Non-Coding RNAs in Muscle Dystrophies

Abstract
:1. Introduction
2. Muscle-Specific and Ubiquitously Expressed miRNAs in Skeletal Muscle
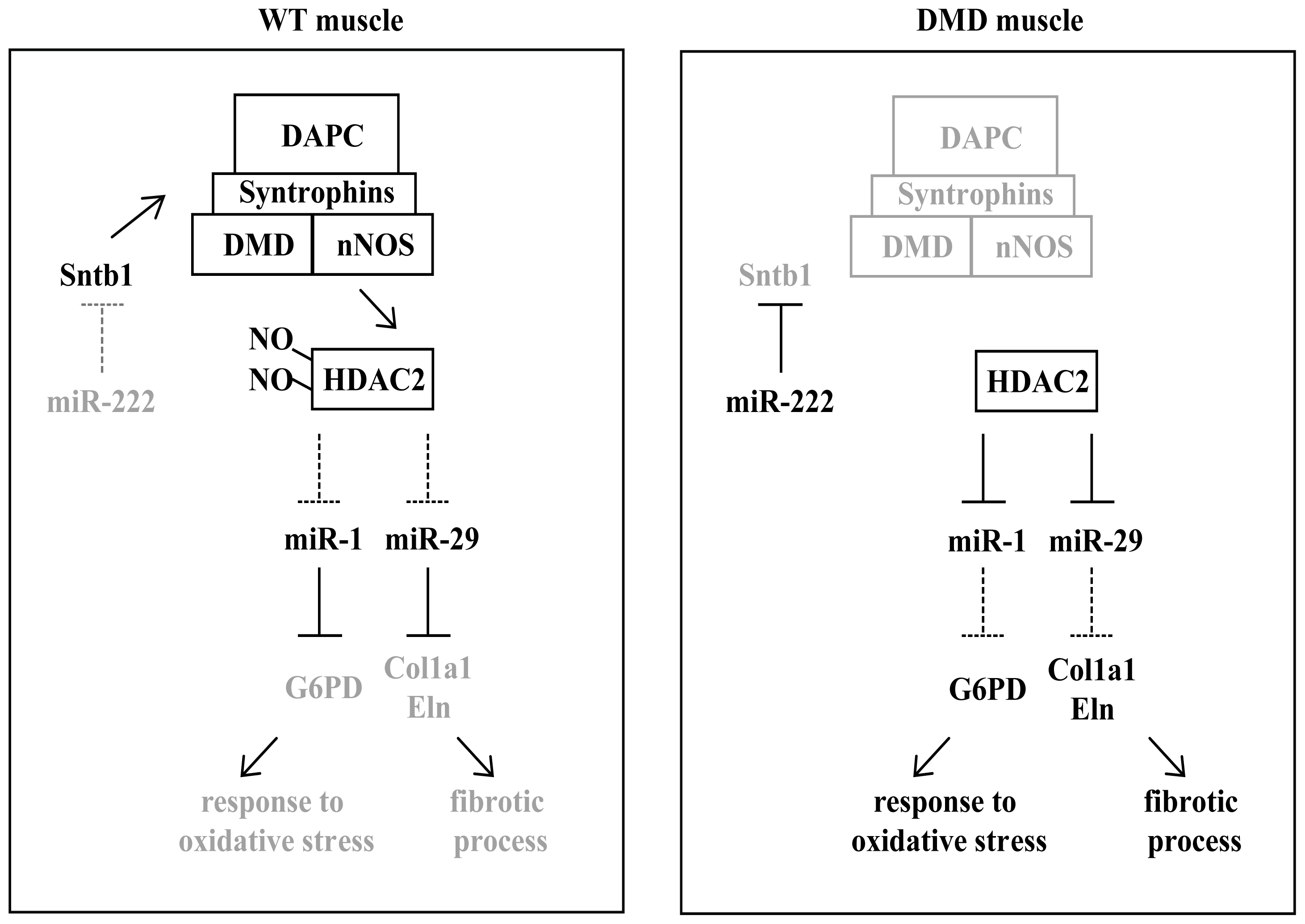
3. miRNAs in Muscular Dystrophies
4. Long Non-Coding RNAs in Skeletal Muscle and Muscular Dystrophies
5. Discussion
6. Conclusions
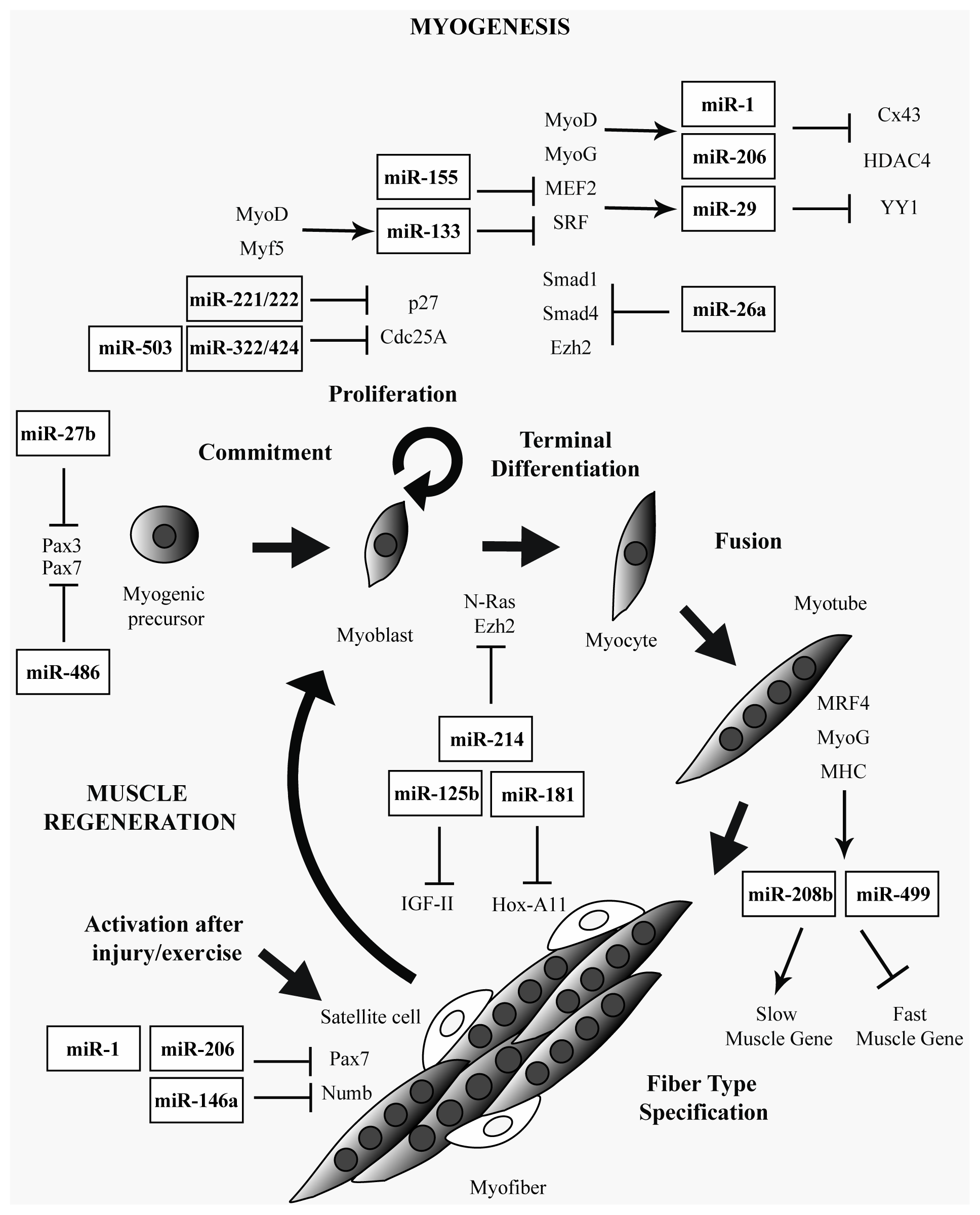

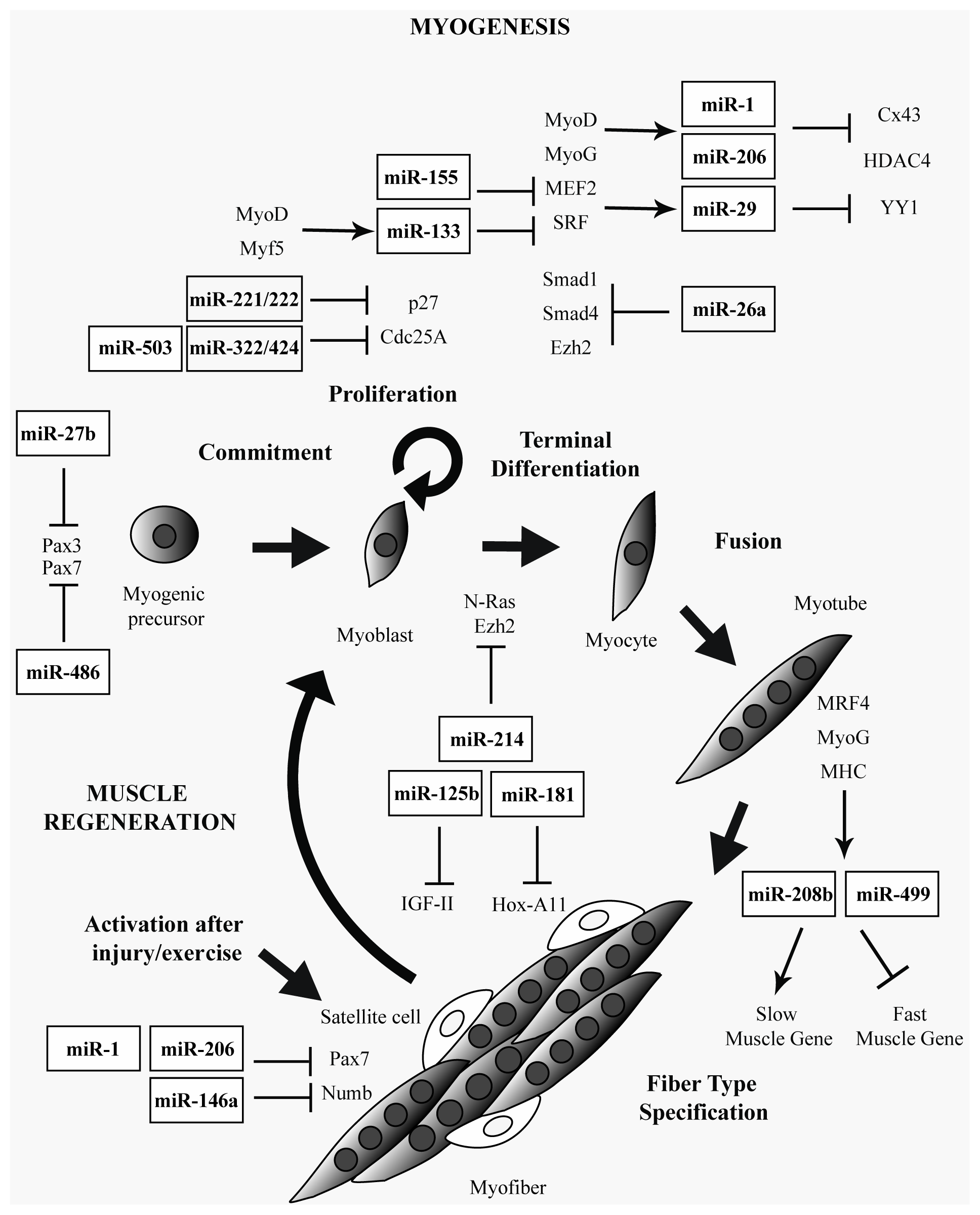{kind=link}
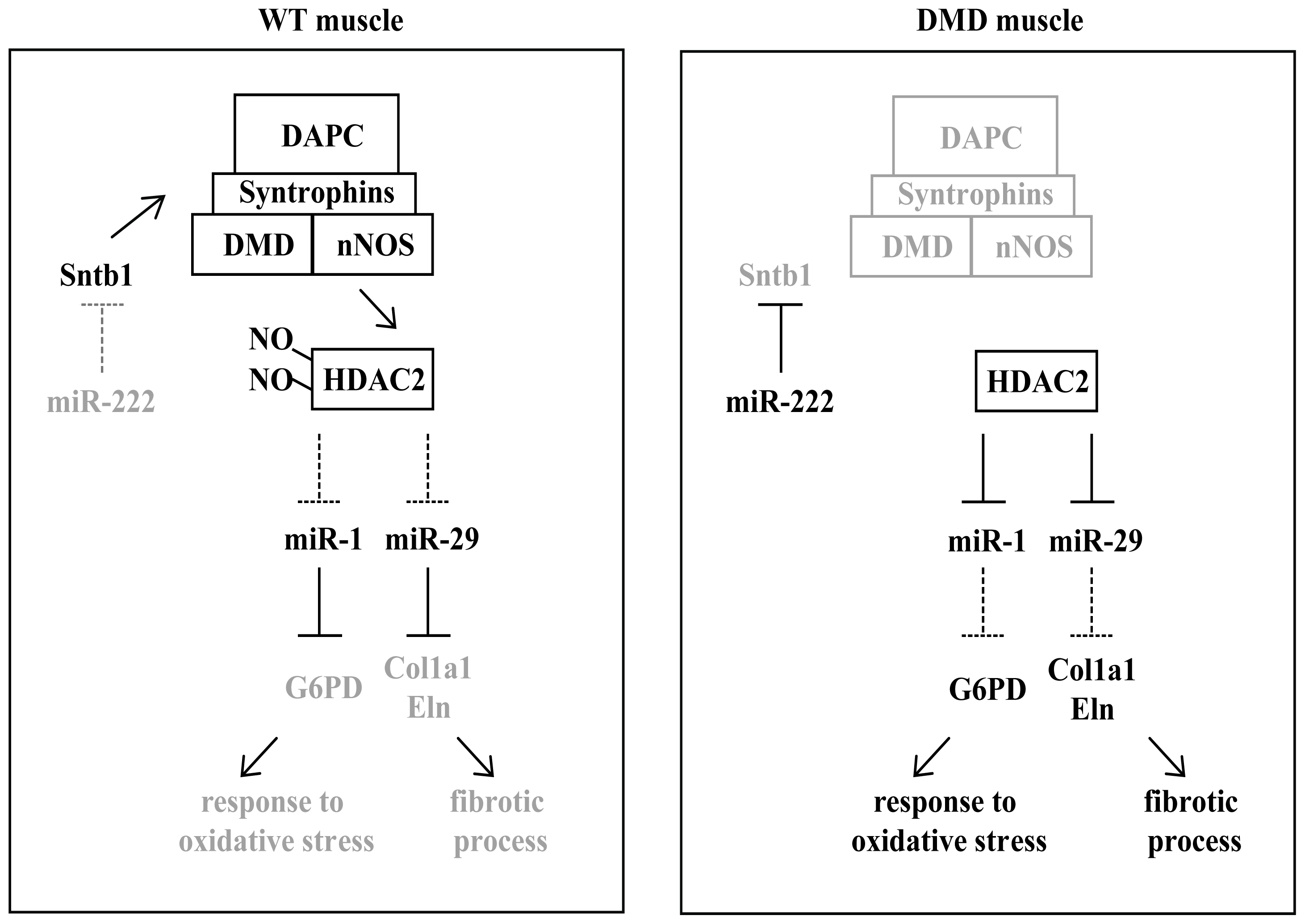{kind=link}
| miRNA | Role in Muscle Metabolism [Refs.] | Tissue Expression |
|---|---|---|
| miR-1 | enhancer of skeletal muscle differentiation [37–40] | muscle-specific |
| miR-133a/b | enhancer of myoblast proliferation [37–40] | muscle-specific |
| miR-206 | enhancer of skeletal muscle differentiation [37–40] | muscle-specific |
| miR-208b | involved in specification of muscle fiber identity [43] | muscle-specific |
| miR-499 | involved in specification of muscle fiber identity [43] | muscle-specific |
| miR-24 | promotes myoblast differentiation [44] | ubiquitous |
| miR-26a | promotes myoblast differentiation [45,46] | ubiquitous |
| miR-27b | promotes entry into differentiation program [47] | ubiquitous |
| miR-29 | enhancer of differentiation [48,49] | ubiquitous |
| miR-125b | negatively contributes to the myoblast differentiation and muscle regeneration [50–52] | ubiquitous |
| miR-155 | represses myoblast differentiation [53] | ubiquitous |
| miR-181 | regulates skeletal muscle differentiation and regeneration after injury [54] | ubiquitous |
| miR-146a | promotes satellite cell differentiation [55,56] | ubiquitous |
| miR-214 | promotes cell cycle exit and differentiation [57] | ubiquitous |
| miR-221/222 | promote cell cycle progression [58] | ubiquitous |
| miR-322/424; miR-503 | promote myogenesis interfering with the progression through the cell cycle [59] | ubiquitous |
| miR-486 | positively regulates myoblast differentiation [60,61] | muscle-enriched |
| miRNA/miRNAs | Deregulated in MDs [References] | Type of Deregulation | Muscular Targets/Process [References] |
|---|---|---|---|
| miR-1 (myomiR) | DMD [65,66]; DM1 [67,68] | down-regulated | HDAC4; Cx43; Pax7; c-Met; G6PD [40] |
| miR-133 (myomiR) | DMD [66] | down-regulated | SRF; nPTB; UCP2 [40] |
| miR-206 (myomiR) | DMD [65]; DM1 [69] | up-regulated | DNApolα; Fstl1; Utrn; Pax7; Cx43; HDAC4; c-Met [40] |
| miR-29b/c | DMD [65,66]; DM1 [67] | down-regulated | YY1; Col1a1; Eln; HDAC4 [40,62,63] |
| miR-135a | DMD [65] | down-regulated | muscle degeneration [65] |
| miR-30c | DMD [66] | down-regulated | - |
| miR-31 | DMD [65,70] | up-regulated | DMD [70] |
| miR-34c; miR-449; miR-494 | DMD [65] | up-regulated | muscle regeneration [65] |
| miR-146b; miR-155 | DMD; BMD; LGMD; FSHD [71] | up-regulated | -; MEF2A [53] |
| miR-214 | DMD; BMD; LGMD; FSHD [71] | up-regulated | Ezh2; N-Ras [40,62,63] |
| miR-221; miR-222 | DMD; BDM; LGMD; FSHD [71] | up-regulated | p27(Cdkn1b/Kip1) [58]; Sntb1 [72] |
| miR-223 | DMD [65] | up-regulated | muscle inflammation [65] |
| miR-335 | DMD [65]; DM1 [67] | up-regulated | muscle regeneration [65] |
| miR-33 | DM1 [67] | down-regulated | - |
| miR-34a-5p; miR-34b-3p; miR-34c-5p; miR-146b-5p; miR-208a; miR-221-3p; miR-381 | DM2 [73] | up-regulated | - |
| miR-125b-5p; miR-193a-3p; miR-193b-3p; miR-378a-3p | DM2 [73] | down-regulated | - |
| lncRNA/lncRNAs [References] | Expression in Muscular Districts | Deregulated in MDs | Activity |
|---|---|---|---|
| linc-MD1 [90] | Expressed in newly regenerating fibers | DMD | natural decoy for miR-133 and -135 (ceRNA) |
| Malat1 [91] | up-regulated during the differentiation of myoblasts into myotubes | ? | regulation of cell growth |
| Men ɛ/β lncRNAs [92–94] | up-regulated upon differentiation of C2C12 myoblats | ? | critical structural/organizational components of paraspeckles |
| SRA ncRNA [95–97] | increased expression during myogenic differentiation | DM1 | co-activator of MYOD transcription factor |
| NRON [98,99] | enriched also in muscle | ? | regulates NFAT’s subcellular localization (scaffold) |
| lncINT44s; lncINT44s2; lncINT55s [14] | transcribed contextually with dystrophin isoforms and upon MYOD-induced myogenic differentiation | ? | negative modulation of endogenous dystrophin full-length isoforms |
| KUCG1 [100] | expressed at low levels in the brain | DMD with mental retardation | possible candidate gene that contribute to develop of mental retardation in the index case |
| DBT-E [101] | not-physiological lncRNA | FSHD | coordinates de-repression of genes located in the 4q35 region |
Acknowledgments
Conflicts of Interest
References
- Carninci, P.; Kasukawa, T.; Katayama, S.; Gough, J.; Frith, M.C.; Maeda, N.; Oyama, R.; Ravasi, T.; Lenhard, B.; Wells, C.; et al. The transcriptional landscape of the mammalian genome. Science 2005, 309, 1559–1563. [Google Scholar]
- Mattick, J.S.; Makunin, I.V. Non-coding RNA. Hum. Mol. Genet 2006, 15, R17–R29. [Google Scholar]
- Kapranov, P.; Cheng, J.; Dike, S.; Nix, D.A.; Duttagupta, R.; Willingham, A.T.; Stadler, P.F.; Hertel, J.; Hackermuller, J.; Hofacker, I.L.; et al. RNA maps reveal new RNA classes and a possible function for pervasive transcription. Science 2007, 316, 1484–1488. [Google Scholar]
- Wang, X.Q.; Crutchley, J.L.; Dostie, J. Shaping the genome with non-coding RNAs. Curr. Genomics 2011, 12, 307–321. [Google Scholar]
- Ponting, C.P.; Oliver, P.L.; Reik, W. Evolution and functions of long noncoding RNAs. Cell 2009, 136, 629–641. [Google Scholar]
- Cirak, S.; Arechavala-Gomeza, V.; Guglieri, M.; Feng, L.; Torelli, S.; Anthony, K.; Abbs, S.; Garralda, M.E.; Bourke, J.; Wells, D.J.; et al. Exon skipping and dystrophin restoration in patients with duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: An open-label, phase 2, dose-escalation study. Lancet 2011, 378, 595–605. [Google Scholar]
- Goemans, N.M.; Tulinius, M.; van den Akker, J.T.; Burm, B.E.; Ekhart, P.F.; Heuvelmans, N.; Holling, T.; Janson, A.A.; Platenburg, G.J.; Sipkens, J.A.; et al. Systemic administration of pro051 in duchenne’s muscular dystrophy. N. Engl. J. Med 2011, 364, 1513–1522. [Google Scholar]
- Evers, M.M.; Pepers, B.A.; van Deutekom, J.C.; Mulders, S.A.; den Dunnen, J.T.; Aartsma-Rus, A.; van Ommen, G.J.; van Roon-Mom, W.M. Targeting several cag expansion diseases by a single antisense oligonucleotide. PLoS One 2011, 6, e24308. [Google Scholar]
- Ferlini, A.; Neri, M.; Gualandi, F. The medical genetics of dystrophinopathies: Molecular genetic diagnosis and its impact on clinical practice. Neuromuscul. Disord. NMD 2013, 23, 4–14. [Google Scholar]
- Mitsuhashi, S.; Kang, P.B. Update on the genetics of limb girdle muscular dystrophy. Semin. Pediatr. Neurol 2012, 19, 211–218. [Google Scholar]
- Pegoraro, E.; Hoffman, E.P. Limb-girdle Muscular Dystrophy Overview. In Genereviews; Pagon, R.A., Adam, M.P., Bird, T.D., Dolan, C.R., Fong, C.T., Stephens, K.,, Eds.; University of Washington: Seattle, WA, USA, 1993. [Google Scholar]
- Davies, K.E.; Nowak, K.J. Molecular mechanisms of muscular dystrophies: Old and new players. Nat. Rev. Mol. Cell Biol 2006, 7, 762–773. [Google Scholar]
- Johnson, N.E.; Heatwole, C.R. Myotonic dystrophy: From bench to bedside. Semin. Neurol 2012, 32, 246–254. [Google Scholar]
- Bovolenta, M.; Erriquez, D.; Valli, E.; Brioschi, S.; Scotton, C.; Neri, M.; Falzarano, M.S.; Gherardi, S.; Fabris, M.; Rimessi, P.; et al. The dmd locus harbours multiple long non-coding RNAs which orchestrate and control transcription of muscle dystrophin mRNA isoforms. PLoS One 2012, 7, e45328. [Google Scholar]
- Graves, P.; Zeng, Y. Biogenesis of mammalian micro RNAs: A global view. Genomics Proteomics Bioinforma 2012, 10, 239–245. [Google Scholar]
- Bentzinger, C.F.; von Maltzahn, J.; Rudnicki, M.A. Extrinsic regulation of satellite cell specification. Stem Cell Res. Ther 2010, 1, 27. [Google Scholar]
- Bentzinger, C.F.; Wang, Y.X.; Rudnicki, M.A. Building muscle: Molecular regulation of myogenesis. Cold Spring Harb Perspect Biol 2012, 4. [Google Scholar] [CrossRef]
- Bismuth, K.; Relaix, F. Genetic regulation of skeletal muscle development. Exp. Cell Res 2010, 316, 3081–3086. [Google Scholar]
- Bryson-Richardson, R.J.; Currie, P.D. The genetics of vertebrate myogenesis. Nat. Rev. Genet 2008, 9, 632–646. [Google Scholar]
- Punch, V.G.; Jones, A.E.; Rudnicki, M.A. Transcriptional networks that regulate muscle stem cell function. Wiley Interdiscip Rev. Syst. Biol. Med 2009, 1, 128–140. [Google Scholar]
- Kassar-Duchossoy, L.; Gayraud-Morel, B.; Gomes, D.; Rocancourt, D.; Buckingham, M.; Shinin, V.; Tajbakhsh, S. Mrf4 determines skeletal muscle identity in myf5:Myod double-mutant mice. Nature 2004, 431, 466–471. [Google Scholar]
- Nabeshima, Y.; Hanaoka, K.; Hayasaka, M.; Esumi, E.; Li, S.; Nonaka, I. Myogenin gene disruption results in perinatal lethality because of severe muscle defect. Nature 1993, 364, 532–535. [Google Scholar]
- Charge, S.B.; Rudnicki, M.A. Cellular and molecular regulation of muscle regeneration. Physiol. Rev 2004, 84, 209–238. [Google Scholar]
- Parker, M.H.; Seale, P.; Rudnicki, M.A. Looking back to the embryo: Defining transcriptional networks in adult myogenesis. Nat. Rev. Genet 2003, 4, 497–507. [Google Scholar]
- Pownall, M.E.; Gustafsson, M.K.; Emerson, C.P., Jr. Myogenic regulatory factors and the specification of muscle progenitors in vertebrate embryos. Annu. Rev. Cell Dev. Biol 2002, 18, 747–783. [Google Scholar]
- Buckingham, M. Skeletal muscle formation in vertebrates. Curr. Opin. Genet. Dev 2001, 11, 440–448. [Google Scholar]
- Hasty, P.; Bradley, A.; Morris, J.H.; Edmondson, D.G.; Venuti, J.M.; Olson, E.N.; Klein, W.H. Muscle deficiency and neonatal death in mice with a targeted mutation in the myogenin gene. Nature 1993, 364, 501–506. [Google Scholar]
- Yokoyama, S.; Asahara, H. The myogenic transcriptional network. Cell Mol. Life Sci 2011, 68, 1843–1849. [Google Scholar]
- O’Rourke, J.R.; Georges, S.A.; Seay, H.R.; Tapscott, S.J.; McManus, M.T.; Goldhamer, D.J.; Swanson, M.S.; Harfe, B.D. Essential role for dicer during skeletal muscle development. Dev. Biol 2007, 311, 359–368. [Google Scholar]
- Zhao, Y.; Samal, E.; Srivastava, D. Serum response factor regulates a muscle-specific microRNA that targets hand2 during cardiogenesis. Nature 2005, 436, 214–220. [Google Scholar]
- Rosenberg, M.I.; Georges, S.A.; Asawachaicharn, A.; Analau, E.; Tapscott, S.J. Myod inhibits fstl1 and utrn expression by inducing transcription of mir-206. J. Cell Biol 2006, 175, 77–85. [Google Scholar]
- Rao, P.K.; Kumar, R.M.; Farkhondeh, M.; Baskerville, S.; Lodish, H.F. Myogenic factors that regulate expression of muscle-specific microRNAs. Proc. Natl. Acad. Sci. USA 2006, 103, 8721–8726. [Google Scholar]
- Liu, N.; Williams, A.H.; Kim, Y.; McAnally, J.; Bezprozvannaya, S.; Sutherland, L.B.; Richardson, J.A.; Bassel-Duby, R.; Olson, E.N. An intragenic mef2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proc. Natl. Acad. Sci. USA 2007, 104, 20844–20849. [Google Scholar]
- Sun, Y.; Ge, Y.; Drnevich, J.; Zhao, Y.; Band, M.; Chen, J. Mammalian target of rapamycin regulates miRNA-1 and follistatin in skeletal myogenesis. J. Cell Biol 2010, 189, 1157–1169. [Google Scholar]
- Weintraub, H. The myod family and myogenesis: Redundancy, networks, and thresholds. Cell 1993, 75, 1241–1244. [Google Scholar]
- Naya, F.J.; Olson, E. Mef2: A transcriptional target for signaling pathways controlling skeletal muscle growth and differentiation. Curr. Opin. Cell Biol 1999, 11, 683–688. [Google Scholar]
- Van Rooij, E.; Liu, N.; Olson, E.N. MicroRNAs flex their muscles. Trends Genet. TIG 2008, 24, 159–166. [Google Scholar]
- Chen, J.F.; Callis, T.E.; Wang, D.Z. MicroRNAs and muscle disorders. J. Cell Sci 2009, 122, 13–20. [Google Scholar]
- Eisenberg, I.; Alexander, M.S.; Kunkel, L.M. miRNAs in normal and diseased skeletal muscle. J. Cell. Mol. Med 2009, 13, 2–11. [Google Scholar]
- Ge, Y.; Chen, J. MicroRNAs in skeletal myogenesis. Cell Cycle 2011, 10, 441–448. [Google Scholar]
- Cacchiarelli, D.; Legnini, I.; Martone, J.; Cazzella, V.; D’Amico, A.; Bertini, E.; Bozzoni, I. miRNAs as serum biomarkers for duchenne muscular dystrophy. EMBO Mol. Med 2011, 3, 258–265. [Google Scholar]
- Mitchell, P.S.; Parkin, R.K.; Kroh, E.M.; Fritz, B.R.; Wyman, S.K.; Pogosova-Agadjanyan, E.L.; Peterson, A.; Noteboom, J.; O’Briant, K.C.; Allen, A.; et al. Circulating microRNAs as stable blood-based markers for cancer detection. Proc. Natl. Acad. Sci. USA 2008, 105, 10513–10518. [Google Scholar]
- Van Rooij, E.; Quiat, D.; Johnson, B.A.; Sutherland, L.B.; Qi, X.; Richardson, J.A.; Kelm, R.J., Jr.; Olson, E.N. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Dev. Cell 2009, 17, 662–673. [Google Scholar]
- Sun, Q.; Zhang, Y.; Yang, G.; Chen, X.; Cao, G.; Wang, J.; Sun, Y.; Zhang, P.; Fan, M.; Shao, N.; et al. Transforming growth factor-beta-regulated mir-24 promotes skeletal muscle differentiation. Nucleic Acids Res 2008, 36, 2690–2699. [Google Scholar]
- Caretti, G.; Di Padova, M.; Micales, B.; Lyons, G.E.; Sartorelli, V. The polycomb ezh2 methyltransferase regulates muscle gene expression and skeletal muscle differentiation. Genes Dev 2004, 18, 2627–2638. [Google Scholar]
- Dey, B.K.; Gagan, J.; Yan, Z.; Dutta, A. Mir-26a is required for skeletal muscle differentiation and regeneration in mice. Genes Dev 2012, 26, 2180–2191. [Google Scholar]
- Crist, C.G.; Montarras, D.; Pallafacchina, G.; Rocancourt, D.; Cumano, A.; Conway, S.J.; Buckingham, M. Muscle stem cell behavior is modified by microRNA-27 regulation of pax3 expression. Proc. Natl. Acad. Sci. USA 2009, 106, 13383–13387. [Google Scholar]
- Wang, H.; Garzon, R.; Sun, H.; Ladner, K.J.; Singh, R.; Dahlman, J.; Cheng, A.; Hall, B.M.; Qualman, S.J.; Chandler, D.S.; et al. Nf-kappab-yy1-mir-29 regulatory circuitry in skeletal myogenesis and rhabdomyosarcoma. Cancer Cell 2008, 14, 369–381. [Google Scholar]
- Li, Z.; Hassan, M.Q.; Jafferji, M.; Aqeilan, R.I.; Garzon, R.; Croce, C.M.; van Wijnen, A.J.; Stein, J.L.; Stein, G.S.; Lian, J.B. Biological functions of mir-29b contribute to positive regulation of osteoblast differentiation. J. Biol. Chem 2009, 284, 15676–15684. [Google Scholar]
- Ge, Y.; Sun, Y.; Chen, J. Igf-ii is regulated by microRNA-125b in skeletal myogenesis. J. Cell Biol 2011, 192, 69–81. [Google Scholar]
- Erbay, E.; Park, I.H.; Nuzzi, P.D.; Schoenherr, C.J.; Chen, J. Igf-ii transcription in skeletal myogenesis is controlled by mtor and nutrients. J. Cell Biol 2003, 163, 931–936. [Google Scholar]
- Ge, Y.; Wu, A.L.; Warnes, C.; Liu, J.; Zhang, C.; Kawasome, H.; Terada, N.; Boppart, M.D.; Schoenherr, C.J.; Chen, J. Mtor regulates skeletal muscle regeneration in vivo through kinase-dependent and kinase-independent mechanisms. Am. J. Physiol. Cell Physiol 2009, 297, C1434–C1444. [Google Scholar]
- Seok, H.Y.; Tatsuguchi, M.; Callis, T.E.; He, A.; Pu, W.T.; Wang, D.Z. Mir-155 inhibits expression of the mef2a protein to repress skeletal muscle differentiation. J. Biol. Chem 2011, 286, 35339–35346. [Google Scholar]
- Naguibneva, I.; Ameyar-Zazoua, M.; Polesskaya, A.; Ait-Si-Ali, S.; Groisman, R.; Souidi, M.; Cuvellier, S.; Harel-Bellan, A. The microRNA mir-181 targets the homeobox protein hox-a11 during mammalian myoblast differentiation. Nat. Cell Biol 2006, 8, 278–284. [Google Scholar]
- Kuang, W.; Tan, J.; Duan, Y.; Duan, J.; Wang, W.; Jin, F.; Jin, Z.; Yuan, X.; Liu, Y. Cyclic stretch induced mir-146a upregulation delays c2c12 myogenic differentiation through inhibition of numb. Biochem. Biophys. Res. Commun 2009, 378, 259–263. [Google Scholar]
- Conboy, I.M.; Rando, T.A. The regulation of notch signaling controls satellite cell activation and cell fate determination in postnatal myogenesis. Dev. Cell 2002, 3, 397–409. [Google Scholar]
- Flynt, A.S.; Li, N.; Thatcher, E.J.; Solnica-Krezel, L.; Patton, J.G. Zebrafish mir-214 modulates hedgehog signaling to specify muscle cell fate. Nat. Genet 2007, 39, 259–263. [Google Scholar]
- Cardinali, B.; Castellani, L.; Fasanaro, P.; Basso, A.; Alema, S.; Martelli, F.; Falcone, G. MicroRNA-221 and microRNA-222 modulate differentiation and maturation of skeletal muscle cells. PLoS One 2009, 4, e7607. [Google Scholar]
- Sarkar, S.; Dey, B.K.; Dutta, A. Mir-322/424 and -503 are induced during muscle differentiation and promote cell cycle quiescence and differentiation by down-regulation of cdc25a. Mol. Biol. Cell 2010, 21, 2138–2149. [Google Scholar]
- Dey, B.K.; Gagan, J.; Dutta, A. Mir-206 and -486 induce myoblast differentiation by downregulating pax7. Mol. Cell Biol 2011, 31, 203–214. [Google Scholar]
- Small, E.M.; O’Rourke, J.R.; Moresi, V.; Sutherland, L.B.; McAnally, J.; Gerard, R.D.; Richardson, J.A.; Olson, E.N. Regulation of pi3-kinase/akt signaling by muscle-enriched microRNA-486. Proc. Natl. Acad. Sci. USA 2010, 107, 4218–4223. [Google Scholar]
- Juan, A.H.; Kumar, R.M.; Marx, J.G.; Young, R.A.; Sartorelli, V. Mir-214-dependent regulation of the polycomb protein ezh2 in skeletal muscle and embryonic stem cells. Mol. Cell 2009, 36, 61–74. [Google Scholar]
- Liu, J.; Luo, X.J.; Xiong, A.W.; Zhang, Z.D.; Yue, S.; Zhu, M.S.; Cheng, S.Y. MicroRNA-214 promotes myogenic differentiation by facilitating exit from mitosis via down-regulation of proto-oncogene n-ras. J. Biol. Chem 2010, 285, 26599–26607. [Google Scholar]
- Marrone, A.K.; Shcherbata, H.R. Dystrophin orchestrates the epigenetic profile of muscle cells via miRNAs. Front. Genet 2011, 2. [Google Scholar] [CrossRef]
- Greco, S.; de Simone, M.; Colussi, C.; Zaccagnini, G.; Fasanaro, P.; Pescatori, M.; Cardani, R.; Perbellini, R.; Isaia, E.; Sale, P.; et al. Common micro-RNA signature in skeletal muscle damage and regeneration induced by duchenne muscular dystrophy and acute ischemia. FASEB J 2009, 23, 3335–3346. [Google Scholar]
- Cacchiarelli, D.; Martone, J.; Girardi, E.; Cesana, M.; Incitti, T.; Morlando, M.; Nicoletti, C.; Santini, T.; Sthandier, O.; Barberi, L.; et al. MicroRNAs involved in molecular circuitries relevant for the duchenne muscular dystrophy pathogenesis are controlled by the dystrophin/nnos pathway. Cell Metab 2010, 12, 341–351. [Google Scholar]
- Perbellini, R.; Greco, S.; Sarra-Ferraris, G.; Cardani, R.; Capogrossi, M.C.; Meola, G.; Martelli, F. Dysregulation and cellular mislocalization of specific miRNAs in myotonic dystrophy type 1. Neuromuscul. Disord. NMD 2011, 21, 81–88. [Google Scholar]
- Rau, F.; Freyermuth, F.; Fugier, C.; Villemin, J.P.; Fischer, M.C.; Jost, B.; Dembele, D.; Gourdon, G.; Nicole, A.; Duboc, D.; et al. Misregulation of mir-1 processing is associated with heart defects in myotonic dystrophy. Nat. Struct. Mol. Biol 2011, 18, 840–845. [Google Scholar]
- Gambardella, S.; Rinaldi, F.; Lepore, S.M.; Viola, A.; Loro, E.; Angelini, C.; Vergani, L.; Novelli, G.; Botta, A. Overexpression of microRNA-206 in the skeletal muscle from myotonic dystrophy type 1 patients. J. Transl. Med 2010, 8. [Google Scholar] [CrossRef]
- Cacchiarelli, D.; Incitti, T.; Martone, J.; Cesana, M.; Cazzella, V.; Santini, T.; Sthandier, O.; Bozzoni, I. Mir-31 modulates dystrophin expression: New implications for duchenne muscular dystrophy therapy. EMBO Rep 2011, 12, 136–141. [Google Scholar]
- Eisenberg, I.; Eran, A.; Nishino, I.; Moggio, M.; Lamperti, C.; Amato, A.A.; Lidov, H.G.; Kang, P.B.; North, K.N.; Mitrani-Rosenbaum, S.; et al. Distinctive patterns of microRNA expression in primary muscular disorders. Proc. Natl. Acad. Sci. USA 2007, 104, 17016–17021. [Google Scholar]
- De Arcangelis, V.; Serra, F.; Cogoni, C.; Vivarelli, E.; Monaco, L.; Naro, F. Beta1-syntrophin modulation by mir-222 in mdx mice. PLoS One 2010, 5, e12098. [Google Scholar]
- Greco, S.; Perfetti, A.; Fasanaro, P.; Cardani, R.; Capogrossi, M.C.; Meola, G.; Martelli, F. Deregulated microRNAs in myotonic dystrophy type 2. PLoS One 2012, 7, e39732. [Google Scholar]
- Williams, A.H.; Valdez, G.; Moresi, V.; Qi, X.; McAnally, J.; Elliott, J.L.; Bassel-Duby, R.; Sanes, J.R.; Olson, E.N. MicroRNA-206 delays als progression and promotes regeneration of neuromuscular synapses in mice. Science 2009, 326, 1549–1554. [Google Scholar]
- Durbeej, M.; Campbell, K.P. Muscular dystrophies involving the dystrophin-glycoprotein complex: An overview of current mouse models. Curr. Opin. Genet. Dev 2002, 12, 349–361. [Google Scholar]
- Ervasti, J.M.; Sonnemann, K.J. Biology of the striated muscle dystrophin-glycoprotein complex. Int. Rev. Cytol 2008, 265, 191–225. [Google Scholar]
- Matsumura, K.; Tome, F.M.; Collin, H.; Leturcq, F.; Jeanpierre, M.; Kaplan, J.C.; Fardeau, M.; Campbell, K.P. Expression of dystrophin-associated proteins in dystrophin-positive muscle fibers (revertants) in duchenne muscular dystrophy. Neuromuscul. Disord. NMD 1994, 4, 115–120. [Google Scholar]
- McKinsey, T.A.; Zhang, C.L.; Lu, J.; Olson, E.N. Signal-dependent nuclear export of a histone deacetylase regulates muscle differentiation. Nature 2000, 408, 106–111. [Google Scholar]
- Puri, P.L.; Iezzi, S.; Stiegler, P.; Chen, T.T.; Schiltz, R.L.; Muscat, G.E.; Giordano, A.; Kedes, L.; Wang, J.Y.; Sartorelli, V. Class i histone deacetylases sequentially interact with myod and prb during skeletal myogenesis. Mol. Cell 2001, 8, 885–897. [Google Scholar]
- Brenman, J.E.; Chao, D.S.; Xia, H.; Aldape, K.; Bredt, D.S. Nitric oxide synthase complexed with dystrophin and absent from skeletal muscle sarcolemma in duchenne muscular dystrophy. Cell 1995, 82, 743–752. [Google Scholar]
- Colussi, C.; Mozzetta, C.; Gurtner, A.; Illi, B.; Rosati, J.; Straino, S.; Ragone, G.; Pescatori, M.; Zaccagnini, G.; Antonini, A.; et al. Hdac2 blockade by nitric oxide and histone deacetylase inhibitors reveals a common target in duchenne muscular dystrophy treatment. Proc. Natl. Acad. Sci. USA 2008, 105, 19183–19187. [Google Scholar]
- Brook, J.D.; McCurrach, M.E.; Harley, H.G.; Buckler, A.J.; Church, D.; Aburatani, H.; Hunter, K.; Stanton, V.P.; Thirion, J.P.; Hudson, T.; et al. Molecular basis of myotonic dystrophy: Expansion of a trinucleotide (ctg) repeat at the 3′ end of a transcript encoding a protein kinase family member. Cell 1992, 68, 799–808. [Google Scholar]
- Fu, Y.H.; Pizzuti, A.; Fenwick, R.G., Jr.; King, J.; Rajnarayan, S.; Dunne, P.W.; Dubel, J.; Nasser, G.A.; Ashizawa, T.; de Jong, P.; et al. An unstable triplet repeat in a gene related to myotonic muscular dystrophy. Science 1992, 255, 1256–1258. [Google Scholar]
- Mahadevan, M.; Tsilfidis, C.; Sabourin, L.; Shutler, G.; Amemiya, C.; Jansen, G.; Neville, C.; Narang, M.; Barcelo, J.; O’Hoy, K.; et al. Myotonic dystrophy mutation: An unstable ctg repeat in the 3′ untranslated region of the gene. Science 1992, 255, 1253–1255. [Google Scholar]
- Liquori, C.L.; Ricker, K.; Moseley, M.L.; Jacobsen, J.F.; Kress, W.; Naylor, S.L.; Day, J.W.; Ranum, L.P. Myotonic dystrophy type 2 caused by a cctg expansion in intron 1 of znf9. Science 2001, 293, 864–867. [Google Scholar]
- Wojciechowska, M.; Krzyzosiak, W.J. Cellular toxicity of expanded RNA repeats: Focus on RNA foci. Hum. Mol. Genet 2011, 20, 3811–3821. [Google Scholar]
- Sicot, G.; Gomes-Pereira, M. RNA toxicity in human disease and animal models: From the uncovering of a new mechanism to the development of promising therapies. Biochim. Biophys. Acta 2013, 1832, 1390–1409. [Google Scholar]
- Udd, B.; Krahe, R. The myotonic dystrophies: Molecular, clinical, and therapeutic challenges. Lancet Neurol 2012, 11, 891–905. [Google Scholar]
- Li, X.; Wu, Z.; Fu, X.; Han, W. Long noncoding RNAs: Insights from biological features and functions to diseases. Med. Res. Rev 2013, 33, 517–553. [Google Scholar]
- Cesana, M.; Cacchiarelli, D.; Legnini, I.; Santini, T.; Sthandier, O.; Chinappi, M.; Tramontano, A.; Bozzoni, I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell 2011, 147, 358–369. [Google Scholar]
- Watts, R.; Johnsen, V.L.; Shearer, J.; Hittel, D.S. Myostatin-induced inhibition of the long noncoding RNA malat1 is associated with decreased myogenesis. Am. J. Physiol. Cell Physiol 2013, 304, C995–C1001. [Google Scholar]
- Sunwoo, H.; Dinger, M.E.; Wilusz, J.E.; Amaral, P.P.; Mattick, J.S.; Spector, D.L. Men epsilon/beta nuclear-retained non-coding RNAs are up-regulated upon muscle differentiation and are essential components of paraspeckles. Genome Res 2009, 19, 347–359. [Google Scholar]
- Sasaki, Y.T.; Ideue, T.; Sano, M.; Mituyama, T.; Hirose, T. Menepsilon/beta noncoding RNAs are essential for structural integrity of nuclear paraspeckles. Proc. Natl. Acad. Sci. USA 2009, 106, 2525–2530. [Google Scholar]
- Clemson, C.M.; Hutchinson, J.N.; Sara, S.A.; Ensminger, A.W.; Fox, A.H.; Chess, A.; Lawrence, J.B. An architectural role for a nuclear noncoding RNA: Neat1 RNA is essential for the structure of paraspeckles. Mol. Cell 2009, 33, 717–726. [Google Scholar]
- Hube, F.; Velasco, G.; Rollin, J.; Furling, D.; Francastel, C. Steroid receptor RNA activator protein binds to and counteracts sra RNA-mediated activation of myod and muscle differentiation. Nucleic Acids Res 2011, 39, 513–525. [Google Scholar]
- Caretti, G.; Schiltz, R.L.; Dilworth, F.J.; Di Padova, M.; Zhao, P.; Ogryzko, V.; Fuller-Pace, F.V.; Hoffman, E.P.; Tapscott, S.J.; Sartorelli, V. The RNA helicases p68/p72 and the noncoding RNA sra are coregulators of myod and skeletal muscle differentiation. Dev. Cell 2006, 11, 547–560. [Google Scholar]
- Caretti, G.; Lei, E.P.; Sartorelli, V. The dead-box p68/p72 proteins and the noncoding RNA steroid receptor activator sra: Eclectic regulators of disparate biological functions. Cell Cycle 2007, 6, 1172–1176. [Google Scholar]
- Willingham, A.T.; Orth, A.P.; Batalov, S.; Peters, E.C.; Wen, B.G.; Aza-Blanc, P.; Hogenesch, J.B.; Schultz, P.G. A strategy for probing the function of noncoding RNAs finds a repressor of nfat. Science 2005, 309, 1570–1573. [Google Scholar]
- Sharma, S.; Findlay, G.M.; Bandukwala, H.S.; Oberdoerffer, S.; Baust, B.; Li, Z.; Schmidt, V.; Hogan, P.G.; Sacks, D.B.; Rao, A. Dephosphorylation of the nuclear factor of activated t cells (nfat) transcription factor is regulated by an RNA-protein scaffold complex. Proc. Natl. Acad. Sci. USA 2011, 108, 11381–11386. [Google Scholar]
- Niland, C.N.; Merry, C.R.; Khalil, A.M. Emerging roles for long non-coding RNAs in cancer and neurological disorders. Front. Genet 2012, 3. [Google Scholar] [CrossRef]
- Cabianca, D.S.; Casa, V.; Bodega, B.; Xynos, A.; Ginelli, E.; Tanaka, Y.; Gabellini, D. A long ncRNA links copy number variation to a polycomb/trithorax epigenetic switch in fshd muscular dystrophy. Cell 2012, 149, 819–831. [Google Scholar]
- Schmidt, L.H.; Spieker, T.; Koschmieder, S.; Schaffers, S.; Humberg, J.; Jungen, D.; Bulk, E.; Hascher, A.; Wittmer, D.; Marra, A.; et al. The long noncoding malat-1 RNA indicates a poor prognosis in non-small cell lung cancer and induces migration and tumor growth. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2011, 6, 1984–1992. [Google Scholar]
- Tripathi, V.; Ellis, J.D.; Shen, Z.; Song, D.Y.; Pan, Q.; Watt, A.T.; Freier, S.M.; Bennett, C.F.; Sharma, A.; Bubulya, P.A.; et al. The nuclear-retained noncoding RNA malat1 regulates alternative splicing by modulating sr splicing factor phosphorylation. Mol. Cell 2010, 39, 925–938. [Google Scholar]
- Wilusz, J.E.; JnBaptiste, C.K.; Lu, L.Y.; Kuhn, C.D.; Joshua-Tor, L.; Sharp, P.A. A triple helix stabilizes the 3′ ends of long noncoding RNAs that lack poly(a) tails. Genes Dev 2012, 26, 2392–2407. [Google Scholar]
- Gutschner, T.; Hammerle, M.; Diederichs, S. Malat1—A paradigm for long noncoding RNA function in cancer. J. Mol. Med 2013, 91, 791–801. [Google Scholar]
- Langley, B.; Thomas, M.; Bishop, A.; Sharma, M.; Gilmour, S.; Kambadur, R. Myostatin inhibits myoblast differentiation by down-regulating myod expression. J. Biol. Chem 2002, 277, 49831–49840. [Google Scholar]
- Rios, R.; Carneiro, I.; Arce, V.M.; Devesa, J. Myostatin is an inhibitor of myogenic differentiation. Am. J. Physiol. Cell Physiol 2002, 282, C993–C999. [Google Scholar]
- Zhang, B.; Arun, G.; Mao, Y.S.; Lazar, Z.; Hung, G.; Bhattacharjee, G.; Xiao, X.; Booth, C.J.; Wu, J.; Zhang, C.; et al. The lncRNA malat1 is dispensable for mouse development but its transcription plays a cis-regulatory role in the adult. Cell Rep 2012, 2, 111–123. [Google Scholar]
- Eissmann, M.; Gutschner, T.; Hammerle, M.; Gunther, S.; Caudron-Herger, M.; Gross, M.; Schirmacher, P.; Rippe, K.; Braun, T.; Zornig, M.; et al. Loss of the abundant nuclear non-coding RNA malat1 is compatible with life and development. RNA Biol 2012, 9, 1076–1087. [Google Scholar]
- Prasanth, K.V.; Prasanth, S.G.; Xuan, Z.; Hearn, S.; Freier, S.M.; Bennett, C.F.; Zhang, M.Q.; Spector, D.L. Regulating gene expression through RNA nuclear retention. Cell 2005, 123, 249–263. [Google Scholar]
- Ahn, A.H.; Kunkel, L.M. The structural and functional diversity of dystrophin. Nat. Genet 1993, 3, 283–291. [Google Scholar]
- Muntoni, F.; Torelli, S.; Ferlini, A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurol 2003, 2, 731–740. [Google Scholar]
- Torelli, S.; Ferlini, A.; Obici, L.; Sewry, C.; Muntoni, F. Expression, regulation and localisation of dystrophin isoforms in human foetal skeletal and cardiac muscle. Neuromuscul. Disord. NMD 1999, 9, 541–551. [Google Scholar]
- Brioschi, S.; Gualandi, F.; Scotton, C.; Armaroli, A.; Bovolenta, M.; Falzarano, M.S.; Sabatelli, P.; Selvatici, R.; D’Amico, A.; Pane, M.; et al. Genetic characterization in symptomatic female dmd carriers: Lack of relationship between x-inactivation, transcriptional dmd allele balancing and phenotype. BMC Med. Genet 2012, 13. [Google Scholar] [CrossRef]
- Tran, T.H.; Zhang, Z.; Yagi, M.; Lee, T.; Awano, H.; Nishida, A.; Okinaga, T.; Takeshima, Y.; Matsuo, M. Molecular characterization of an x(p21.2;q28) chromosomal inversion in a duchenne muscular dystrophy patient with mental retardation reveals a novel long non-coding gene on xq28. J. Hum. Genet 2013, 58, 33–39. [Google Scholar]
- Lee, J.T. Lessons from x-chromosome inactivation: Long ncRNA as guides and tethers to the epigenome. Genes Dev 2009, 23, 1831–1842. [Google Scholar]
- Rinn, J.L.; Kertesz, M.; Wang, J.K.; Squazzo, S.L.; Xu, X.; Brugmann, S.A.; Goodnough, L.H.; Helms, J.A.; Farnham, P.J.; Segal, E.; et al. Functional demarcation of active and silent chromatin domains in human hox loci by noncoding RNAs. Cell 2007, 129, 1311–1323. [Google Scholar]
- Rukov, J.L.; Shomron, N. MicroRNA pharmacogenomics: Post-transcriptional regulation of drug response. Trends Mol. Med 2011, 17, 412–423. [Google Scholar]
- Mishra, P.J. The miRNA-drug resistance connection: A new era of personalized medicine using noncoding RNA begins. Pharmacogenomics 2012, 13, 1321–1324. [Google Scholar]
- Giovannetti, E.; van der Velde, A.; Funel, N.; Vasile, E.; Perrone, V.; Leon, L.G.; de Lio, N.; Avan, A.; Caponi, S.; Pollina, L.E.; et al. High-throughput microRNA (mirRNA) arrays unravel the prognostic role of mir-211 in pancreatic cancer. PLoS One 2012, 7, e49145. [Google Scholar]
- Gagan, J.; Dey, B.K.; Dutta, A. MicroRNAs regulate and provide robustness to the myogenic transcriptional network. Curr. Opin. Pharmacol 2012, 12, 383–388. [Google Scholar]
- Twayana, S.; Legnini, I.; Cesana, M.; Cacchiarelli, D.; Morlando, M.; Bozzoni, I. Biogenesis and function of non-coding RNAs in muscle differentiation and in duchenne muscular dystrophy. Biochem. Soc. Trans 2013, 41, 844–849. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Erriquez, D.; Perini, G.; Ferlini, A. Non-Coding RNAs in Muscle Dystrophies. Int. J. Mol. Sci. 2013, 14, 19681-19704. https://doi.org/10.3390/ijms141019681
Erriquez D, Perini G, Ferlini A. Non-Coding RNAs in Muscle Dystrophies. International Journal of Molecular Sciences. 2013; 14(10):19681-19704. https://doi.org/10.3390/ijms141019681
Chicago/Turabian StyleErriquez, Daniela, Giovanni Perini, and Alessandra Ferlini. 2013. "Non-Coding RNAs in Muscle Dystrophies" International Journal of Molecular Sciences 14, no. 10: 19681-19704. https://doi.org/10.3390/ijms141019681
APA StyleErriquez, D., Perini, G., & Ferlini, A. (2013). Non-Coding RNAs in Muscle Dystrophies. International Journal of Molecular Sciences, 14(10), 19681-19704. https://doi.org/10.3390/ijms141019681