Vitamin D and Death by Sunshine

Abstract
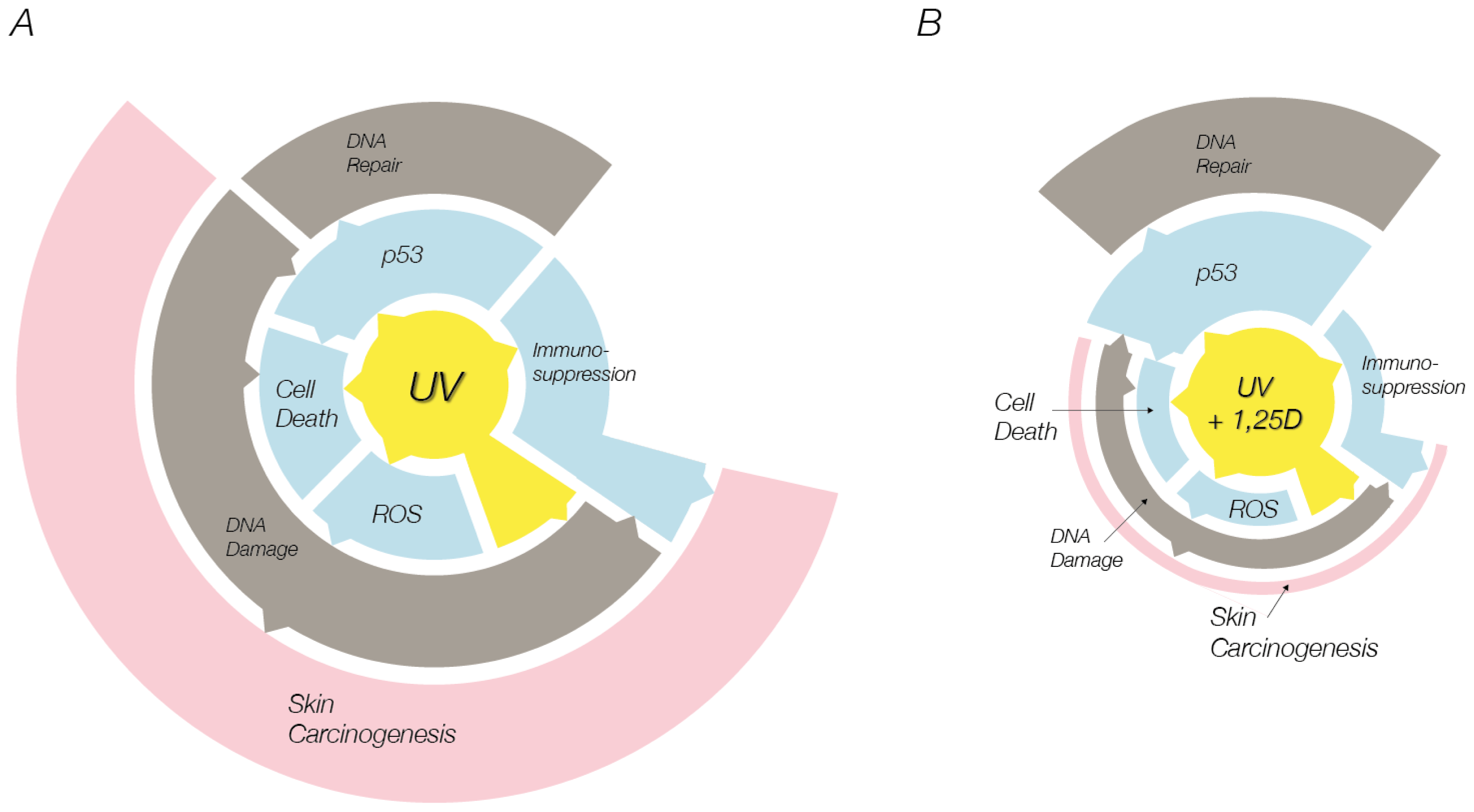
:1. Introduction
2. Vitamin D and Cell Death
3. UV-Induced Skin Cell Death: Mechanisms of Photoprotection by Vitamin D Compounds
3.1. Vitamin D Reduces UV-Induced DNA Damage
3.2. Effects of Vitamin D on p53
3.3. Vitamin D Compounds Reduce Nitric Oxide Derivatives
3.4. Vitamin D and Antioxidant Systems
3.5. Vitamin D and MAPK Signaling
3.6. Vitamin D and Akt Signaling
4. Conclusions
Acknowledgments
- Conflict of InterestThe authors declare no conflict of interest.
References
- Matsumura, Y.; Ananthaswamy, H.N. Toxic effects of ultraviolet radiation on the skin. Toxicol. Appl. Pharmacol 2004, 195, 298–308. [Google Scholar]
- Dixon, K.M.; Sequeira, V.B.; Camp, A.J.; Mason, R.S. Vitamin D-fence. Photochem. Photobiol. Sci 2010, 9, 564–570. [Google Scholar]
- Holick, M.F. The cutaneous photosynthesis of previtamin D3: A unique photoendocrine system. J. Invest. Dermatol 1981, 77, 51–58. [Google Scholar]
- Bikle, D.D.; Nemanic, M.K.; Whitney, J.O.; Elias, P.W. Neonatal human foreskin keratinocytes produce 1,25-dihydroxyvitamin D3. Biochemistry 1986, 25, 1545–1548. [Google Scholar]
- Lehmann, B.; Rudolph, T.; Pietzsch, J.; Meurer, M. Conversion of vitamin D3 to 1alpha,25-dihydroxyvitamin D3 in human skin equivalents. Exp. Dermatol 2000, 9, 97–103. [Google Scholar]
- Pattison, D.I.; Davies, M.J. Actions of ultraviolet light on cellular structures. Cancer 2006, 131–157. [Google Scholar]
- Deliconstantinos, G.; Villiotou, V.; Stravrides, J.C. Release by ultraviolet b (U.V.B) radiation of nitric oxide (no) from human keratinocytes: A potential role for nitric oxide in erythema production. Br. J. Pharmacol 1995, 114, 1257–1265. [Google Scholar]
- Bruch-Gerharz, D.; Ruzicka, T.; Kolb-Bachofen, V. Nitric oxide in human skin: Current status and future prospects. J. Invest. Dermatol 1998, 110, 1–7. [Google Scholar]
- Cals-Grierson, M.M.; Ormerod, A.D. Nitric oxide function in the skin. Nitric Oxide 2004, 10, 179–193. [Google Scholar]
- Paunel, A.N.; Dejam, A.; Thelen, S.; Kirsch, M.; Horstjann, M.; Gharini, P.; Murtz, M.; Kelm, M.; de Groot, H.; Kolb-Bachofen, V.; et al. Enzyme-independent nitric oxide formation during UVA challenge of human skin: Characterization, molecular sources, and mechanisms. Free Radic. Biol. Med 2005, 38, 606–615. [Google Scholar]
- Mowbray, M.; McLintock, S.; Weerakoon, R.; Lomatschinsky, N.; Jones, S.; Rossi, A.G.; Weller, R.B. Enzyme-independent no stores in human skin: Quantification and influence of UV radiation. J. Invest. Dermatol 2009, 129, 834–842. [Google Scholar]
- Hess, D.T.; Matsumoto, A.; Nudelman, R.; Stamler, J.S. S-nitrosylation: Spectrum and specificity. Nat. Cell Biol 2001, 3, E46–E49. [Google Scholar]
- Halliday, G.M. Inflammation, gene mutation and photoimmunosuppression in response to UVR-induced oxidative damage contributes to photocarcinogenesis. Mutat. Res 2005, 571, 107–120. [Google Scholar]
- Ohshima, H.; Sawa, T.; Akaike, T. 8-nitroguanine, a product of nitrative DNA damage caused by reactive nitrogen species: Formation, occurrence, and implications in inflammation and carcinogenesis. Antioxid. Redox Signal 2006, 8, 1033–1045. [Google Scholar]
- Pacher, P.; Beckman, J.S.; Liaudet, L. Nitric oxide and peroxynitrite in health and disease. Physiol. Rev 2007, 87, 315–424. [Google Scholar]
- Virág, L.; Szabó, C. The therapeutic potential of poly(adp-ribose) polymerase inhibitors. Physiol. Rev 2002, 54, 375–429. [Google Scholar]
- Ouhtit, A.; Muller, H.K.; Gorny, A.; Ananthaswamy, H.N. UVB-induced experimental carcinogenesis: Dysregulation of apoptosis and p53 signalling pathway. Redox Report 2000, 5, 128–129. [Google Scholar]
- Jaiswal, M.; LaRusso, N.F.; Burgart, L.J.; Gores, G.J. Inflammatory cytokines induce DNA damage and inhibit DNA repair in cholangiocarcinoma cells by a nitric oxide-dependent mechanism. Cancer Res 2000, 60, 184–190. [Google Scholar]
- Bau, D.T.; Gurr, J.R.; Jan, K.Y. Nitric oxide is involved in arsenite inhibition of pyrimidine dimer excision. Carcinogenesis 2001, 22, 709–716. [Google Scholar]
- Saelens, X.; Festjens, N.; Walle, L.V.; van Gurp, M.; van Loo, G.; Vandenabeele, P. Toxic proteins released from mitochondria in cell death. Oncogene 2004, 23, 2861–2874. [Google Scholar]
- Simboli-Campbell, M.; Narvaez, C.J.; Tenniswood, M.; Welsh, J. 1,25-dihydroxyvitamin D3 induces morphological and biochemical markers of apoptosis in mcf-7 breast cancer cells. J. Steroid. Biochem. Mol. Biol 1996, 58, 367–376. [Google Scholar]
- Guzey, M.; Kitada, S.; Reed, J.C. Apoptosis induction by 1α,25-dihydroxyvitamin D3 in prostate cancer. Mol. Cancer Ther 2002, 1, 667–677. [Google Scholar]
- Vandewalle, B.; Wattez, N.; Lefebvre, J. Effects of vitamin D3 derivatives on growth, differentiation and apoptosis in tumoral colonic ht 29 cells: Possible implication of intracellular calcium. Cancer Lett 1995, 97, 99–106. [Google Scholar]
- Mason, R.S.; Holliday, C.J. 1,25dihydroxyVitamin D Contributes to Photoprotection in Skin Cells. In Vitamin D Endocrine System: Structural, Biological, Genetic and Clinical Aspects; Norman, A., Bouillon, R., Thomasset, M., Eds.; University of California: Riverside, CA, USA, 2000; pp. 605–608. [Google Scholar]
- Mason, R.S.; Holliday, C.J.; Gupta, R. 1,25 Dihydroxyvitamin D and Photoprotection in Skin Cells. In Modern Trends in Skin Pharmacology, 1st ed; Tsambos, D., Merk, H., Eds.; Parissianos Medical Publications S.A. Athens: Athens, Greece, 2002; pp. 59–66. [Google Scholar]
- De Haes, P.; Garmyn, M.; Degreef, H.; Vantieghem, K.; Bouillon, R.; Segaert, S. 1,25-dihydroxyvitamin D3 inhibits ultraviolet B-induced apoptosis, jun kinase activation, and interleukin-6 production in primary human keratinocytes. J. Cell Biochem 2003, 89, 663–673. [Google Scholar]
- De Haes, P.; Garmyn, M.; Verstuyf, A.; de Clercq, P.; Vandewalle, M.; Vantieghem, K.; Degreef, H.; Bouillon, R.; Segaert, S. Two 14-epi analogues of 1,25-dihydroxyvitamin D3 protect human keratinocytes against the effects of UVB. Arch. Dermatol. Res 2004, 295, 527–534. [Google Scholar]
- Wong, G.; Gupta, R.; Dixon, K.M.; Deo, S.S.; Choong, S.M.; Halliday, G.M.; Bishop, J.E.; Ishizuka, S.; Norman, A.W.; Posner, G.H.; et al. 1,25-dihydroxyvitamin D and three low-calcemic analogs decrease UV-induced DNA damage via the rapid response pathway. J. Steroid. Biochem. Mol. Biol. 2004, 89–90, 567–570. [Google Scholar]
- Gupta, R.; Dixon, K.M.; Deo, S.S.; Holliday, C.J.; Slater, M.; Halliday, G.M.; Reeve, V.E.; Mason, R.S. Photoprotection by 1,25 dihydroxyvitamin D3 is associated with an increase in p53 and a decrease in nitric oxide products. J. Invest. Dermatol 2007, 127, 707–715. [Google Scholar]
- Manggau, M.; Kim, D.S.; Ruwisch, L.; Vogler, R.; Korting, H.C.; Schafer-Korting, M.; Kleuser, B. 1α,25-dihydroxyvitamin D-3 protects human keratinocytes from apoptosis by the formation of sphingosine-1-phosphate. J. Invest. Dermatol 2001, 117, 1241–1249. [Google Scholar]
- Lee, J.; Youn, J.I. The photoprotective effect of 1,25-dihydroxyvitamin D3 on ultraviolet light B-induced damage in keratinocyte and its mechanism of action. J. Dermatol. Sci 1998, 18, 11–18. [Google Scholar]
- Benassi, L.; Ottani, D.; Fantini, F.; Marconi, A.; Chiodino, C.; Giannetti, A.; Pincelli, C. 1,25-dihydroxyvitamin D3, transforming growth factor beta1, calcium, and ultraviolet B radiation induce apoptosis in cultured human keratinocytes. J. Invest. Dermatol 1997, 109, 276–282. [Google Scholar]
- Youn, J.I.; Park, B.S.; Chung, J.H.; Lee, J.H. Photoprotective effect of calcipotriol upon skin photoreaction to UVA and UVB. Photodermatol. Photoimmunol. Photomed 1997, 13, 109–114. [Google Scholar]
- Dixon, K.M.; Sequeira, V.B.; Deo, S.S.; Mohan, R.; Posner, G.H.; Mason, R.S. Differential photoprotective effects of 1,25-dihydroxyvitamin D3 and a low calcaemic deltanoid. Photochem. Photobiol. Sci 2012, 11, 1825–1830. [Google Scholar]
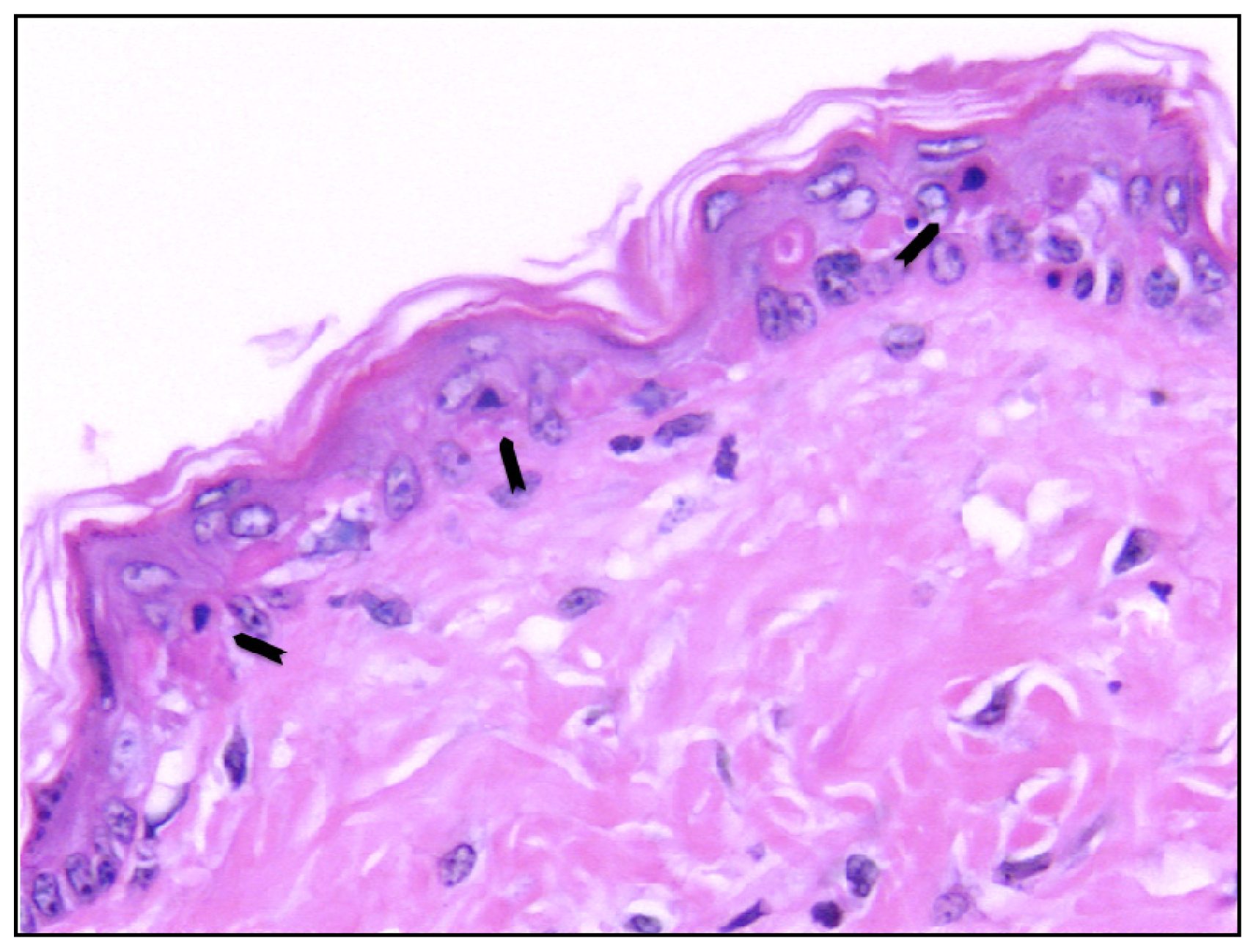
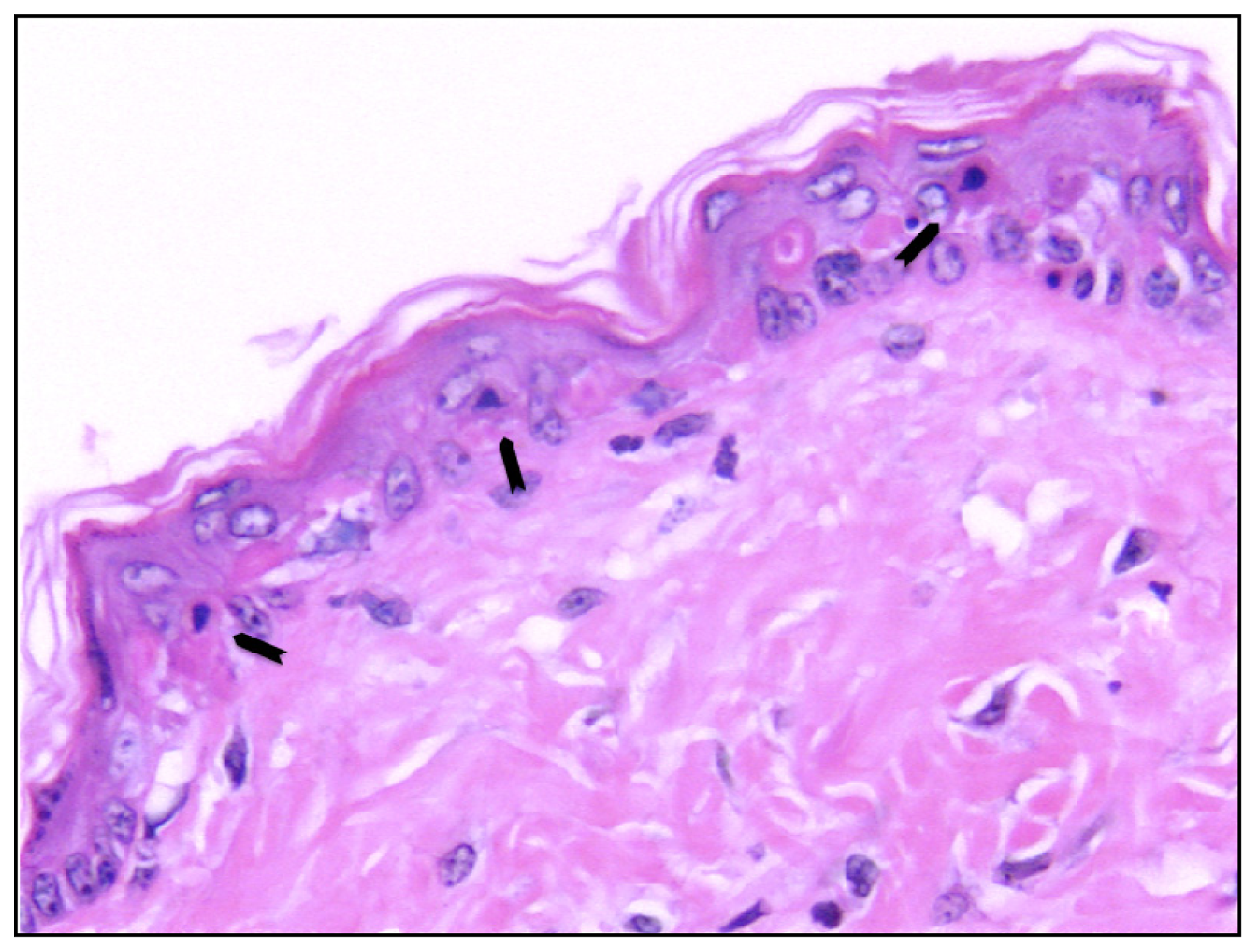
- Sheehan, J.M.; Young, J.R. The sunburn cell revisited: An update on mechanistic aspects. Photochem. Photobiol. Sci 2002, 1, 365–377. [Google Scholar]
- Claerhout, S.; van Laethem, A.; Agostinis, P.; Garmyn, M. Pathways involved in sunburn cell formation: Deregulation in skin cancer. Photochem. Photobiol. Sci 2006, 5, 199–207. [Google Scholar]
- Hanada, K.; Sawamura, D.; Nakano, H.; Hashimoto, I. Possible role of 1,25-dihydroxyvitamin D3-induced metallothionein in photoprotection against UVB injury in mouse skin and cultured rat keratinocytes. J. Dermatol. Sci 1995, 9, 203–208. [Google Scholar]
- Dixon, K.M.; Deo, S.S.; Norman, A.W.; Bishop, J.E.; Halliday, G.M.; Reeve, V.E.; Mason, R.S. In vivo relevance for photoprotection by the vitamin D rapid response pathway. J. Steroid. Biochem. Mol. Biol 2007, 103, 451–456. [Google Scholar]
- Dixon, K.M.; Norman, A.W.; Sequeira, V.B.; Mohan, R.; Rybchyn, M.S.; Reeve, V.E.; Halliday, G.M.; Mason, R.S. 1α,25(OH)2-vitamin D and a nongenomic vitamin D analogue inhibit ultraviolet radiation-induced skin carcinogenesis. Cancer Prev. Res 2011, 4, 1485–1494. [Google Scholar]
- Damian, D.L.; Kim, Y.J.; Dixon, K.M.; Halliday, G.M.; Javeri, A.; Mason, R.S. Topical calcitriol protects from UV-induced genetic damage but suppresses cutaneous immunity in humans. Exp. Dermatol 2010, 19, E23–E30. [Google Scholar]
- Mason, R.S.; Sequeira, V.B.; Dixon, K.M.; Gordon-Thomson, C.; Pobre, K.; Dilley, A.; Mizwicki, M.T.; Norman, A.W.; Feldman, D.; Halliday, G.M.; et al. Photoprotection by 1α,25-dihydroxyvitamin D and analogs: Further studies on mechanisms and implications for UV-damage. J. Steroid. Biochem. Mol. Biol 2010, 121, 164–168. [Google Scholar]
- Mason, R.S.; Sequeira, V.B.; Gordon-Thomson, C. Vitamin D: The light side of sunshine. Eur. J. Clin. Nut 2011, 65, 986–993. [Google Scholar]
- Song, E.J.; Gordon-Thomson, C.; Coleb, L.; Sternc, H.; Halliday, G.M.; Damian, D.L.; Reeve, V.E.; Mason, R.S. 1α,25-dihydroxyvitamin D3 reduces several types of UV-induced DNA damage and contributes to photoprotection. J. Steroid. Biochem. Mol. Biol. 2012. submitted for publication. [Google Scholar]
- Gordon-Thomson, C.; Gupta, R.; Tongkao-on, W.; Ryan, A.; Halliday, G.M.; Mason, R.S. 1α,25 dihydroxyvitamin D3 enhances cellular defences against UV-induced oxidative and other forms of DNA damage in skin. Photochem. Photobiol. Sci. 2012. [Google Scholar] [CrossRef]
- Sequeira, V.B.; Rybchyn, M.S.; Tongkao-On, W.; Gordon-Thomson, C.; Malloy, P.J.; Nemere, I.; Norman, A.W.; Reeve, V.E.; Halliday, G.M.; Feldman, D.; et al. The role of the vitamin D receptor and ERp57 in photoprotection by 1α,25-dihydroxyvitamin D-3. Mol. Endocrinol 2012, 26, 574–582. [Google Scholar]
- Douki, T.; Court, M.; Sauvaigo, S.; Odin, F.; Cadet, J. Formation of the main UV-induced thymine dimeric lesions within isolated and cellular DNA as measured by high performance liquid chromatography-tandem mass spectrometry. J. Biol. Chem 2000, 275, 11678–11685. [Google Scholar]
- Cooke, M.S.; Podmore, I.D.; Mistry, N.; Evans, M.D.; Herbert, K.E.; Griffiths, H.R.; Lunec, J. Immunochemical detection of UV-induced DNA damage and repair. J. Immunol. Methods 2003, 280, 125–133. [Google Scholar]
- Mouret, S.; Baudouin, C.; Charveron, M.; Favier, A.; Cadet, J.; Douki, T. Cyclobutane pyrimidine dimers are predominant DNA lesions in whole human skin exposed to UVA radiation. Proc. Natl. Acad. Sci. USA 2006, 103, 13765–13770. [Google Scholar]
- Kvam, E.; Tyrrell, R.M. Induction of oxidative DNA base damage in human skin cells by UV and near visible radiation. Carcinogenesis 1997, 18, 2379–2384. [Google Scholar]
- Agar, N.S.; Halliday, G.M.; Barnetson, R.S.; Ananthaswamy, H.N.; Wheeler, M.; Jones, A.M. The basal layer in human squamous tumors harbors more UVA than UVB fingerprint mutations: A role for UVA in human skin carcinogenesis. Proc. Natl. Acad. Sci. USA 2004, 101, 4954–4959. [Google Scholar]
- De Haes, P.; Garmyn, M.; Verstuyf, A.; De Clercq, P.; Vandewalle, M.; Degreef, H.; Vantieghem, K.; Bouillon, R.; Segaert, S. 1,25-dihydroxyvitamin D3 and analogues protect primary human keratinocytes against UVB-induced DNA damage. J. Photochem. Photobiol. B 2005, 78, 141–148. [Google Scholar]
- Dixon, K.M.; Deo, S.S.; Wong, G.; Slater, M.; Norman, A.W.; Bishop, J.E.; Posner, G.H.; Ishizuka, S.; Halliday, G.M.; Reeve, V.E.; et al. Skin cancer prevention: A possible role of 1,25dihydroxyvitamin D3 and its analogs. J. Steroid. Biochem. Mol. Biol 2005, 97, 137–143. [Google Scholar]
- Sequeira, V.B.; Rybchyn, M.S.; Gordon-Thomson, C.; Tongkao-On, W.; Mizwicki, M.T.; Norman, A.W.; Reeve, V.E.; Halliday, G.M.; Mason, R.S. Opening of chloride channels by 1α,25-dihydroxyvitamin D(3) contributes to photoprotection against UVR-induced thymine dimers in keratinocytes. J. Invest. Dermatol. 2012. [Google Scholar] [CrossRef]
- Ellison, T.I.; Smith, M.K.; Gilliam, A.C.; MacDonald, P.N. Inactivation of the vitamin D receptor enhances susceptibility of murine skin to UV-induced tumorigenesis. J. Invest. Dermatol 2008, 128, 2508–2517. [Google Scholar]
- Zanello, L.P.; Norman, A. 1α,25(OH)2 vitamin D3 actions on ion channels in osteoblasts. Steroids 2006, 71, 291–297. [Google Scholar]
- Hall, P.A.; McKee, P.H.; Menage, H.D.; Dover, R.; Lane, D.P. High levels of p53 protein in UV-irradiated normal human skin. Oncogene 1993, 8, 203–207. [Google Scholar]
- Yamaguchi, Y.; Coelho, S.G.; Zmudzka, B.Z.; Takahashi, K.; Beer, J.Z.; Hearing, V.J.; Miller, S.A. Cyclobutane pyrimidine dimer formation and p53 production in human skin after repeated UV irradiation. Exp. Dermatol 2008, 17, 916–924. [Google Scholar]
- Moll, P.R.; Sander, V.; Frischauf, A.M.; Richter, K. Expression profiling of vitamin D treated primary human keratinocytes. J. Cell Biochem 2007, 100, 574–592. [Google Scholar]
- Fitch, M.E.; Cross, I.V.; Ford, J.M. P53 responsive nucleotide excision repair gene products p48 and xpc, but not p53, localize to sites of UV-irradiation-induced DNA damage, in vivo. Carcinogenesis 2003, 24, 843–850. [Google Scholar]
- Paunel-Gorgulu, A.; Dejam, A.; Thelen, S.; Kirsch, M.; Horstjann, M.; Gharini, P.; Murtz, M.; Kelm, M.; de Groot, H.; Kolb-Bachofen, V.; et al. UVA induces immediate and enzyme-independent nitric oxide formation in healthy human skin leading to no-specific signalling. Eur. J. Cell Biol 2005, 84, 37–38. [Google Scholar]
- Hiraku, Y.; Kawanishi, S. Immunohistochemical Analysis of 8-Nitroguanine, a Nitrative DNA Lesion, in Relation to Inflammation-Associated Carcinogenesis. In Inflammation and Cancer; Kozlov, S., Ed.; Humana Press: New York, NY, USA, 2009; Volume 512, pp. 3–14. [Google Scholar]
- Leccia, M.T.; Yaar, M.; Allen, N.; Gleason, M.; Gilchrest, B.A. Solar simulated irradiation modulates gene expression and activity of antioxidant enzymes in cultured human dermal fibroblasts. Exp. Dermatol 2001, 10, 272–279. [Google Scholar]
- Ravid, A.; Rubinstein, E.; Gamady, A.; Rotem, C.; Liberman, U.A.; Koren, R. Vitamin D inhibits the activation of stress-activated protein kinases by physiological and environmental stresses in keratinocytes. J. Endocrinol 2002, 173, 525–532. [Google Scholar]
- Diker-Cohen, T.; Koren, R.; Liberman, U.A.; Ravid, A. Vitamin D protects keratinocytes from apoptosis induced by osmotic shock, oxidative stress, and tumor necrosis factor. Ann. N. Y. Acad. Sci 2003, 1010, 350–353. [Google Scholar]
- Hanada, K.; Gange, R.W.; Siebert, E.; Hasan, T. Protective effects of cadmium chloride against UVB injury in mouse skin and in cultured human cells: A possible role of cadmium-induced metallothionein. Photodermatol. Photoimmunol. Photomed 1991, 8, 111–115. [Google Scholar]
- Hanada, K.; Baba, T.; Hashimoto, I.; Fukui, R.; Watanabe, S. Possible role of cutaneous metallothionein in protection against photo-oxidative stress-epidermal localization and scavenging activity for superoxide and hydroxyl radicals. Photodermatol. Photoimmunol. Photomed 1992, 9, 209–213. [Google Scholar]
- Reeve, V.E.; Nishimura, N.; Bosnic, M.; Michalska, A.E.; Choo, K.H.A. Lack of metallothionein-i and -ii exacerbates the immunosuppressive effect of ultraviolet B radiation and cis-urocanic acid in mice. Immunology 2000, 100, 399–404. [Google Scholar]
- Widyarini, S.; Allanson, M.; Gallagher, N.L.; Pedley, J.; Boyle, G.M.; Parsons, P.G.; Whiteman, D.C.; Walker, C.; Reeve, V.E. Isoflavonoid photoprotection in mouse and human skin is dependent on metallothionein. J. Invest. Dermatol 2006, 126, 198–204. [Google Scholar]
- Karasawa, M.; Hosoi, J.; Hashiba, H.; Nose, K.; Tohyama, C.; Abe, E.; Suda, T.; Kuroki, T. Regulation of metallothionein gene expression by 1α,25-dihydroxyvitamin D3 in cultured cells and in mice. Proc. Natl. Acad. Sci. USA 1987, 84, 8810–8813. [Google Scholar]
- Assefa, Z.; van Laethem, A.; Garmyn, M.; Agostinis, P. Ultraviolet radiation-induced apoptosis in keratinocytes: On the role of cytosolic factors. Biochim. Biophys. Acta 2005, 1755, 90–106. [Google Scholar]
- Bode, A.M.; Dong, Z. Mitogen-activated protein kinase activation in UV-induced signal transduction. Sci. STKE 2003, 2003, RE2. [Google Scholar]
- Gardner, J. Mechanisms of photoprotection by 1a,25-dihydroxyvitamin D3 in keratinocytes. Honours Thesis, University of Sydney, Sydney, NSW, Australia, 2005. [Google Scholar]
- Bennett, B.L.; Sasaki, D.T.; Murray, B.W.; O’Leary, E.C.; Sakata, S.T.; Xu, W.; Leisten, J.C.; Motiwala, A.; Pierce, S.; Satoh, Y.; et al. Sp600125, an anthrapyrazolone inhibitor of jun N-terminal kinase. Proc. Natl. Acad. Sci. USA 2001, 98, 13681–13686. [Google Scholar]
- Peus, D.; Vasa, R.A.; Beyerle, A.; Meves, A.; Krautmacher, C.; Pittelkow, M.R. UVB activates erk1/2 and p38 signaling pathways via reactive oxygen species in cultured keratinocytes. J. Invest. Dermatol 1999, 112, 751–756. [Google Scholar]
- Wang, H.Q.; Quan, T.; He, T.; Franke, T.F.; Voorhees, J.J.; Fisher, G.J. Epidermal growth factor receptor-dependent, NF-κB-independent activation of the phosphatidylinositol 3-kinase/akt pathway inhibits ultraviolet irradiation-induced caspases-3, -8, and -9 in human keratinocytes. J. Biol. Chem 2003, 278, 45737–45745. [Google Scholar]
- Diker-Cohen, T.; Koren, R.; Ravid, A. Programmed cell death of stressed keratinocytes and its inhibition by vitamin D: The role of death and survival signaling pathways. Apoptosis 2006, 11, 519–534. [Google Scholar]
- De Haes, P.; Garmyn, M.; Carmeliet, G.; Degreef, H.; Vantieghem, K.; Bouillon, R.; Segaert, S. Molecular pathways involved in the anti-apoptotic effect of 1,25-dihydroxyvitamin D3 in primary human keratinocytes. J. Cell Biochem 2004, 93, 951–967. [Google Scholar]
- Sequeira, V.B. Studies on the mechanisms of vitamin D compounds. Ph.D. Thesis, University of Sydney, Sydney, NSW, Australia, 2011. [Google Scholar]
- Rybchyn, M.S.; Slater, M.; Conigrave, A.D.; Mason, R.S. An akt-dependent increase in canonical wnt signaling and a decrease in sclerostin protein levels are involved in strontium ranelate-induced osteogenic effects in human osteoblasts. J. Biol. Chem 2011, 286, 23771–23779. [Google Scholar]
- Bikle, D.D. Vitamin D metabolism and function in the skin. Mol. Cell Endocrinol 2011, 347, 80–89. [Google Scholar]
- Kripke, M.L.; Cox, P.A.; Alas, L.G.; Yarosh, D.B. Pyrimidine dimers in DNA initiate systemic immunosuppression in UV-irradiated mice. Proc. Natl. Acad. Sci. USA 1992, 89, 7516–7520. [Google Scholar]

{kind=link}
{kind=link}
| References | UVB (mJ/cm2) | UVA (mJ/cm2) | 1,25D Dose (nm) | Cell type |
|---|---|---|---|---|
| Gupta et al. 2007 J. Invest. Derm. ([29]) | 200 | 1173 | 1–100 | Keratinocytes |
| Wong et al. 2004 J. Ster. Biochem. Mol. Biol. ([28]) | 200 | 1173 | 0.01–10 | Fibroblasts |
| Lee & Youn 1998 J. Derm. Sci. ([31]) | 50 | - | 1.2 & 12 | Keratinocytes |
| Manggau et al. 2001 J. Invest. Derm. ([30]) | 11.76 | - | 100 | Keratinocytes |
| De Haes et al. 2003 J. Cell. Biochem. ([26]) | 32 | - | 100–1000 | Keratinocytes |
© 2013 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dixon, K.M.; Tongkao-On, W.; Sequeira, V.B.; Carter, S.E.; Song, E.J.; Rybchyn, M.S.; Gordon-Thomson, C.; Mason, R.S. Vitamin D and Death by Sunshine. Int. J. Mol. Sci. 2013, 14, 1964-1977. https://doi.org/10.3390/ijms14011964
Dixon KM, Tongkao-On W, Sequeira VB, Carter SE, Song EJ, Rybchyn MS, Gordon-Thomson C, Mason RS. Vitamin D and Death by Sunshine. International Journal of Molecular Sciences. 2013; 14(1):1964-1977. https://doi.org/10.3390/ijms14011964
Chicago/Turabian StyleDixon, Katie M., Wannit Tongkao-On, Vanessa B. Sequeira, Sally E. Carter, Eric J. Song, Mark S. Rybchyn, Clare Gordon-Thomson, and Rebecca S. Mason. 2013. "Vitamin D and Death by Sunshine" International Journal of Molecular Sciences 14, no. 1: 1964-1977. https://doi.org/10.3390/ijms14011964
APA StyleDixon, K. M., Tongkao-On, W., Sequeira, V. B., Carter, S. E., Song, E. J., Rybchyn, M. S., Gordon-Thomson, C., & Mason, R. S. (2013). Vitamin D and Death by Sunshine. International Journal of Molecular Sciences, 14(1), 1964-1977. https://doi.org/10.3390/ijms14011964