Structural Insights into a Novel Interkingdom Signaling Circuit by Cartography of the Ligand-Binding Sites of the Homologous Quorum Sensing LuxR-Family


Abstract
:1. Introduction
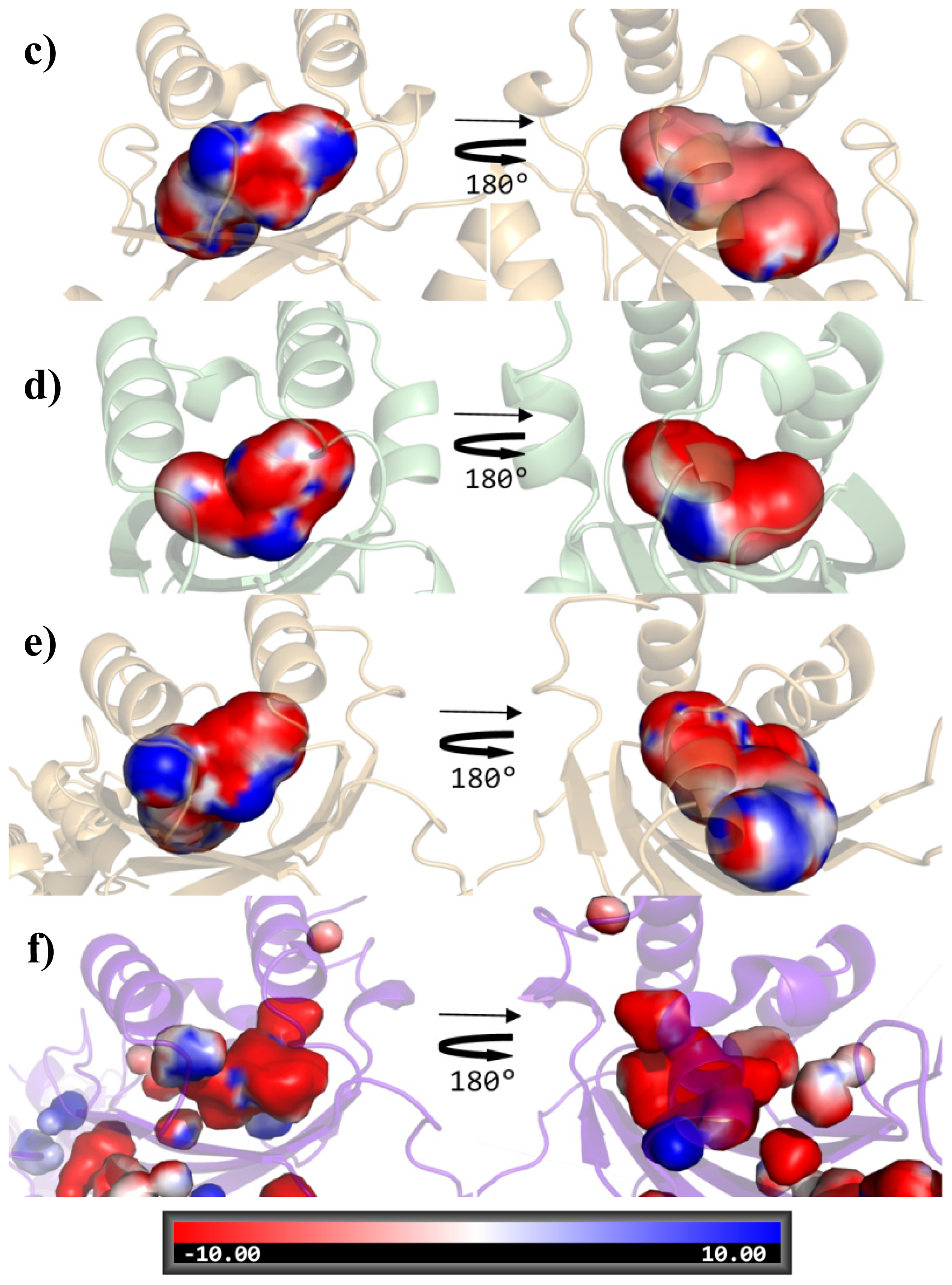
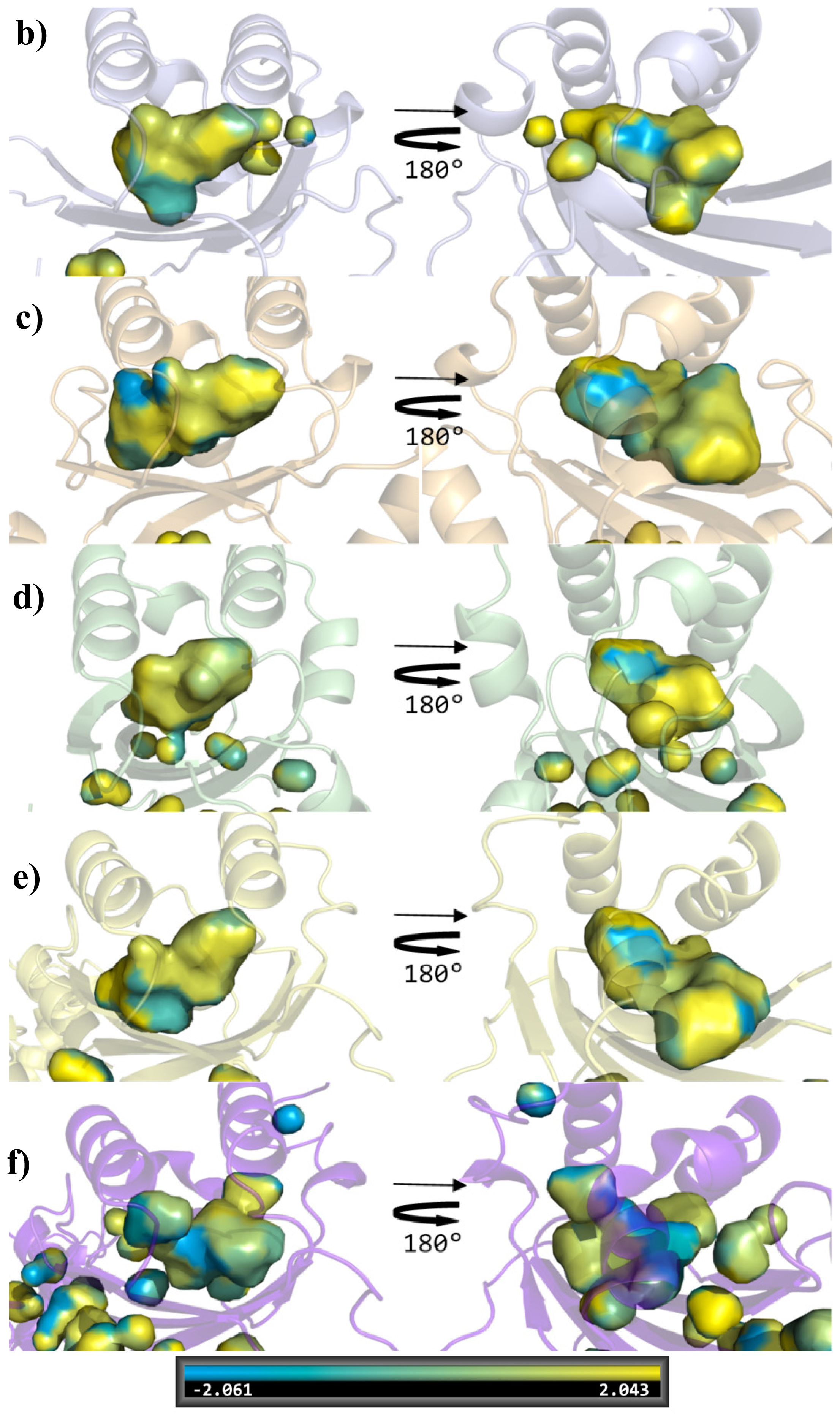
2. Results and Discussion
3. Experimental Section
4. Conclusions






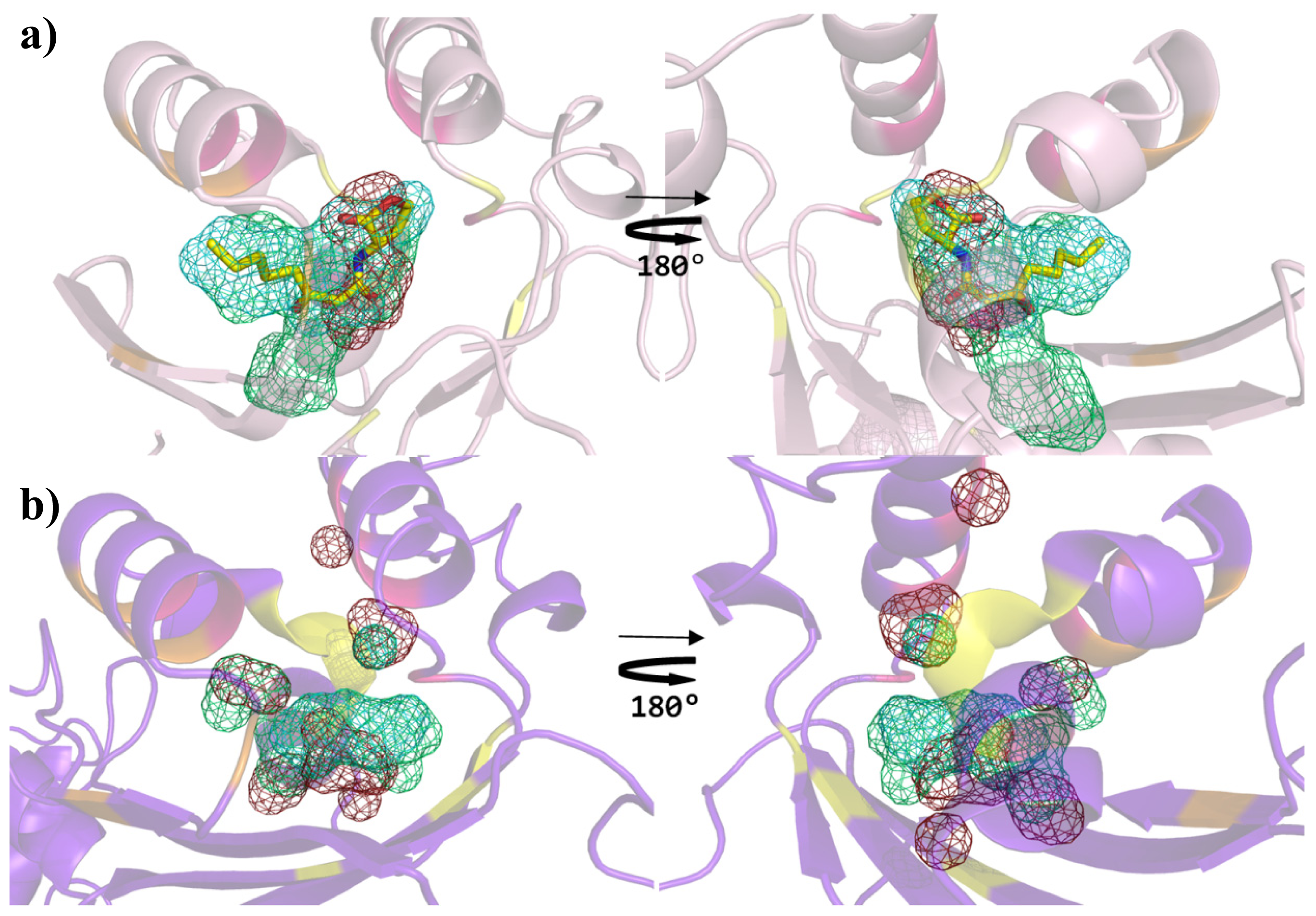

{kind=link}
{kind=link}
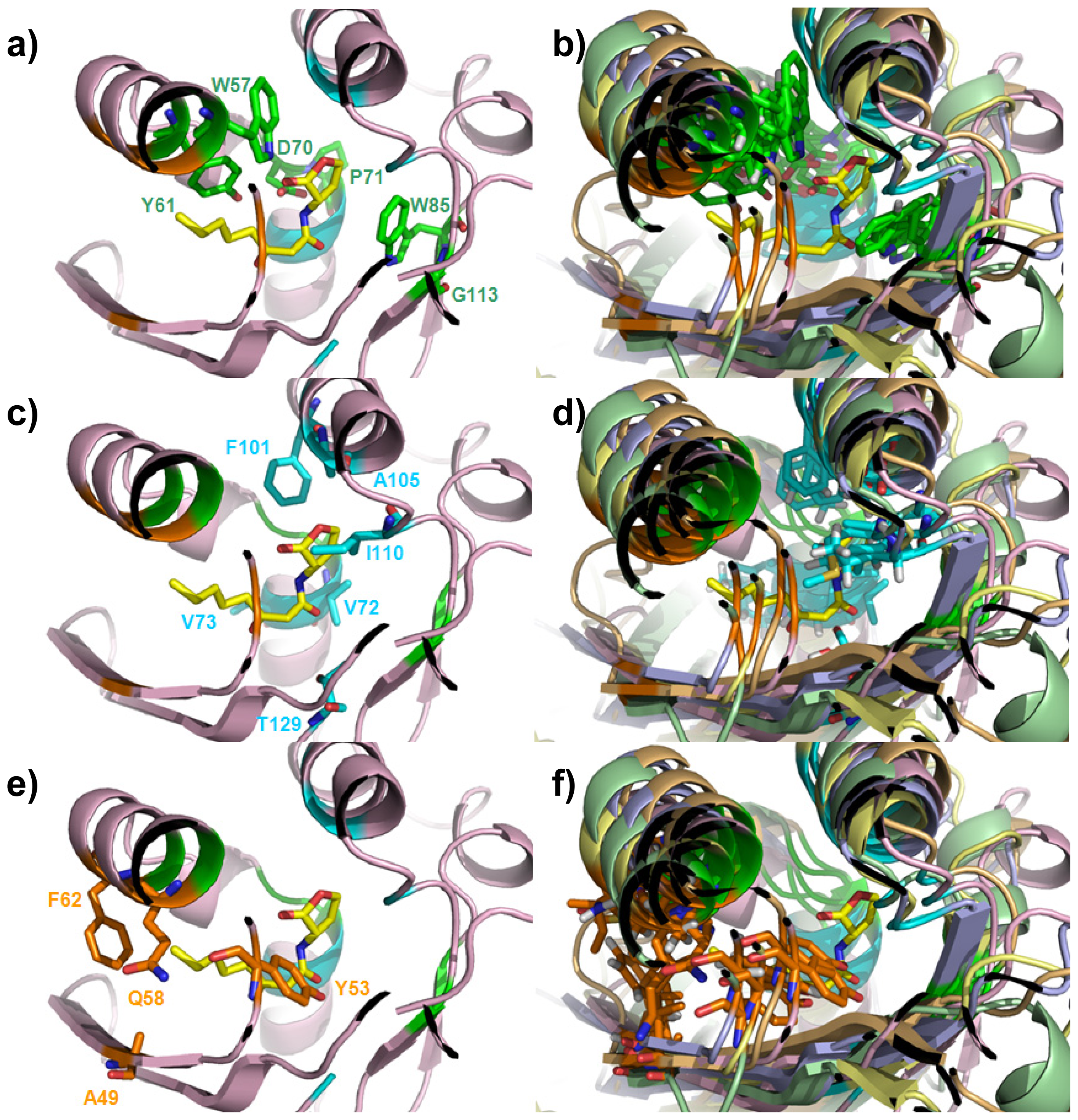{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
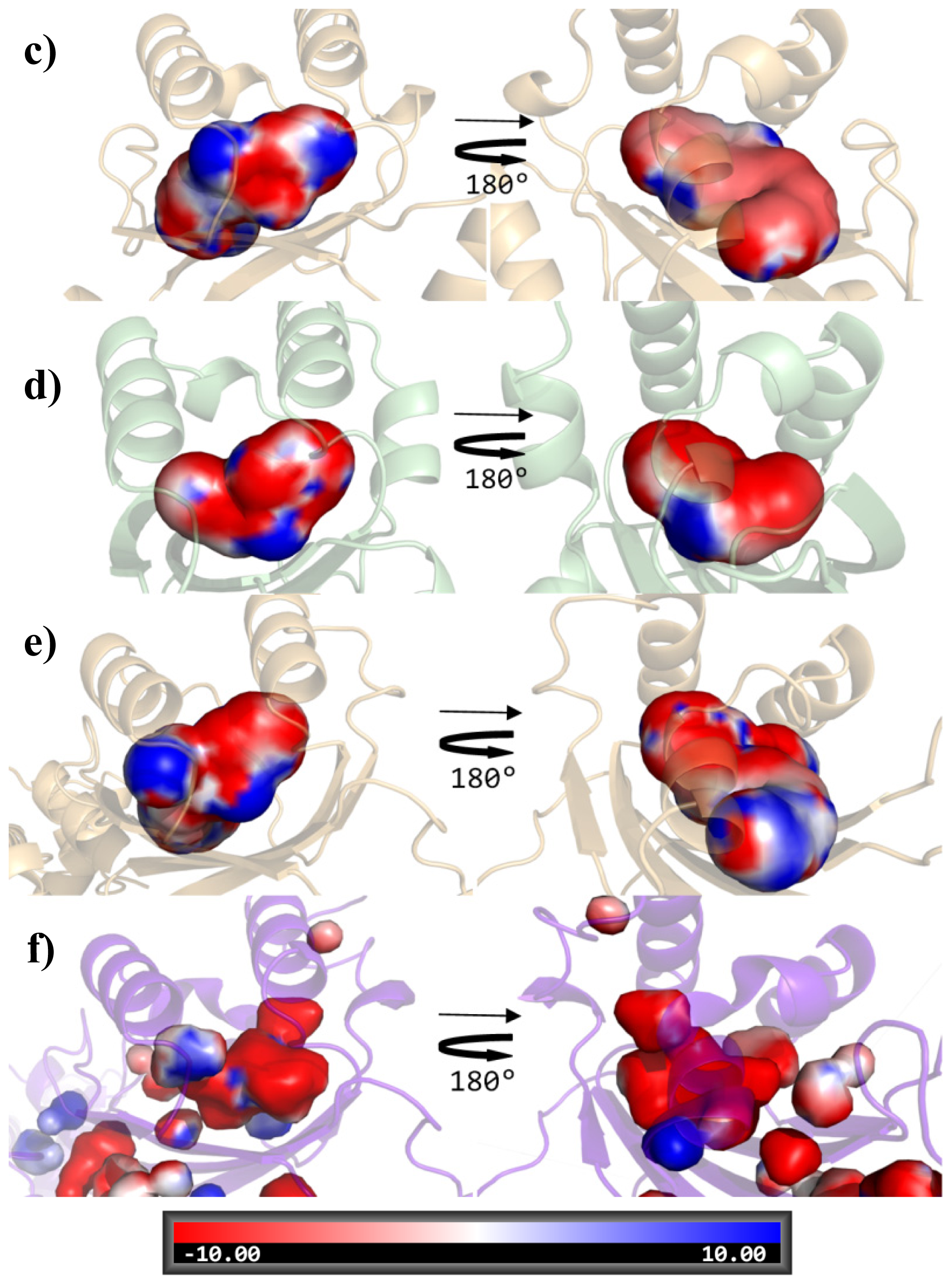{kind=link}
{kind=link}
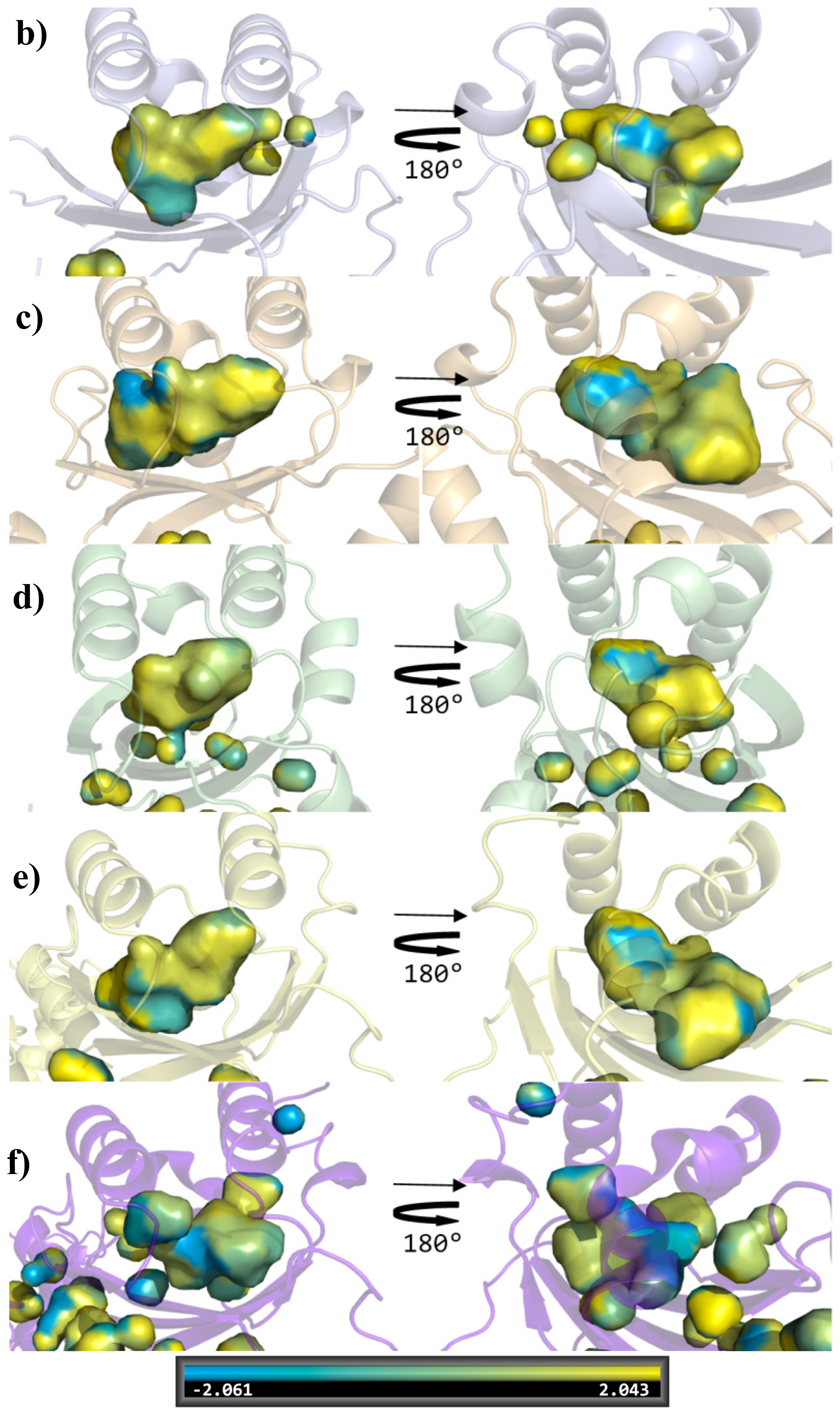{kind=link}
{kind=link}
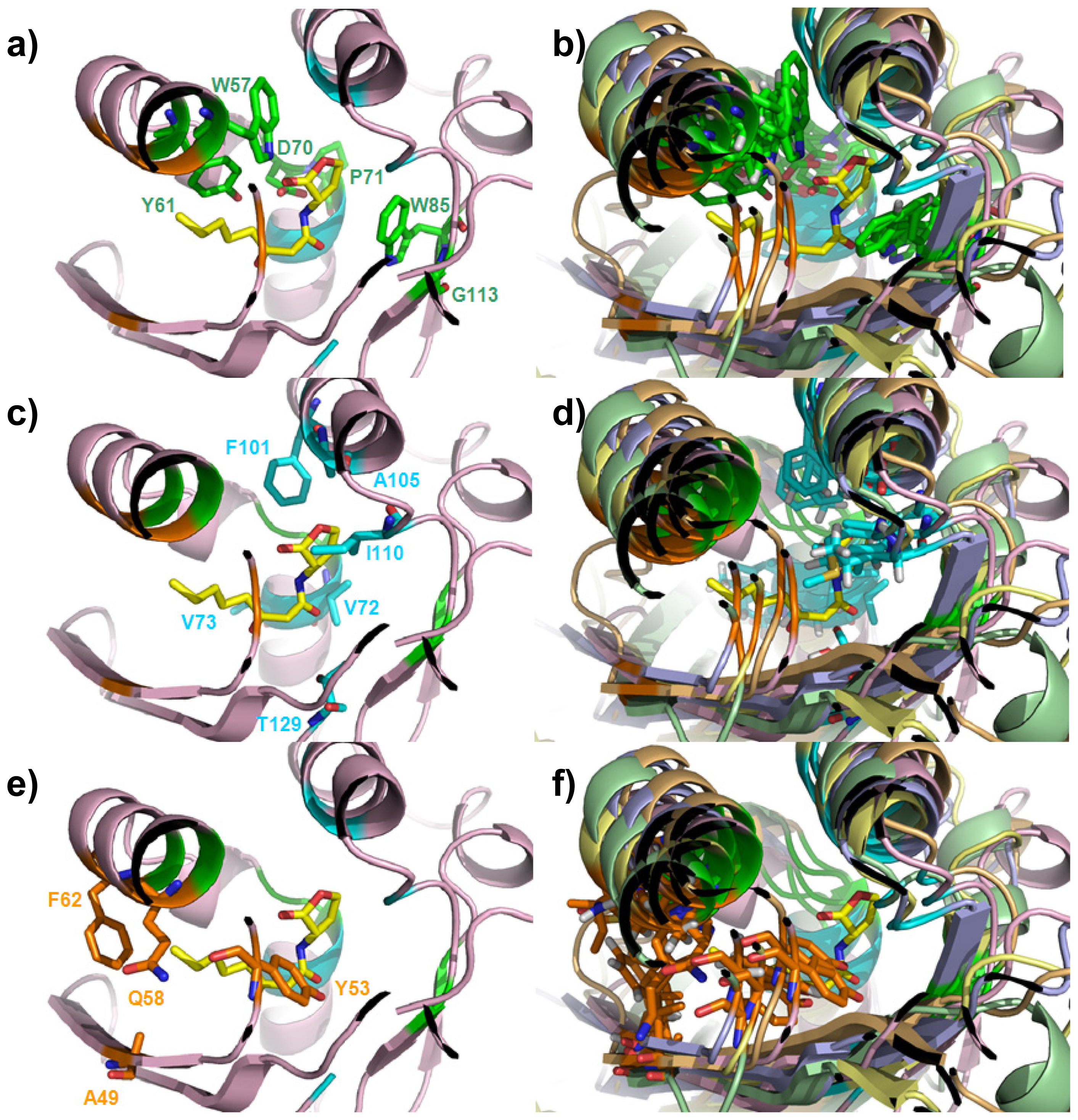
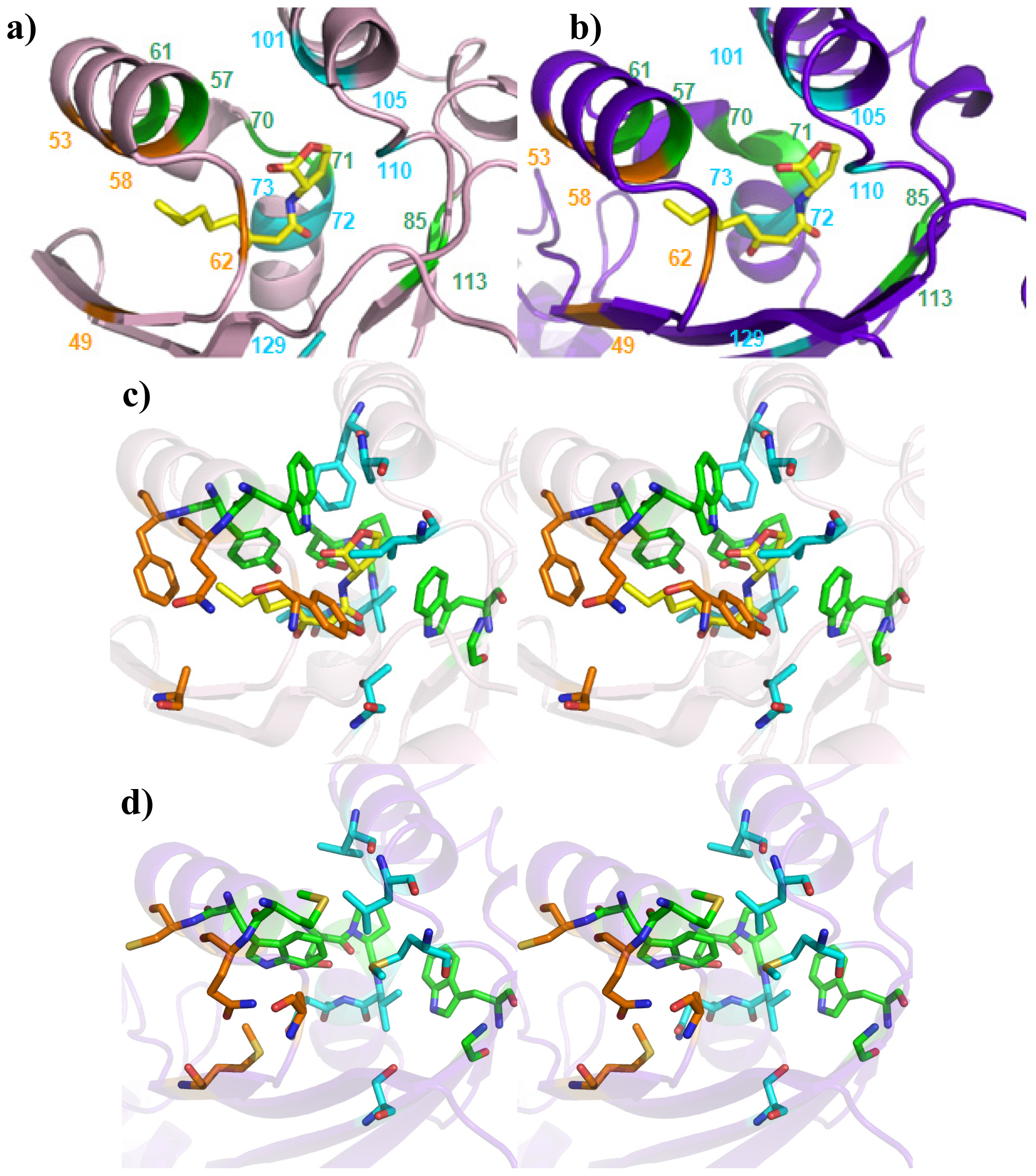
| Cluster | Position | QS LuxRs: residue and frequency | QS LuxRs: ligand binding frequency | PAB LuxR solos: residue and frequency |
|---|---|---|---|---|
| 1 | 57 | W (6/6) | 16/16 | M (13/18), W (5/18) |
| 1 | 61 | Y (6/6) | 16/16 | W (18/18) |
| 1 | 70 | D (6/6) | 2/16 | D (18/18) |
| 1 | 71 | P (6/6) | 16/16 | P (18/18) |
| 1 | 85 | W (6/6) | 0/16 | W (18/18) |
| 1 | 113 | G (6/6) | 16/16 | G (18/18) |
| 2 | 72 | V (3/6), I (2/6), T (1/6) | 16/16 | V (18/18) |
| 2 | 73 | V (3/6), L (3/6) | 16/16 | Q (18/18) |
| 2 | 101 | F (5/6), L (1/6) | 16/16 | V (18/18) |
| 2 | 105 | A (6/6) | 16/16 | L (17/18), M (1/18) |
| 2 | 110 | I (3/6), L (2/6), M (1/6) | 16/16 | M (15/18), L (2/18), V (1/18) |
| 2 | 129 | S (4/6), T (2/6) | 16/16 | S (6/18), T (12/18) |
| 3 | 49 | F (2/6), I (1/6), V (1/6), T (1/6), A (1/6) | 16/16 | M (5/18), K (5/18), E (2/18), S (2/18), L (1/18), V (1/18), Q (1/18), R (1/18) |
| 3 | 53 | Y (6/6) | 16/16 | A (6/18), T (5/18), I (4/18), L (2/18), V (1/18) |
| 3 | 58 | E (1/6), K (1/6), Q (1/6), R (1/6), L (1/6), V (1/6) | 14/16 | H (8/18), Q (5/18), R (2/18), L (2/18), A (1/18) |
| 3 | 62 | M (1/6), F (1/6), Q (1/6), D(1/6), L (1/6), I (1/6) | 14/16 | C (12/18), F (4/18), S (1/18), L (1/18) |
| Roof | Distal Wall | Floor | Proximal Wall | |
|---|---|---|---|---|
| Structural determinants | Two anti-parallel α-helices | Two perpendicular α-helices fragments | Four anti-parallel β-strands | Loops |
| Cluster 1 | W57, Y61 | D70, P71 | W85, G113 | - |
| Cluster 2 | F101, A105 | V72, V73 | T129 | I110 |
| Cluster 3 | Q58, F62 | - | A49 | Y53 |
Acknowledgments
Conflicts of Interest
References
- Fuqua, W.C.; Winans, S.C.; Greenberg, E.P. Quorum sensing in bacteria: The LuxR-LuxI family of cell density-responsive transcriptional regulators. J. Bacteriol 1994, 176, 269–275. [Google Scholar]
- Patankar, A.V.; Gonzalez, J.E. An orphan LuxR homolog of Sinorhizobium meliloti affects stress adaptation and competition for nodulation. Appl. Environ. Microbiol 2009, 75, 946–955. [Google Scholar]
- Subramoni, S.; Venturi, V. LuxR-family “solos”: Bachelor sensors/regulators of signalling molecules. Microbiology 2009, 155, 1377–1385. [Google Scholar]
- González, J.F.; Venturi, V. A novel widespread interkingdom signalling circuit. Trends Plant Sci 2013, 18, 167–174. [Google Scholar]
- Venturi, V.; Fuqua, W.C. Chemical signaling between plants and plant-pathogenic bacteria. Annu. Rev. Phytopathol 2013, 51, 17–37. [Google Scholar]
- Fuqua, C.; Greenberg, E.P. Listening in on bacteria: Acylhomoserine lactone signalling. Nat. Rev. Mol. Cell. Biol 2002, 3, 685–695. [Google Scholar]
- Slock, J.; VanRiet, D.; Kolibachuk, D.; Greenberg, E.P. Critical regions of the Vibrio fischeri LuxR protein defined by mutational analysis. J. Bacteriol 1990, 172, 3974–3979. [Google Scholar]
- Ferluga, S.; Bigirimana, J.; Hofte, M.; Venturi, V. A LuxR homologue of Xanthomonas oryzae pv. oryzae is required for optimal rice virulence. Mol. Plant. Pathol 2007, 8, 529–538. [Google Scholar]
- Ferluga, S.; Venturi, V. OryR is a LuxR-family protein involved in interkingdom signaling between pathogenic Xanthomonas. oryzae pv. oryzae and rice. J. Bacteriol 2009, 191, 890–897. [Google Scholar]
- Zhang, L.; Jia, Y.; Wang, L.; Fang, R. A proline iminopeptidase gene upregulated in planta by a LuxR homologue is essential for pathogenicity of Xanthomonas. campestris pv. campestris. Mol. Microbiol 2007, 65, 121–136. [Google Scholar]
- Subramoni, S.; Gonzalez, J.F.; Johnson, A.; Pechy-Tarr, M.; Rochat, L.; Paulsen, I.; Loper, J.E.; Keel, C.; Venturi, V. Bacterial subfamily of LuxR regulators that respond to plant compounds. Appl. Environ. Microbiol 2011, 77, 4579–4588. [Google Scholar]
- Chatnaparat, T.; Prathuangwong, S.; Ionescu, M.; Lindow, S.E. XagR, a LuxR homolog, contributes to the virulence of Xanthomonas. axonopodis pv. glycines to soybean. Mol. Plant Microbe Interact 2012, 25, 1104–1117. [Google Scholar]
- Shadel, G.S.; Young, R.; Baldwin, T.O. Use of regulated cell lysis in a lethal genetic selection in Escherichia coli: Identification of the autoinducer-binding region of the LuxR protein from Vibrio fischeri ATCC 7744. J. Bacteriol 1990, 172, 3980–3987. [Google Scholar]
- Choi, S.H.; Greenberg, E.P. Genetic dissection of DNA binding and luminescence gene activation by the Vibrio fischeri LuxR protein. J. Bacteriol 1992, 174, 4064–4069. [Google Scholar]
- Choi, S.H.; Greenberg, E.P. The C-terminal region of the Vibrio fischeri LuxR protein contains an inducer-independent lux gene activating domain. Proc. Natl. Acad. Sci. USA 1991, 88, 11115–11119. [Google Scholar]
- Zhu, J.; Winans, S.C. The quorum-sensing transcriptional regulator TraR requires its cognate signaling ligand for protein folding, protease resistance, and dimerization. Proc. Natl. Acad. Sci. USA 2001, 98, 1507–1512. [Google Scholar]
- Welch, M.; Todd, D.E.; Whitehead, N.A.; McGowan, S.J.; Bycroft, B.W.; Salmond, G.P.C. N-acyl homoserine lactone binding to the CarR receptor determines quorum-sensing specificity in Erwinia. EMBO J 2000, 19, 631–641. [Google Scholar]
- Nasser, W.; Reverchon, S. New insights into the regulatory mechanisms of the LuxR family of quorum sensing regulators. Anal. Bioanal. Chem 2007, 387, 381–390. [Google Scholar]
- Whitehead, N.A.; Barnard, A.M.; Slater, H.; Simpson, N.J.; Salmond, G.P. Quorum-sensing in Gram-negative bacteria. FEMS Microbiol. Rev 2001, 25, 365–404. [Google Scholar]
- Fuqua, C.; Winans, S.C.; Greenberg, E.P. Census and consensus in bacterial ecosystems: The LuxR-LuxI family of quorum-sensing transcriptional regulators. Annu. Rev. Microbiol 1996, 50, 727–751. [Google Scholar]
- Soulère, L.; Frezza, M.; Queneau, Y.; Doutheau, A. Exploring the active site of acyl homoserine lactones-dependent transcriptional regulators with bacterial quorum sensing modulators using molecular mechanics and docking studies. J. Mol. Graph. Model 2007, 26, 581–590. [Google Scholar]
- Ahumedo, M.; Díaz, A.; Vivas-Reyes, R. Theoretical and structural analysis of the active site of the transcriptional regulators LasR and TraR, using molecular docking methodology for identifying potential analogues of acyl homoserine lactones (AHLs) with anti-quorum sensing activity. Eur. J. Med. Chem 2010, 45, 608–615. [Google Scholar]
- Zhang, R.G.; Pappas, K.M.; Brace, J.L.; Miller, P.C.; Oulmassov, T.; Molyneaux, J.M.; Anderson, J.C.; Bashkin, J.K.; Winans, S.C.; Joachimiak, A. Structure of a bacterial quorum-sensing transcription factor complexed with pheromone and DNA. Nature 2002, 417, 971–974. [Google Scholar]
- Vannini, A.; Volpari, C.; Gargioli, C.; Muraglia, E.; Cortese, R.; De Francesco, R.; Neddermann, P.; Marco, S.D. The crystal structure of the quorum sensing protein TraR bound to its autoinducer and target DNA. EMBO J 2002, 21, 4393–4401. [Google Scholar]
- Chen, G.; Jeffrey, P.D.; Fuqua, C.; Shi, Y.; Chen, L. Structural basis for antiactivation in bacterial quorum sensing. Proc. Natl. Acad. Sci. USA 2007, 104, 16474–16479. [Google Scholar]
- Bottomley, M.J.; Muraglia, E.; Bazzo, R.; Carfì, A. Molecular insights into quorum sensing in the human pathogen Pseudomonas aeruginosa from the structure of the virulence regulator LasR bound to its autoinducer. J. Biol. Chem 2007, 282, 13592–13600. [Google Scholar]
- Zou, Y.; Nair, S.K. Molecular basis for the recognition of structurally distinct autoinducer mimics by the Pseudomonas aeruginosa LasR quorum-sensing signaling receptor. Chem. Biol 2009, 16, 961–970. [Google Scholar]
- Lintz, M.J.; Oinuma, K.; Wysoczynski, C.L.; Greenberg, E.P.; Churchill, M.E. Crystal structure of QscR, a Pseudomonas aeruginosa quorum sensing signal receptor. Proc. Natl. Acad. Sci. USA 2011, 108, 15763–15768. [Google Scholar]
- Chen, G.; Swem, L.R.; Swem, D.L.; Stauff, D.L.; O’Loughlin, C.T.; Jeffrey, P.D.; Bassler, B.L.; Hughson, F.M. A strategy for antagonizing quorum sensing. Mol. Cell 2011, 42, 199–209. [Google Scholar]
- Yao, Y.; Martinez-Yamout, M.A.; Dickerson, T.J.; Brogan, A.P.; Wright, P.E.; Dyson, H.J. Structure of the Escherichia coli quorum sensing protein SdiA: Activation of the folding switch by acyl homoserine lactones. J. Mol. Biol 2006, 355, 262–273. [Google Scholar]
- Armougom, F.; Moretti, S.; Poirot, O.; Audic, S.; Dumas, P.; Schaeli, B.; Keduas, V.; Notredame, C. Expresso: Automatic incorporation of structural information in multiple sequence alignments using 3D-Coffee. Nucleic Acids Res 2006, 34, W604–W608. [Google Scholar]
- Berman, H.M.; Westbrook, J.; Feng, Z.; Gilliland, G.; Bhat, T.N.; Weissig, H.; Shindyalov, I.N.; Bourne, P.E. The Protein Data Bank. Nucleic Acids Res 2000, 28, 235–242. [Google Scholar]
- The PyMOL Molecular Graphics System, Version 1.5.0.4; Schrödinger, LLC: Palo Alto, CA, USA, 2002.
- Krissinel, E.; Henrick, K. Inference of macromolecular assemblies from crystalline state. J. Mol. Biol 2007, 372, 774–797. [Google Scholar]
- Patankar, A.V.; González, J.E. Orphan LuxR regulators of quorum sensing. FEMS Microbiol. Rev 2009, 33, 739–756. [Google Scholar]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinforma 2008, 9, 40–47. [Google Scholar]
- Hernandez, M.; Ghersi, D.; Sanchez, R. SITEHOUND-web: A server for ligand binding site identification in protein structures. Nucleic Acids Res. 2009, 37, W413–W416. [Google Scholar]
- Taylor, W.R. Protein structure comparison using SAP. Methods Mol. Biol 2000, 143, 19–32. [Google Scholar]
- Zhang, Y.; Skolnick, J. TM-align: A protein structure alignment algorithm based on TM-score. Nucleic Acids Res. 2005, 33, 2302–2309. [Google Scholar]
- Arnold, K.; Bordoli, L.; Kopp, J.; Schwede, T. The SWISS-MODEL workspace: A web-based environment for protein structure homology modelling. Bioinformatics 2006, 22, 195–201. [Google Scholar]
- Huang, X.; Miller, W. A time-efficient, linear-space local similarity algorithm. Adv. Appl. Math 1991, 12, 337–357. [Google Scholar]
- Eswar, N.; John, B.; Mirkovic, N.; Fiser, A.; Ilyin, V.A.; Pieper, U.; Stuart, A.C.; Marti-Renom, M.A.; Madhusudhan, M.S.; Yerkovich, B.; et al. Tools for comparative protein structure modeling and analysis. Nucleic Acids Res 2003, 31, 3375–3380. [Google Scholar]
- Martí-Renom, M.A.; Stuart, A.C.; Fiser, A.; Sánchez, R.; Melo, F.; Sali, A. Comparative protein structure modeling of genes and genomes. Annu. Rev. Biophys. Biomol. Struct 2000, 29, 291–325. [Google Scholar]
- Pieper, U.; Eswar, N.; Stuart, A.C.; Ilyin, V.A; Sali, A. MODBASE, a database of annotated comparative protein structure models. Nucleic Acids Res 2002, 30, 255–259. [Google Scholar]
- Sanchez, R.; Sali, A. Large-scale protein structure modeling of the Saccharomyces cerevisiae genome. Proc. Natl. Acad. Sci. USA 1998, 95, 13597–13602. [Google Scholar]
- Fernandez-Fuentes, N.; Madrid-Aliste, C.J.; Rai, B.K.; Fajardo, J.E.; Fiser, A. M4T: A comparative protein structure modeling server. Nucleic Acids Res 2007, 35, 363–368. [Google Scholar]
- Rai, B.K; Fiser, A. Multiple mapping method: A novel approach to the sequence-to-structure alignment problem in comparative protein structure modeling. Proteins 2006, 63, 644–661. [Google Scholar]
- Biegert, A.; Mayer, C.; Remmert, M.; Soding, J.; Lupas, A.N. The MPI Bioinformatics Toolkit for protein sequence analysis. Nucleic Acids Res 2006, 34, W335–W339. [Google Scholar]
- Wallner, B.; Elofsson, A. Can correct protein models be identified? Protein Sci 2003, 12, 1073–1086. [Google Scholar]
- Mereghetti, P.; Ganadu, M.L.; Papaleo, E.; Fantucci, P.; De Gioia, L. Validation of protein models by a neural network approach. BMC Bioinforma 2008, 9, 66–76. [Google Scholar]
- Dolinsky, T.J.; Nielsen, J.E.; McCammon, J.A.; Baker, N.A. PDB2PQR: An automated pipeline for the setup, execution, and analysis of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res 2004, 32, W665–W667. [Google Scholar]
- Black, S.D.; Mould, D.R. Amino acid scale: Hydrophobicity of physiological L-alpha amino acids. Anal. Biochem. 1991, 193, 72–82. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Covaceuszach, S.; Degrassi, G.; Venturi, V.; Lamba, D. Structural Insights into a Novel Interkingdom Signaling Circuit by Cartography of the Ligand-Binding Sites of the Homologous Quorum Sensing LuxR-Family. Int. J. Mol. Sci. 2013, 14, 20578-20596. https://doi.org/10.3390/ijms141020578
Covaceuszach S, Degrassi G, Venturi V, Lamba D. Structural Insights into a Novel Interkingdom Signaling Circuit by Cartography of the Ligand-Binding Sites of the Homologous Quorum Sensing LuxR-Family. International Journal of Molecular Sciences. 2013; 14(10):20578-20596. https://doi.org/10.3390/ijms141020578
Chicago/Turabian StyleCovaceuszach, Sonia, Giuliano Degrassi, Vittorio Venturi, and Doriano Lamba. 2013. "Structural Insights into a Novel Interkingdom Signaling Circuit by Cartography of the Ligand-Binding Sites of the Homologous Quorum Sensing LuxR-Family" International Journal of Molecular Sciences 14, no. 10: 20578-20596. https://doi.org/10.3390/ijms141020578
APA StyleCovaceuszach, S., Degrassi, G., Venturi, V., & Lamba, D. (2013). Structural Insights into a Novel Interkingdom Signaling Circuit by Cartography of the Ligand-Binding Sites of the Homologous Quorum Sensing LuxR-Family. International Journal of Molecular Sciences, 14(10), 20578-20596. https://doi.org/10.3390/ijms141020578