Scholar]
- Warmann, S.; Göhring, G.; Teichmann, B.; Geerlings, H.; Pietsch, T.; Fuchs, J. P-glycoprotein modulation improves in vitro chemosensitivity in malignant pediatric liver tumors. Anticancer Res 2003, 23, 4607–4611. [Google Scholar]
- Warmann, S.W.; Heitmann, H.; Teichmann, B.; Gratz, K.F.; Ruck, P.; Hunger, M.; Fuchs, J. Effects of P-glycoprotein modulation on the chemotherapy of xenotransplanted human hepatoblastoma. Pediatr. Hematol. Oncol 2005, 22, 373–386. [Google Scholar]
- Dean, M.; Fojo, T.; Bates, S. Tumour stem cells and drug resistance. Nat. Rev. Cancer 2005, 5, 275–284. [Google Scholar]
- Eicher, C.; Dewerth, A.; Kirchner, B.; Warmann, S.W.; Fuchs, J.; Armeanu-Ebinger, S. Development of a drug resistance model for hepatoblastoma. Int. J. Oncol 2011, 38, 447–454. [Google Scholar]
- Clarke, M.F.; Dick, J.E.; Dirks, P.B.; Eaves, C.J.; Jamieson, C.H.; Jones, D.L.; Visvader, J.; Weissman, I.L.; Wahl, G.M. Cancer stem cells—Perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 2006, 66, 9339–9344. [Google Scholar]
- Gires, O. Lessons from common markers of tumor-initiating cells in solid cancers. Cell. Mol. Life Sci 2011, 68, 4009–4022. [Google Scholar]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: Accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar]
- Tirino, V.; Desiderio, V.; Paino, F.; De Rosa, A.; Papaccio, F.; La Noce, M.; Laino, L.; de Francesco, F.; Papaccio, G. Cancer stem cells in solid tumors: An overview and new approaches for their isolation and characterization. FASEB J 2013, 27, 13–24. [Google Scholar]
- Dela Cruz, F.S. Cancer stem cells in pediatric sarcomas. Front. Oncol 2013, 3, 168. [Google Scholar]
- Tirino, V.; Desiderio, V.; D’Aquino, R.; De Francesco, F.; Pirozzi, G.; Graziano, A.; Galderisi, U.; Cavaliere, C.; De Rosa, A.; Papaccio, G.; Giordano, A. Detection and characterization of CD133+ cancer stem cells in human solid tumours. PLoS One 2008, 3, e3469. [Google Scholar]
- Tirino, V.; Desiderio, V.; Paino, F.; De Rosa, A.; Papaccio, F.; Fazioli, F.; Pirozzi, G.; Papaccio, G. Human primary bone sarcomas contain CD133+ cancer stem cells displaying high tumorigenicity in vivo. FASEB J 2011, 25, 2022–2030. [Google Scholar]
- Walter, D.; Satheesha, S.; Albrecht, P.; Bornhauser, B.C.; D’Alessandro, V.; Oesch, S.M.; Rehrauer, H.; Leuschner, I.; Koscielniak, E.; Gengler, C.; et al. CD133 positive embryonal rhabdomyosarcoma stem-like cell population is enriched in rhabdospheres. PLoS One 2011, 6, e19506. [Google Scholar]
- Goodell, M.A.; Rosenzweig, M.; Kim, H.; Marks, D.F.; DeMaria, M.; Paradis, G.; Grupp, S.A.; Sieff, C.A.; Mulligan, R.C.; Johnson, R.P. Dye efflux studies suggest that hematopoietic stem cells expressing low or undetectable levels of CD34 antigen exist in multiple species. Nat. Med 1997, 3, 1337–1345. [Google Scholar]
- Sales-Pardo, I.; Avendaño, A.; Martinez-Muñoz, V.; García-Escarp, M.; Celis, R.; Whittle, P.; Barquinero, J.; Domingo, J.C.; Marin, P.; Petriz, J. Flow cytometry of the side population: Tips and tricks. Cell. Oncol 2006, 28, 37–53. [Google Scholar]
- Hosonuma, S.; Kobayashi, Y.; Kojo, S.; Wada, H.; Seino, K.; Kiguchi, K.; Ishizuka, B. Clinical significance of side population in ovarian cancer cells. Hum. Cell 2011, 24, 9–12. [Google Scholar]
- Newton, T.C.; Wolcott, K.; Roberts, S.S. Comparison of the side populations in pretreatment and postrelapse neuroblastoma cell lines. Transl. Oncol 2010, 3, 246–251. [Google Scholar]
- Bez, A.; Corsini, E.; Curti, D.; Biggiogera, M.; Colombo, A.; Nicosia, R.F.; Pagano, S.F.; Parati, E.A. Neurosphere and neurosphere-forming cells: Morphological and ultrastructural characterization. Brain Res 2003, 993, 18–29. [Google Scholar]
- Coulon, A.; Flahaut, M.; Mühlethaler-Mottet, A.; Meier, R.; Liberman, J.; Balmas-Bourloud, K.; Nardou, K.; Yan, P.; Tercier, S.; Joseph, J.M.; et al. Functional sphere profiling reveals the complexity of neuroblastoma tumor-initiating cell model. Neoplasia 2011, 13, 991–1004. [Google Scholar]
- Gibbs, C.P.; Kukekov, V.G.; Reith, J.D.; Tchigrinova, O.; Suslov, O.N.; Scott, E.W.; Ghivizzani, S.C.; Ignatova, T.N.; Steindler, D.A. Stem-like cells in bone sarcomas: Implications for tumorigenesis. Neoplasia 2005, 7, 967–976. [Google Scholar]
- Singh, S.K.; Clarke, I.D.; Terasaki, M.; Bonn, V.E.; Hawkins, C.; Squire, J.; Dirks, P. Identification of a cancer stem cell in human brain tumors. Cancer Res 2003, 63, 5821–5828. [Google Scholar]
- Walton, J.D.; Kattan, D.R.; Thomas, S.K.; Spengler, B.A.; Guo, H.F.; Biedler, J.L.; Cheung, N.K.; Ross, R.A. Characteristics of stem cells from human neuroblastoma cell lines and in tumors. Neoplasia 2004, 6, 838–845. [Google Scholar]
- Tong, Q.S.; Zheng, L.D.; Tang, S.T.; Ruan, Q.L.; Liu, Y.; Li, S.W.; Jiang, G.S.; Cai, J.B. Expression and clinical significance of stem cell marker CD133 in human neuroblastoma. World J. Pediatr 2008, 4, 58–62. [Google Scholar]
- Takenobu, H.; Shimozato, O.; Nakamura, T.; Ochiai, H.; Yamaguchi, Y.; Ohira, M.; Nakagawara, A.; Kamijo, T. CD133 suppresses neuroblastoma cell differentiation via signal pathway modification. Oncogene 97–105.
- Kamijo, T.; Nakagawara, A. Molecular and genetic bases of neuroblastoma. Int. J. Clin. Oncol 2012, 17, 190–195. [Google Scholar]
- Hansford, L.M.; McKee, A.E.; Zhang, L.; George, R.E.; Gerstle, J.T.; Thorner, P.S.; Smith, K.M.; Look, A.T.; Yeger, H.; Miller, F.D.; et al. Neuroblastoma cells isolated from bone marrow metastases contain a naturally enriched tumor-initiating cell. Cancer Res 2007, 67, 11234–11243. [Google Scholar]
- Cournoyer, S.; Nyalendo, C.; Addioui, A.; Belounis, A.; Beaunoyer, M.; Aumont, A.; Teira, P.; Duval, M.; Fernandes, K.; Fetni, R.; et al. Genotype analysis of tumor initiating cells expressing CD133 in neuroblastoma. Genes Chromosomes Cancer 2012, 51, 792–804. [Google Scholar]
- Castelo-Branco, P.; Zhang, C.; Lipman, T.; Fujitani, M.; Hansford, L.; Clarke, I.; Harley, C.B.; Tressler, R.; Malkin, D.; Walker, E.; et al. Neural tumor-initiating cells have distinct telomere maintenance and can be safely targeted for telomerase inhibition. Clin. Cancer Res 2011, 17, 111–121. [Google Scholar]
- Wan, F.; Zhang, S.; Xie, R.; Gao, B.; Campos, B.; Herold-Mende, C.; Lei, T. The utility and limitations of neurosphere assay, CD133 immunophenotyping and side population assay in glioma stem cell research. Brain Pathol 2010, 20, 877–889. [Google Scholar]
- Chen, Y.C.; Hsu, H.S.; Chen, Y.W.; Tsai, T.H.; How, C.K.; Wang, C.Y.; Hung, S.C.; Chang, Y.L.; Tsai, M.L.; Lee, Y.Y.; et al. Oct-4 expression maintained cancer stem-like properties in lung cancer-derived CD133-positive cells. PLoS One 2008, 3, e2637. [Google Scholar]
- Hirschmann-Jax, C.; Foster, A.E.; Wulf, G.G.; Goodell, M.A.; Brenner, M.K. A distinct “side population” of cells in human tumor cells: Implications for tumor biology and therapy. Cell Cycle 2005, 4, 203–205. [Google Scholar]
- Vangipuram, S.D.; Wang, Z.J.; Lyman, W.D. Resistance of stem-like cells from neuroblastoma cell lines to commonly used chemotherapeutic agents. Pediatr. Blood Cancer 2010, 54, 361–368. [Google Scholar]
- Ruck, P.; Wichert, G.; Handgretinger, R.; Kaiserling, E. Ep-CAM in malignant liver tumours. J. Pathol 2000, 191, 102–103. [Google Scholar]
- Ward, S.C.; Thung, S.N.; Lim, K.H.; Tran, T.T.; Hong, T.K.; Hoang, P.L.; Jang, J.J.; Park, Y.N.; Abe, K. Hepatic progenitor cells in liver cancers from Asian children. Liver Int 2010, 30, 102–111. [Google Scholar]
- Yun, W.J.; Shin, E.; Lee, K.; Jung, H.Y.; Kim, S.H.; Park, Y.N.; Yu, E.; Jang, J.J. Clinicopathologic implication of hepatic progenitor cell marker expression in hepatoblastoma. Pathol. Res. Pract 2013, 209, 568–573. [Google Scholar]
- Armeanu-Ebinger, S.; Hoh, A.; Wenz, J.; Fuchs, J. Targeting EpCAM (CD326) for immunotherapy in hepatoblastoma. Oncoimmunology 2013, 2, e22620. [Google Scholar]
- Akita, M.; Tanaka, K.; Murai, N.; Matsumoto, S.; Fujita, K.; Takaki, T.; Nishiyama, H. Detection of CD133 (prominin-1) in a human hepatoblastoma cell line (HuH-6 clone 5). Microsc. Res. Tech 2013, 76, 844–852. [Google Scholar]
- Sanchez-Diaz, P.C.; Chen, T.L.; Meyers, R.; Malogolowkin, M.H.; Hung, J.Y.; Tomlinson, G.E. Sorafenib, gamma-secretase inhibitor, and bortezomib as potential therapeutic agents for hepatoblastoma. J. Clin. Oncol 2010, 28, 9550. [Google Scholar]
- Cairo, S.; Armengol, C.; De Reyniès, A.; Wei, Y.; Thomas, E.; Renard, C.A.; Goga, A.; Balakrishnan, A.; Semeraro, M.; Gresh, L.; et al. Hepatic stem-like phenotype and interplay of Wnt/beta-catenin and Myc signaling in aggressive childhood liver cancer. Cancer Cell 2008, 14, 471–484. [Google Scholar]
- Lingala, S.; Cui, Y.Y.; Chen, X.; Ruebner, B.H.; Qian, X.F.; Zern, M.A.; Wu, J. Immunohistochemical staining of cancer stem cell markers in hepatocellular carcinoma. Exp. Mol. Pathol 2010, 89, 27–35. [Google Scholar]
- Ruck, P.; Xiao, J.C.; Pietsch, T.; Von, S.D.; Kaiserling, E. Hepatic stem-like cells in hepatoblastoma: Expression of cytokeratin 7, albumin and oval cell associated antigens detected by OV-1 and OV-6. Histopathology 1997, 31, 324–329. [Google Scholar]
- Badve, S.; Logdberg, L.; Lal, A.; de Davila, M.T.; Greco, M.A.; Mitsudo, S.; Saxena, R. Small cells in hepatoblastoma lack “oval” cell phenotype. Mod. Pathol 2003, 16, 930–936. [Google Scholar]
- Pang, R.W.; Poon, R.T. Cancer stem cell as a potential therapeutic target in hepatocellular carcinoma. Curr. Cancer Drug Targets 2012, 12, 1081–1094. [Google Scholar]
- Fiegel, H.C.; Glüer, S.; Roth, B.; Rischewski, J.; von Schweinitz, D.; Ure, B.; Lambrecht, W.; Kluth, D. Stem-like cells in human hepatoblastoma. J. Histochem. Cytochem 2004, 52, 1495–1501. [Google Scholar]
- Liu, C.; Ma, Z.; Hou, J.; Zhang, H.; Liu, R.; Wu, W.; Liu, W.; Lu, Y. Germline traits of human hepatoblastoma cells associated with growth and metastasis. Biochem. Biophys. Res. Commun 2013, 437, 120–126. [Google Scholar]
- Hayashi, S.; Fujita, K.; Matsumoto, S.; Akita, M.; Satomi, A. Isolation and identification of cancer stem cells from a side population of a human hepatoblastoma cell line, HuH-6 clone-5. Pediatr. Surg. Int 2011, 27, 9–16. [Google Scholar]
- Chow, E.K.; Fan, L.L.; Chen, X.; Bishop, J.M. Oncogene-specific formation of chemoresistant murine hepatic cancer stem cells. Hepatology 2012, 56, 1331–1341. [Google Scholar]
- Holohan, C.; Van Schaeybroeck, S.; Longley, D.B.; Johnston, P.G. Cancer drug resistance: An evolving paradigm. Nat. Rev. Cancer 2013, 13, 714–726. [Google Scholar]
- Science Daily. Available online: http://www.sciencedaily.com/releases/2012/09/120910122114.htm (accessed on 10 October 2013).
- LaBarge, M.A. The difficulty of targeting cancer stem cell niches. Clin. Cancer Res 2010, 16, 3121–3129. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Alisi, A.; Cho, W.C.; Locatelli, F.; Fruci, D. Multidrug Resistance and Cancer Stem Cells in Neuroblastoma and Hepatoblastoma. Int. J. Mol. Sci. 2013, 14, 24706-24725. https://doi.org/10.3390/ijms141224706
Alisi A, Cho WC, Locatelli F, Fruci D. Multidrug Resistance and Cancer Stem Cells in Neuroblastoma and Hepatoblastoma. International Journal of Molecular Sciences. 2013; 14(12):24706-24725. https://doi.org/10.3390/ijms141224706
Chicago/Turabian StyleAlisi, Anna, William C. Cho, Franco Locatelli, and Doriana Fruci. 2013. "Multidrug Resistance and Cancer Stem Cells in Neuroblastoma and Hepatoblastoma" International Journal of Molecular Sciences 14, no. 12: 24706-24725. https://doi.org/10.3390/ijms141224706
APA StyleAlisi, A., Cho, W. C., Locatelli, F., & Fruci, D. (2013). Multidrug Resistance and Cancer Stem Cells in Neuroblastoma and Hepatoblastoma. International Journal of Molecular Sciences, 14(12), 24706-24725. https://doi.org/10.3390/ijms141224706