α-Tocopherol at Nanomolar Concentration Protects PC12 Cells from Hydrogen Peroxide-Induced Death and Modulates Protein Kinase Activities

Abstract
:1. Introduction
2. Results and Discussion
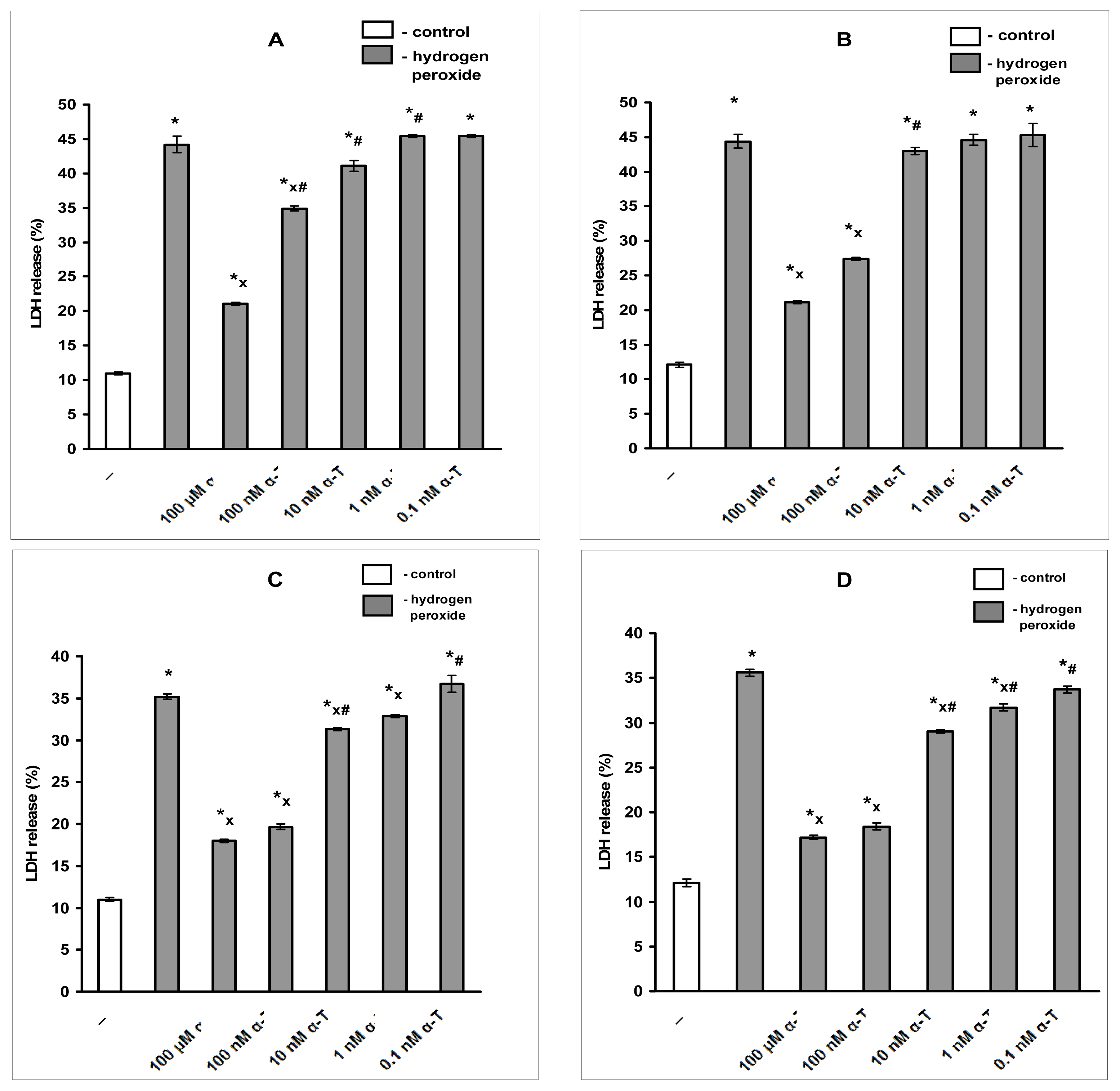
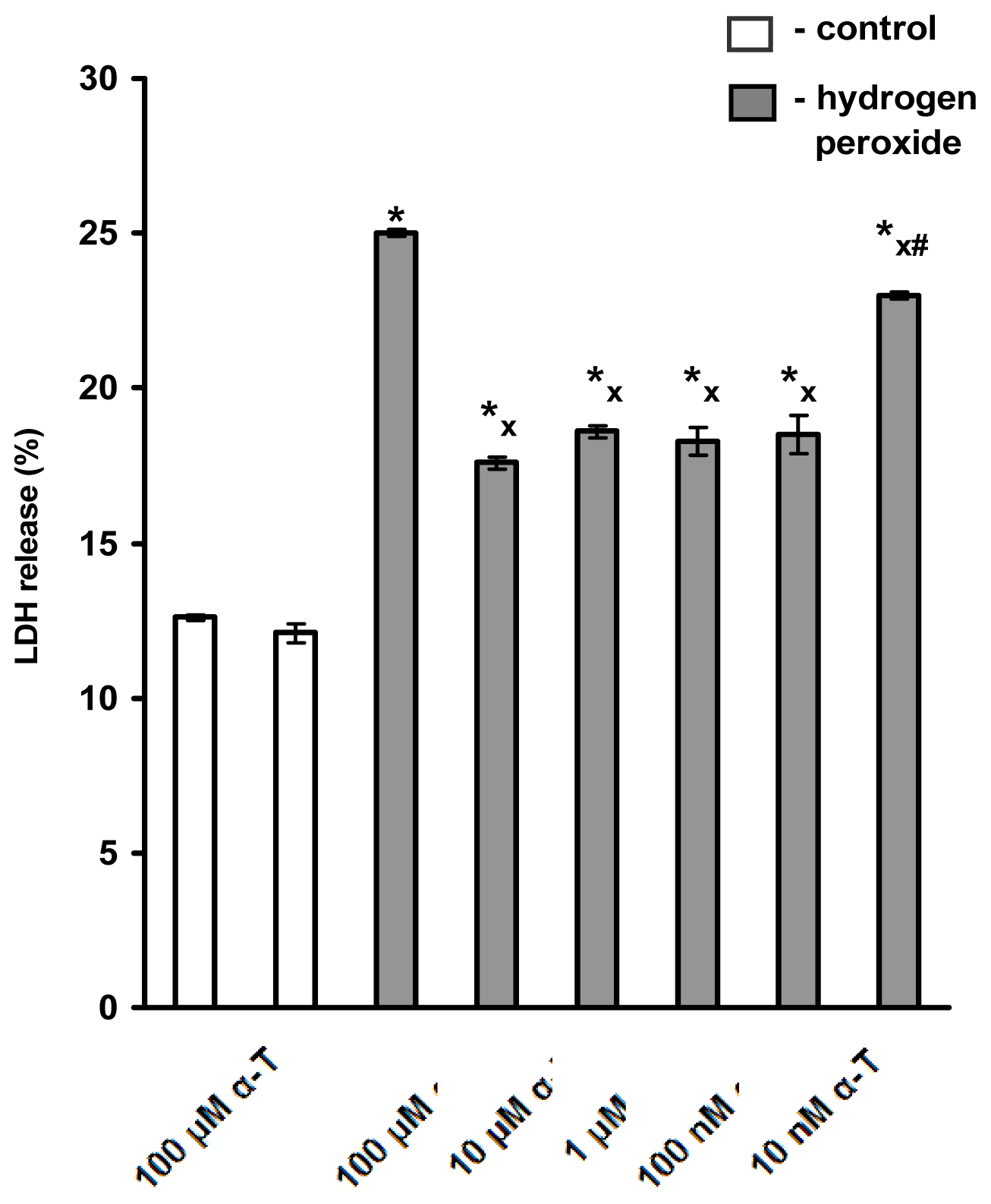
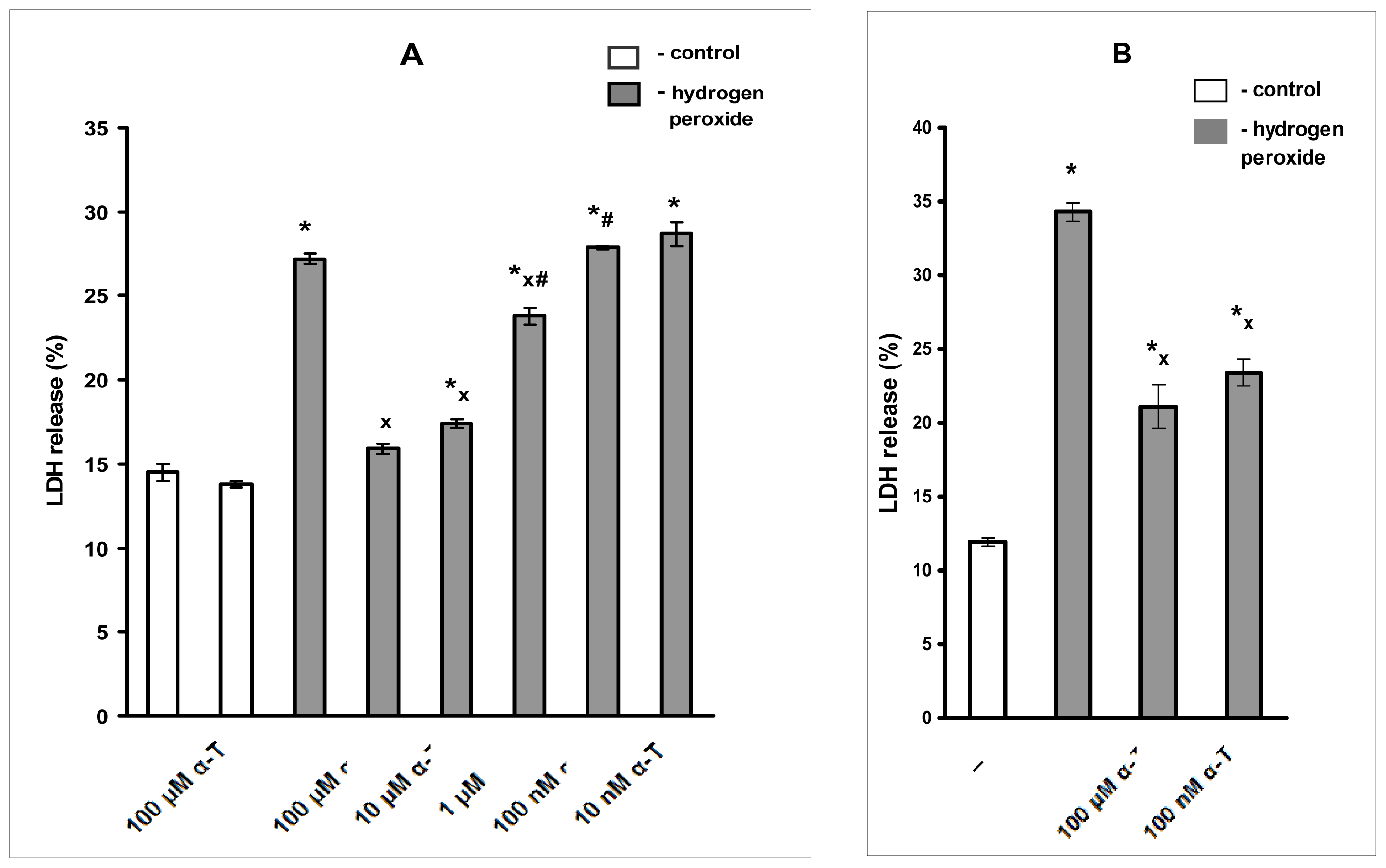
2.1. Pre-Incubation with Nanomolar α-T for 3–18 h Protects PC12 Cells against H2O2-Induced Death; the Protective Effect of α-T Is Concentration-Dependent in the Nanomolar Range (1 nM < 10 nM < 100 nM) if Pre-Incubation of PC12 Cells with It Is Performed for 18 h
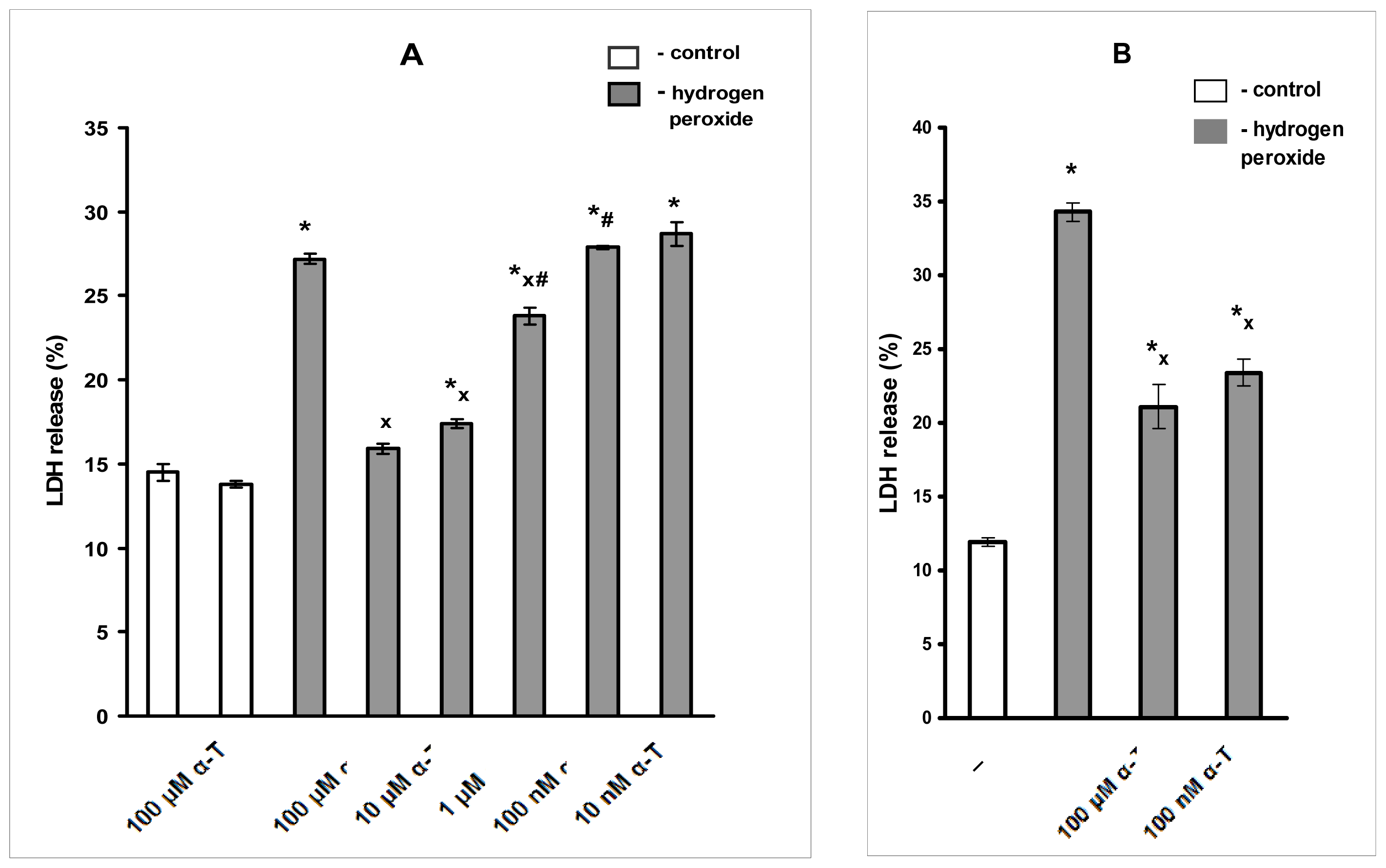
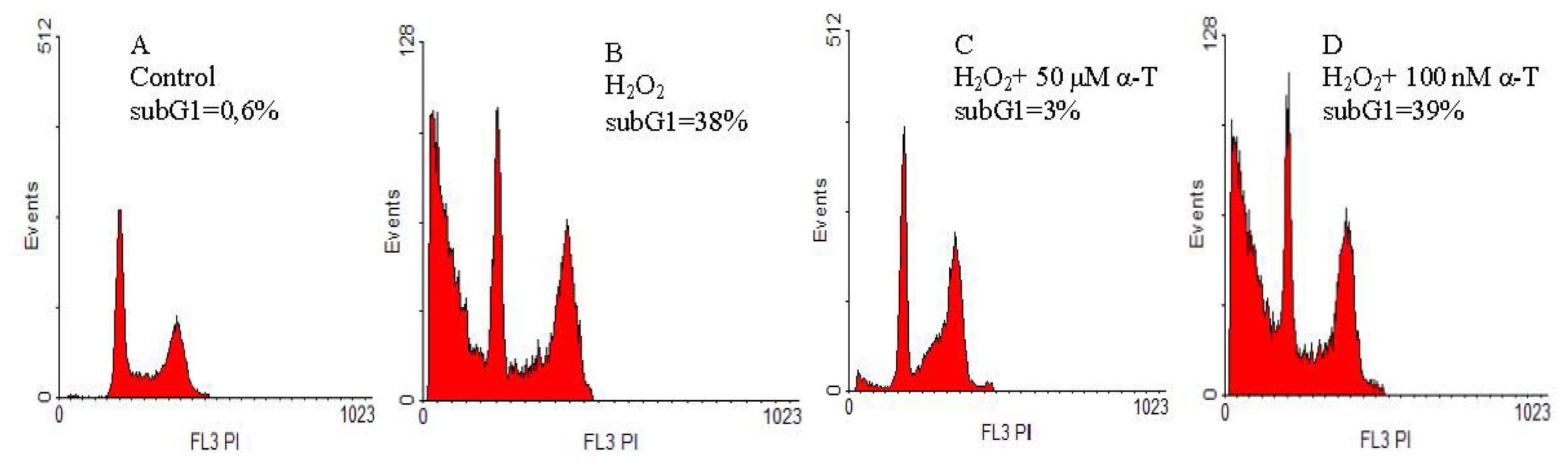
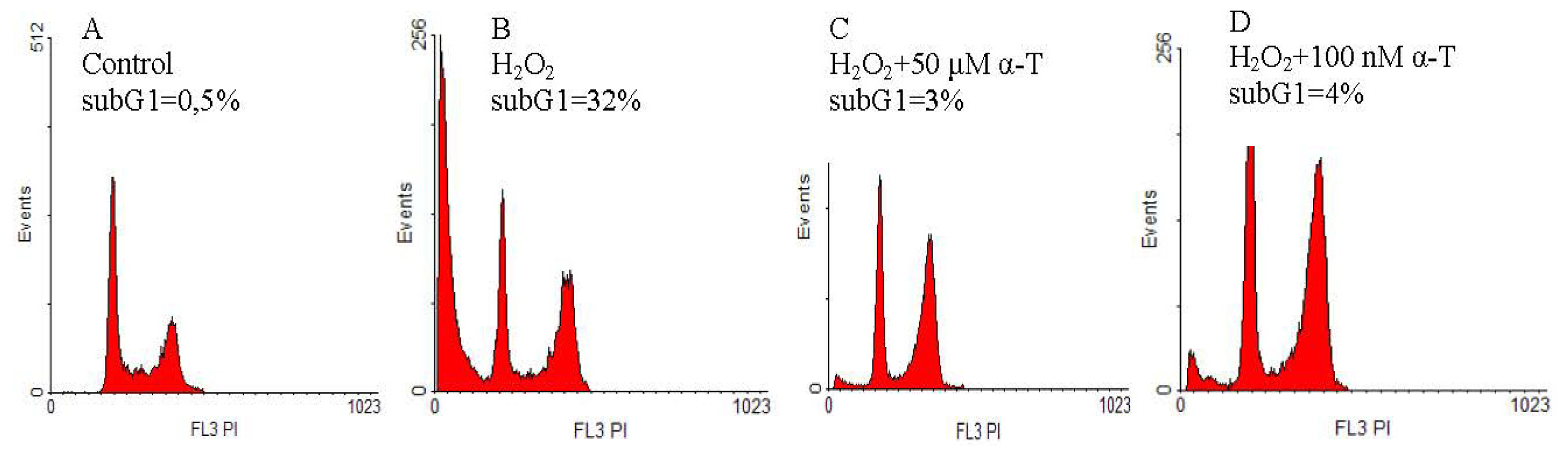
2.2. Anti-Apoptotic Effect of α-T at Nanomolar and Micromolar Concentrations on PC12 Cells Exposed to Hydrogen Peroxide
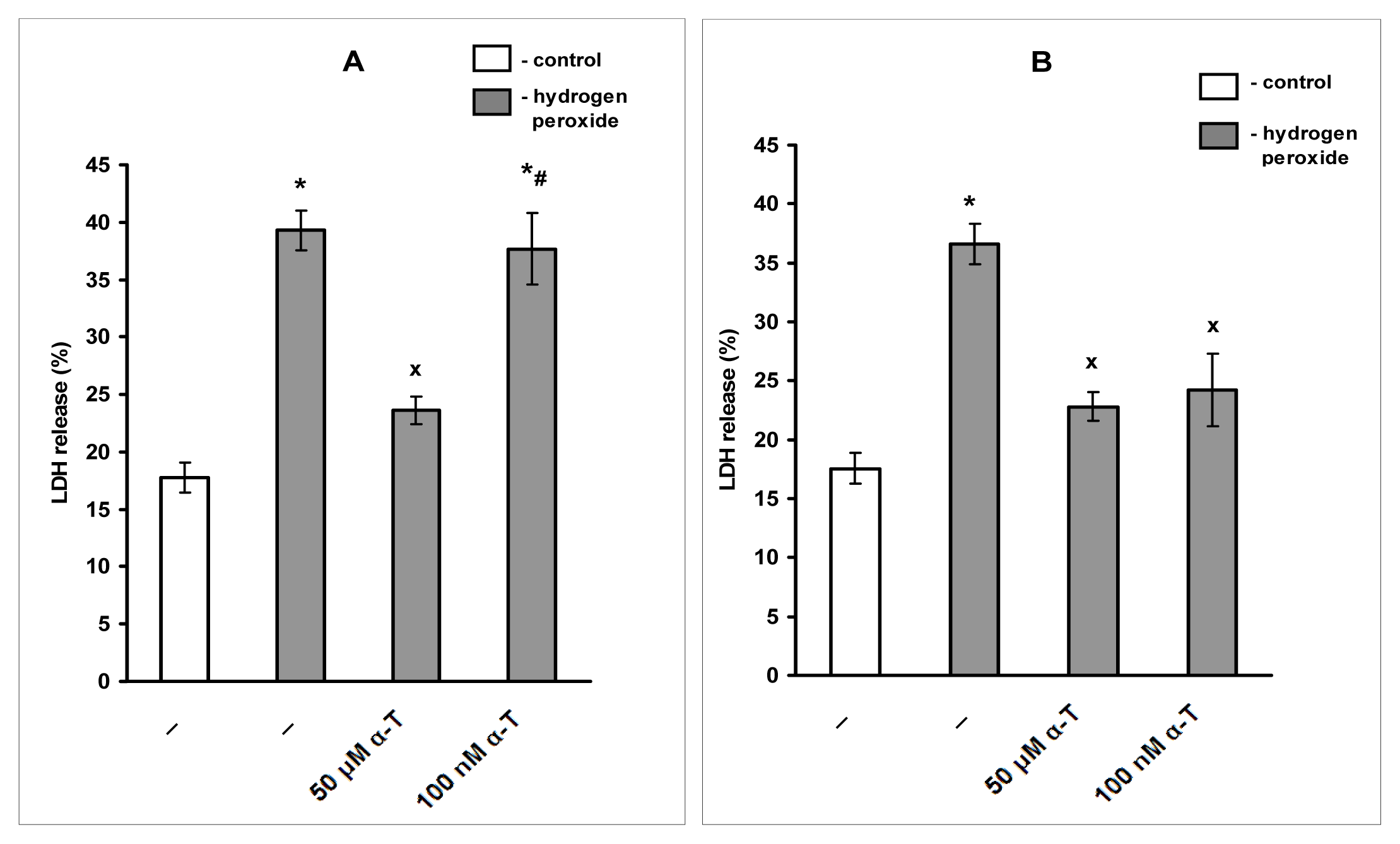
2.3. Pre-Incubation with α-T at Nanomolar Concentrations for 18 h (But Not for 0.5 h) Protects Immature Cortical Neurons from H2O2-Induced Cell Death
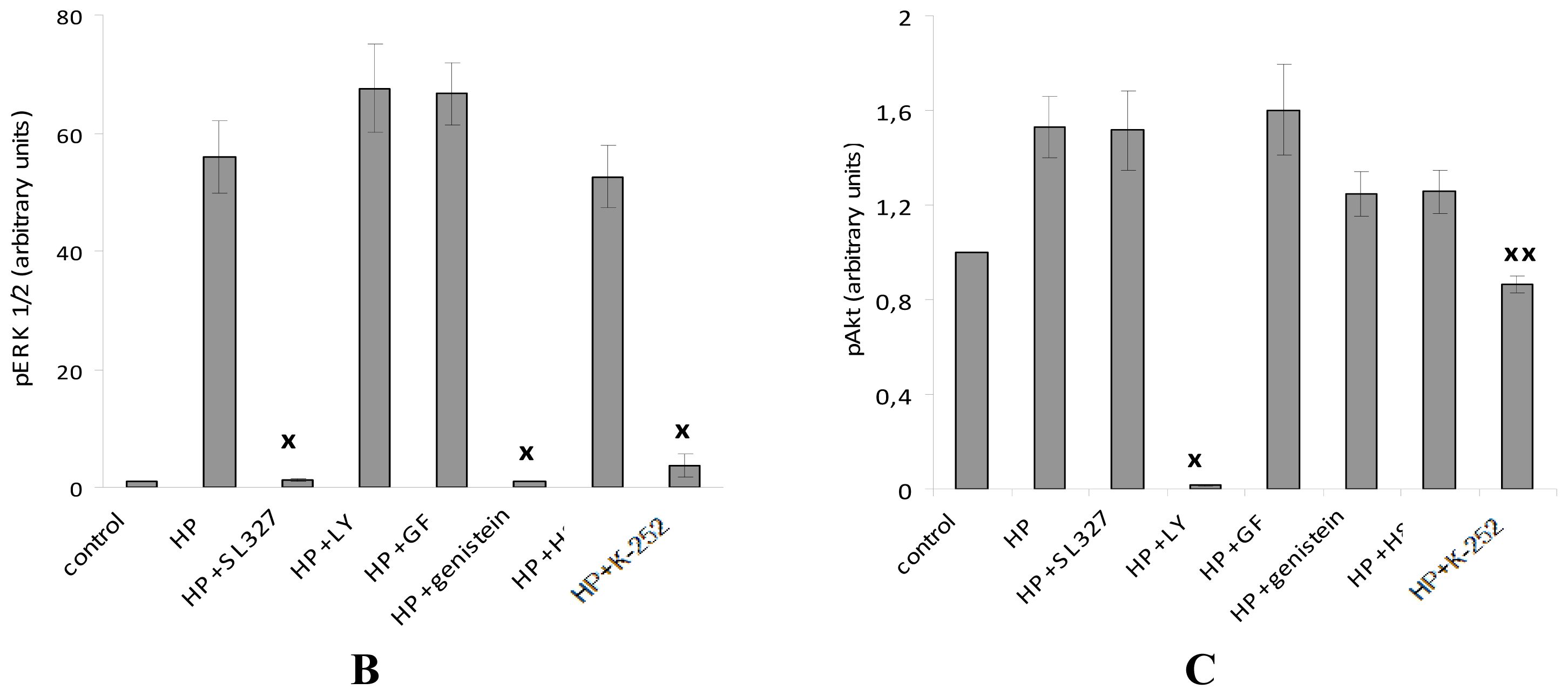
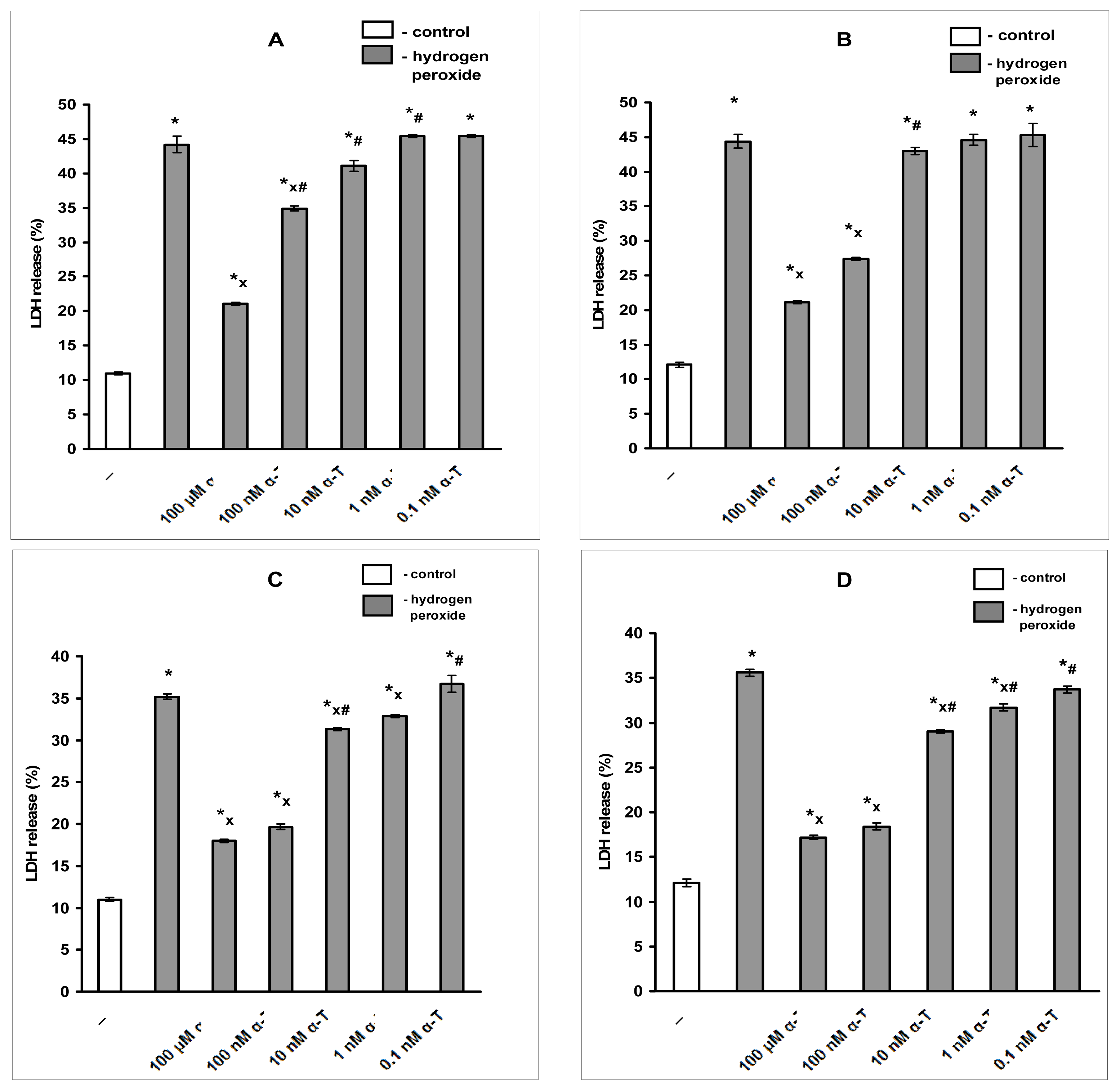
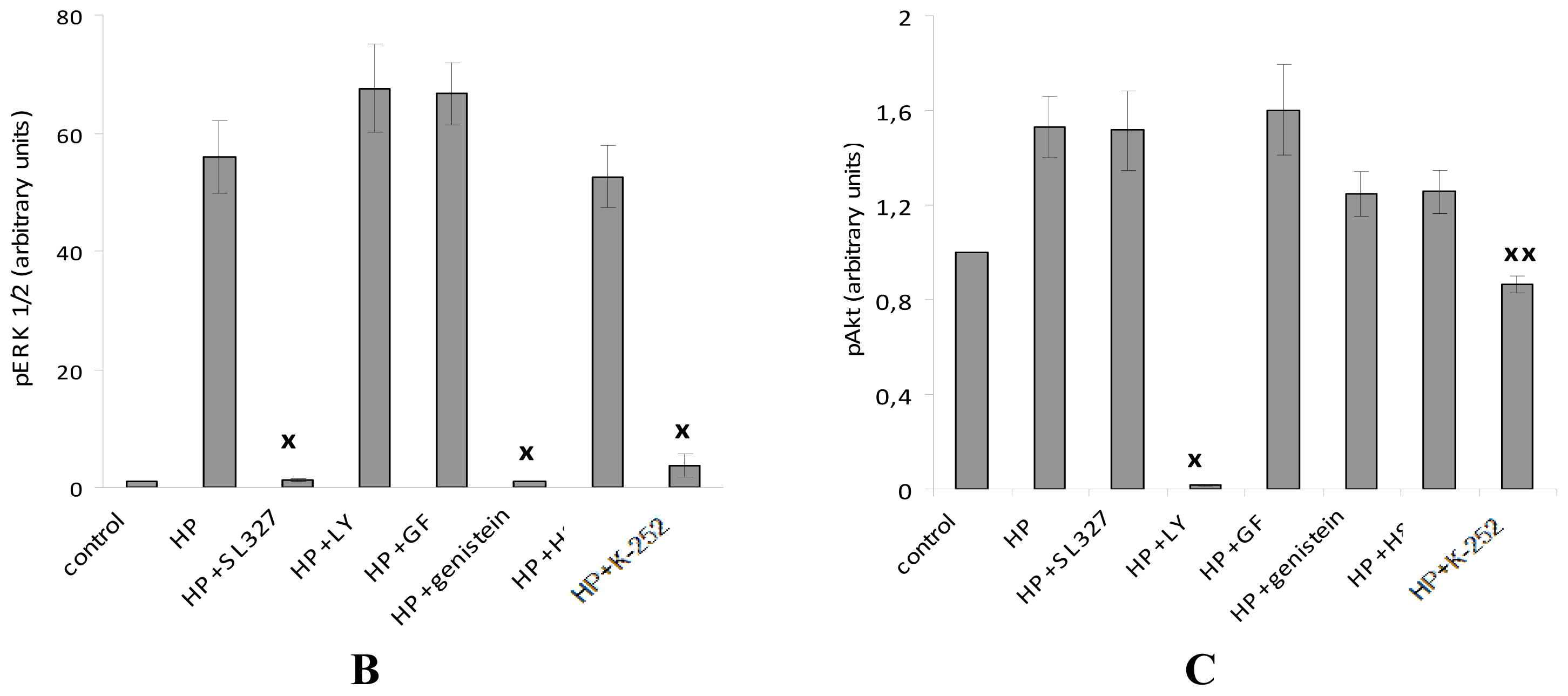
2.4. The Protective Effect of Pre-Incubation with α-T for 18 h against H2O2-Induced Toxicity in PC12 Cells Is Diminished or Abolished in the Presence of the Inhibitors of PKC, MEK 1/2 And PI 3-Kinase (GF109203X, SL327 and LY294002, Respectively)
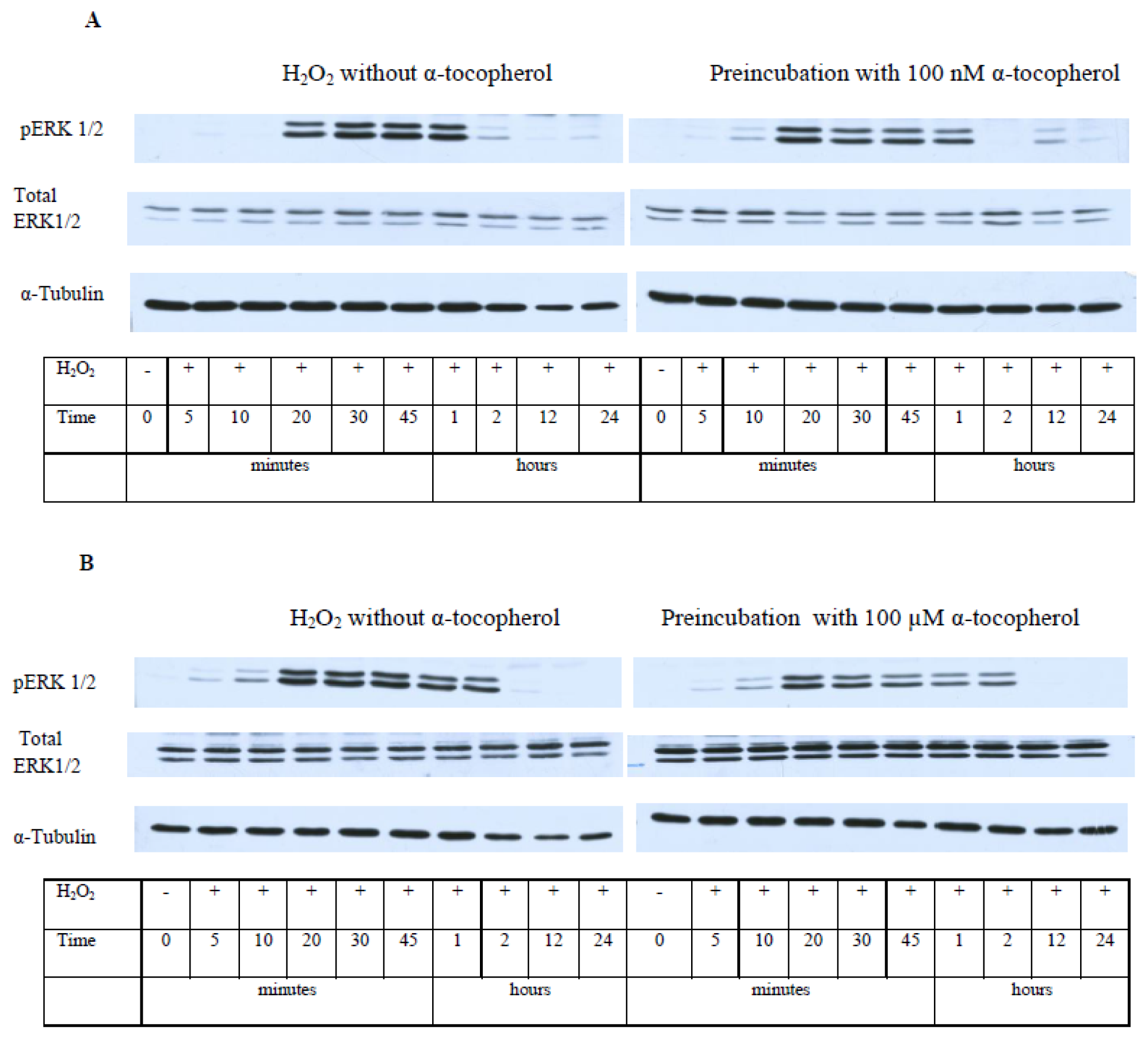
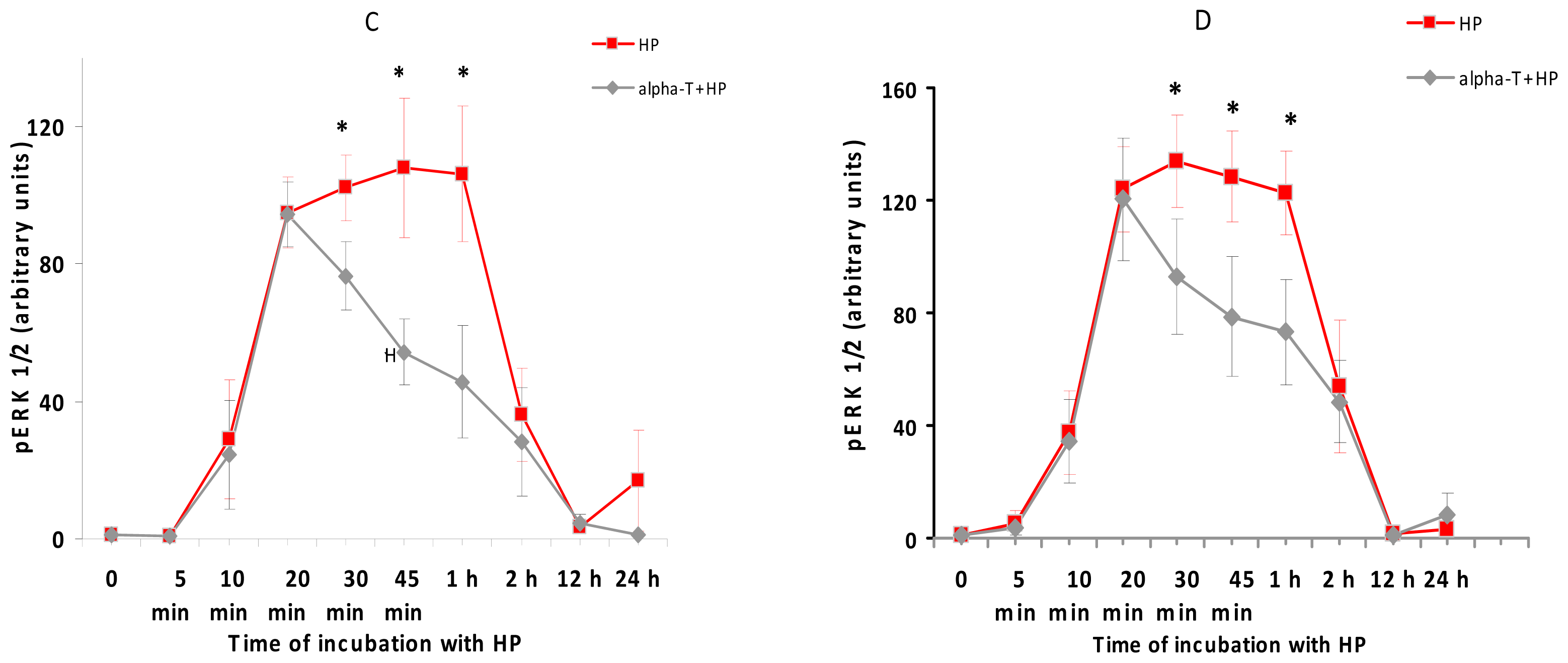
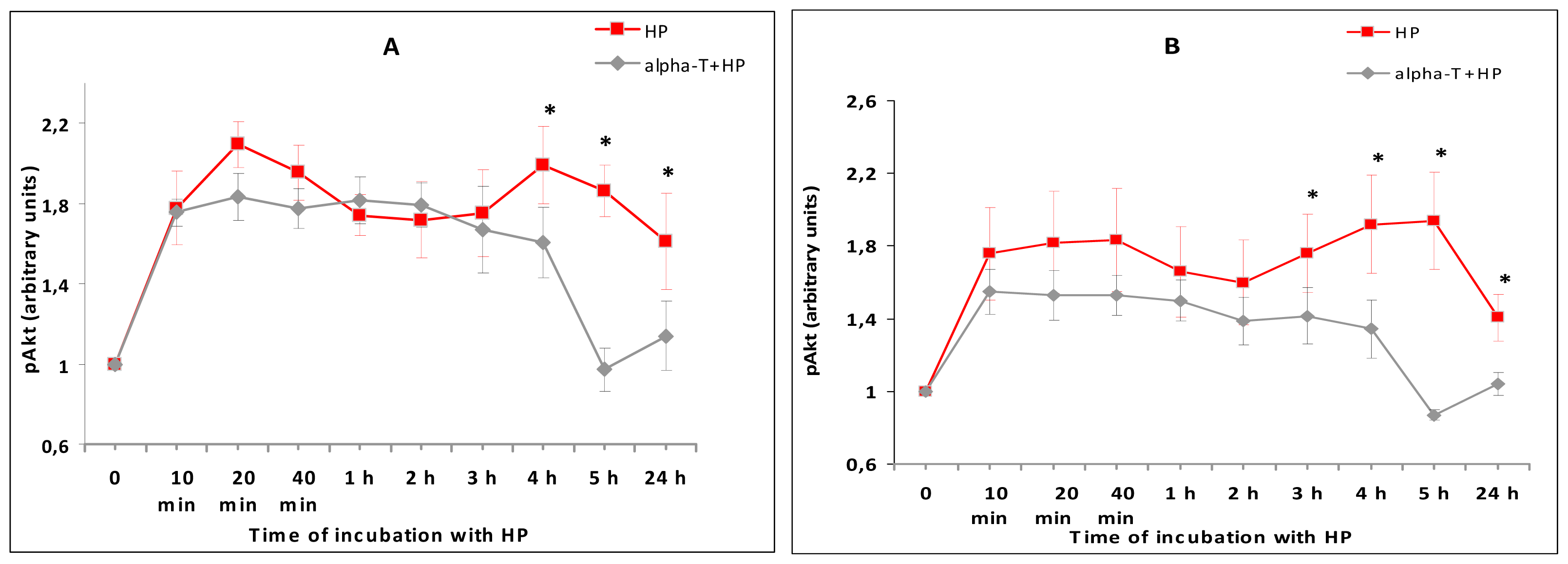
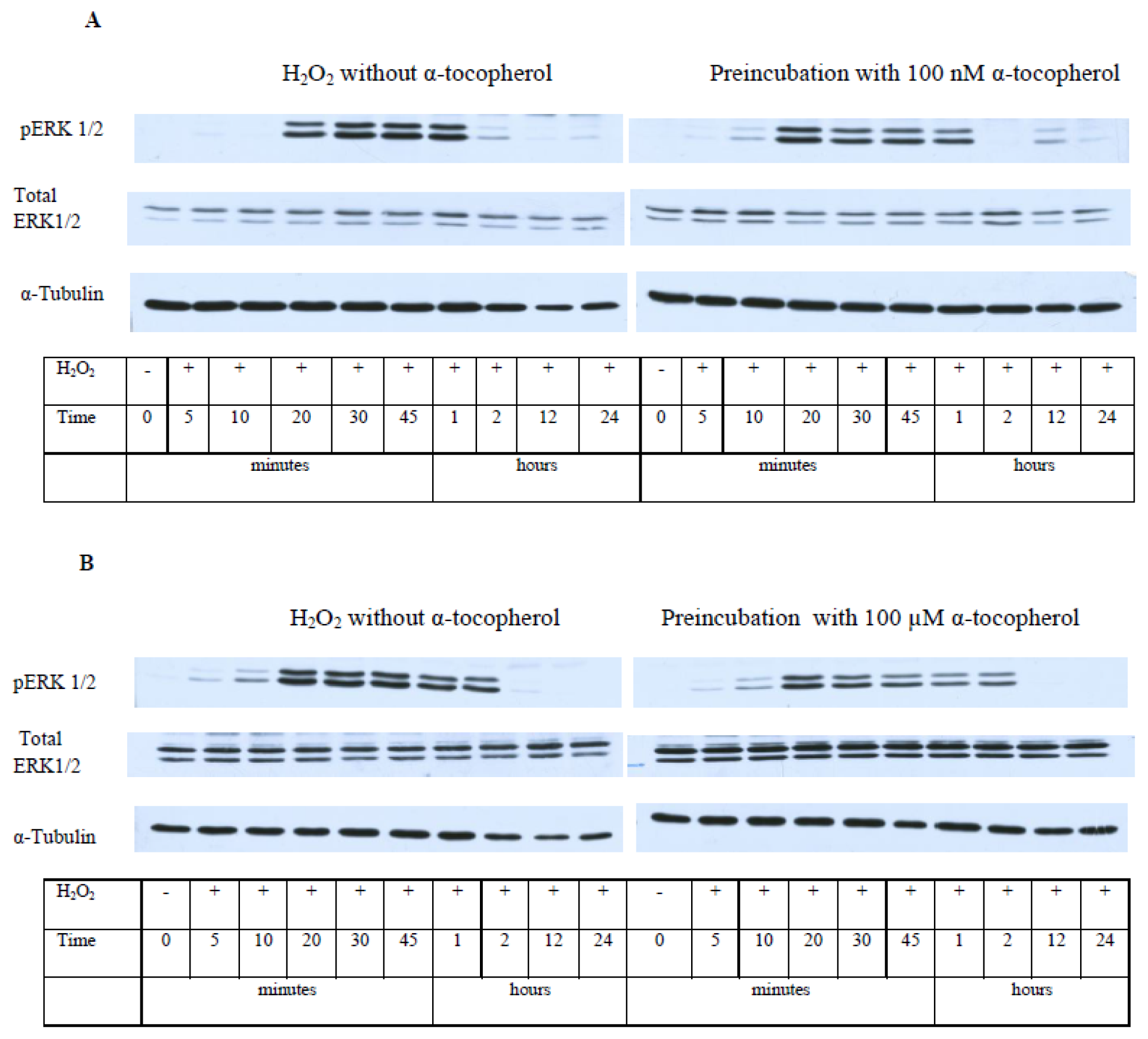
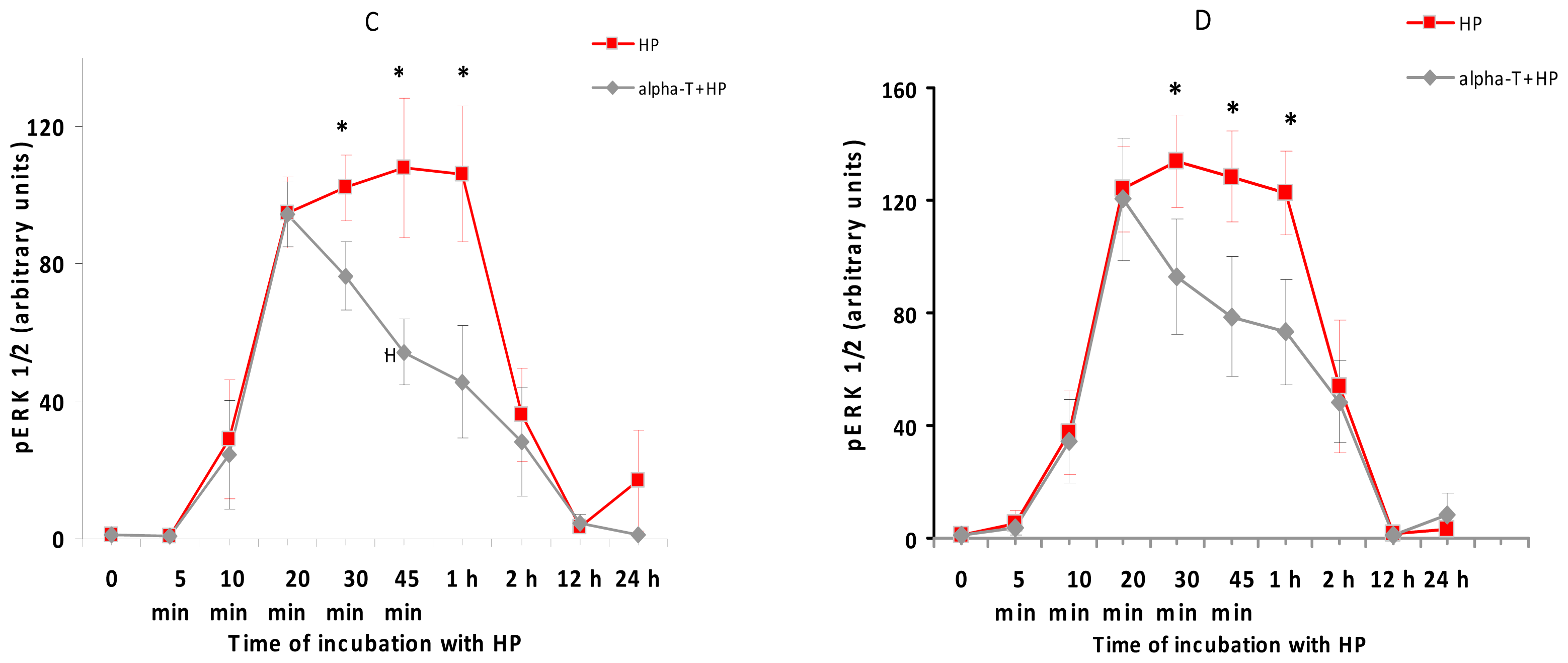
2.5. While α-T Decreases the Time of Maximal Activation of ERK 1/2 and Akt in PC12 Cells Initiated by H2O2, the Effect of 100 nM and 100 μM α-T Is Similar
2.6. Discussion
2.6.1. α-T and the Function of the Central Nervous System
2.6.2. α-T at Nanomolar Concentration Protects PC12 Neuronal Cell Line and Cultured Immature Cortical Neurons from H2O2-Induced Cell Death If Pre-Incubation Is Long, Nanomolar α-T Has Anti-Apoptotic Effect on PC12 Cells
2.6.3. The Modulation of ERK 1/2, Akt and PKC Activity by Nanomolar and Micromolar α-T and Its Contribution to α-T Protective Effect in PC12 Neuronal Cell Line
3. Experimental Procedure
3.1. Materials
3.2. PC12 Cells in Culture
3.3. Assessment of Cell Viability Using the Lactate Dehydrogenase (LDH) Method
3.4. Isolation of Embryonic Rat Cortical Neurons
3.5. Flow Cytometric Analysis
3.6. Immunoblotting
3.7. Statistical Analysis
4. Conclusion
Acknowledgements
Abbreviations
| α-T | α-tocopherol |
| ERK 1/2 | extracellular signal-regulated kinase 1/2 |
| MEK 1/2 | extracellular signal-regulated kinase kinase 1/2 |
| Akt | protein kinase B |
| PI 3-kinase | phosphoinositide 3-kinase |
| PKC | protein kinase C |
References
- De Jesus Ferreira, M.C.; Crouzin, N.; Barbanel, G.; Cohen-Solal, C.; Recasens, M.; Vignes, M.; Guiramand, J. A transient treatment of hippocampal neurons with alpha-tocopherol induces a long-lasting protection against oxidative damage via a genomic action. Free Radic. Biol. Med 2005, 39, 1009–1020. [Google Scholar]
- Ferri, P.; Cecchini, T.; Ambrogini, P.; Betti, M.; Cuppini, R.; Del Grande, P.; Ciaroni, S. α-Tocopherol affects neuronal plasticity in adult rat dentate gyrus: The possible role of PKCδ. J. Neurobiol 2006, 66, 793–810. [Google Scholar]
- Azzi, A. Molecular mechanism of α-tocopherol action. Free Radic. Biol. Med 2007, 43, 16–21. [Google Scholar]
- Zingg, J.M. Modulation of signal transduction by vitamin E. Mol. Aspects Med 2007, 28, 481–506. [Google Scholar]
- Engin, K.N. Alpha-Tocopherol: Looking beyond an antioxidant. Mol. Vis 2009, 15, 855–860. [Google Scholar]
- Brigelius-Flohe, R. Vitamin E: The shrew waiting to be tamed. Free Radic. Biol. Med 2009, 46, 543–554. [Google Scholar]
- Crouzin, N.; Ferreira, M.C.; Cohen-Solal, C.; Barbanel, G.; Guiramand, J.; Vignes, M. Neuroprotection induced by vitamin E against oxidative stress in hippocampal neurons: Involvement of TRPV1 channels. Mol. Nutr. Food Res 2010, 54, 496–505. [Google Scholar]
- Galli, F.; Azzi, A. Present trends in vitamin E research. Biofactors 2010, 36, 33–42. [Google Scholar]
- Traber, M.G.; Atkinson, J. Viamin E, antioxidant and nothing more. Free Radic. Biol. Med 2007, 43, 4–15. [Google Scholar]
- Cuddihy, S.L.; Ali, S.S.; Musiec, E.S.; Lucero, J.; Kopp, S.J.; Morrow, J.D.; Dugan, L.L. Prolonged α-tocopherol deficiency decreases oxidative stress and unmasks α-tocopherol-dependent regulation of mitochondrial function in the brain. J. Biol. Chem 2008, 283, 6915–6924. [Google Scholar]
- Vatassery, G.T.; Adityanjee; Quach, H.T.; Smith, W.E.; Kuskowski, M.A.; Melnyk, D. Alpha and gamma tocopherols in cerebrospinal fluid and serum from older, male, human subjects. J. Am. Coll. Nutr. 2004, 23, 233–238. [Google Scholar]
- De Bustos, F.; Jimenez-Jimenez, F.J.; Molina, J.A.; Esteban, J.; Guerrero-Sola, A.; Zurdo, M.; Orti-Pareja, M.; Tallon-Barranco, A.; Gomez-Escalonilla, C.; Ramirez-Ramos, C.; et al. Cerebrospinal fluid levels of alpha-tocopherol in amyotrophic lateral sclerosis. J. Neural Transm 1998, 105, 703–708. [Google Scholar]
- Schippling, S.; Kontush, A.; Arit, S.; Buhmann, C.; Sturenburg, H.J.; Mann, U.; Muller-Thomsen, T.; Beisiegel, U. Increased lipoprotein oxidation in Alzheimer’s disease. Free Radic. Biol. Med 2000, 28, 351–360. [Google Scholar]
- Khanna, S.; Roy, S.; Ryu, H.; Bahaddun, P.; Swaan, P.W.; Ratian, R.R.; Sen, C.K. Molecular basis of vitamin E action: Tocotrienol modulates 12-lipoxygenase, a key mediator of glutamate-induced neurodegeneration. J. Biol. Chem 2003, 278, 43508–43515. [Google Scholar]
- Khanna, S.; Parinandi, N.L.; Kotha, S.R.; Roy, S.; Rink, C.; Bibus, D.; Sen, C.K. Nanomolar vitamin E α-tocotrienol inhibits glutamate-induced activation of phospholipase A2 and causes neuroprotection. J. Neurochem 2010, 112, 1249–1260. [Google Scholar]
- Saito, Y.; Nishio, K.; Akazawa, Y.O.; Yamanaka, K.; Miyama, A.; Yoshida, Y.; Noguchi, N.; Niki, E. Cytoprotective effects of vitamin E homologues against glutamate-induced cell death in immature primary cortical neuron cultures: Tocopherols and tocotrienols exert similar effects by antioxidant function. Free Radic. Biol. Med 2010, 49, 1542–1549. [Google Scholar]
- Numakawa, Y.; Numakawa, T.; Matsumoto, T.; Yagasaki, Y.; Kumamaru, E.; Kunugi, H.; Taguchi, T.; Niki, E. Vitamin E protected cultured cortical neurons from oxidative stress-induced cell death through the activation of mitogen-activated protein kinase and phosphatidylinositol 3-kinase. J. Neurochem 2006, 97, 1191–1202. [Google Scholar]
- Kondo, K.; Obitsu, S.; Ohta, S.; Matsunami, K.; Otsuka, H.; Teshima, R. Poly(ADP-ribose) polymerase (PARP)-1-independent apoptosis-inducing factor (AIF) release and cell death are induced by eleostearic acid and blocked by α-tocopherol and MEK inhibition. J. Biol. Chem 2010, 285, 13079–13091. [Google Scholar]
- Cerezo-Guisado, M.I.; Garcia-Marin, L.J.; Lorenzo, M.J.; Bragado, M.J. Lovastatin inhibits the growth and survival pathways of phosphoinositide 3-kinase/protein kinase B in immortalized rat brain neuroblasts. J. Neurochem 2005, 94, 1277–1287. [Google Scholar]
- Di Donato, I.; Bianchi, S.; Federico, A. Ataxia with vitamin E deficiency: Update of molecular diagnosis. Neurol. Sci 2010, 31, 511–515. [Google Scholar]
- Ames, D.; Ritchie, C. Antioxidants and Alzheimer’s disease: Time to stop feeding vitamin E to dementia patients? Int. Psychogeriatr 2007, 19, 1–8. [Google Scholar]
- Miller, E.R.; Pastor-Barriuso, R.; Dalal, D.; Riemersma, R.; Appel, L.J.; Guallar, E. Meta-analysis: High-Dosage vitamin E supplementation may increase all-cause mortality. Ann. Intern. Med 2005, 142, 37–46. [Google Scholar]
- Bjelakovic, G.; Nikolova, D.; Gluud, L.L.; Simonetti, R.G.; Gluud, C. Mortality in randomized trials of antioxidant supplements for primary and secondary prevention: Systematic review and meta-analysis. J. Am. Med. Assoc 2007, 297, 842–857. [Google Scholar]
- Proskuryakov, S.Y.; Konoplyannikov, A.G.; Gabai, V.L. Necrosis: A specific form of programmed cell death? Exp. Cell Res 2003, 283, 1–16. [Google Scholar]
- Krysko, D.V.; Vanden Berghe, T.; D’Herde, K.; Vandenabeele, P. Apoptosis and necrosis: Detection, discrimination and phagocytosis. Methods 2008, 44, 205–221. [Google Scholar]
- Cole, K.K.; Perez-Polo, J.R. Poly (ADP-ribose) polymerase inhibition prevents both apoptotic-like delayed neuronal death and necrosis after H2O2 injury. J. Neurochem 2002, 82, 19–29. [Google Scholar]
- Luo, Y.; DeFranco, D.B. Opposing roles for ERK 1/2 in neuronal oxidative toxicity: Distinct mechanisms of ERK 1/2 action at early versus late phases of oxidative stress. J. Biol. Chem 2006, 281, 16436–16442. [Google Scholar]
- Chen, L.; Liu, L.; Yin, J.; Luo, Y.; Huang, S. Hydrogen peroxide-induced neuronal apoptosis is associated with inhibition of protein phosphatase 2A and 5, leading to activation of MAPK pathway. Int. J. Biochem. Cell Biol 2009, 41, 1284–1295. [Google Scholar]
- Kulebyakin, K.; Karpova, L.; Lakonsteva, E.; Krasavin, M.; Boldyrev, A. Carnosine protects neurons against oxidative stress and modulates the time profile of MAPK cascade signaling. Amino Acids 2012, 43, 91–96. [Google Scholar]
- Azzi, A.; Breyer, I.; Feher, M.; Ricciarelli, R.; Stocker, A.; Zimmer, S.; Zingg, J. Nonantioxidant functions of alpha-tocopherol in smooth muscle cells. J. Nutr 2001, 131, 378S–381S. [Google Scholar]
- Yano, T.; Yajima, S.; Hagiwara, K.; Kumadaki, I.; Yano, Y.; Otani, S.; Uchida, M.; Ichikawa, T. Vitamin E inhibits cell proliferation and the activation of extracellular signal-regulated kinase during the promotion phase of lung tumorigenesis irrespective of antioxidative effect. Carcinogenesis 2000, 21, 2129–2133. [Google Scholar]
- He, K.; Aizenman, E. ERK signaling leads to mitochondrial dysfunction in extracellular zinc-induced neurotoxicity. J. Neurochem 2010, 114, 452–461. [Google Scholar]
- Ho, Y.; Samarasinghe, R.; Knoch, M.E.; Lewis, M.E.; Aizenman, E.; DeFranco, D.B. Selective inhibition of mitogen-activated protein kinase phosphatases by zinc accounts for extracellar signal-regulated kinase 1/2-dependent oxidative neuronal cell death. Mol. Pharmacol 2008, 74, 1141–1151. [Google Scholar]
- Ziegler, C.G.; Sicard, F.; Sperber, S.; Ehrhart-Bomstein, M.; Bornstein, S.R.; Krug, A.W. DHEA reduces NGF-mediated cell survival in serum-deprived PC12 cells. Ann. N. Y. Acad. Sci 2006, 1073, 306–311. [Google Scholar]
- Vauzour, D.; Vafeiadou, K.; Rice-Evans, C.; Williams, R.J.; Spencer, J.P. Activation of pro-survival Akt and ERK 1/2 signaling pathways underlie the anti-apoptotic effects of flavanones in cortical neurons. J. Neurochem 2007, 103, 1355–1367. [Google Scholar]
- Namura, S.; Iihara, K.; Takami, S.; Nagata, I.; Kikuchi, H.; Matsushita, K.; Moskowitz, M.A.; Bonventre, J.V.; Alessandrini, A. Intravenous administration of MEK inhibitor UO126 affords brain protection against forebrain ischemia and focal cerebral ischemia. Proc. Natl. Acad. Sci. USA 2001, 98, 11569–11574. [Google Scholar]
- Wang, X.; Wang, H.; Xu, L.; Rozanski, D.J.; Sugawara, T.; Chan, P.H.; Trzaskos, J.M.; Feuerstein, G.Z. Significant neuroprotection against ischemic brain injury by inhibition of the MEK1 protein kinase in mice: Exploration of potential mechanism associated with apoptosis. J. Pharmacol. Exp. Ther 2003, 304, 172–178. [Google Scholar]
- Lu, K.; Liang, C.-L.; Liliang, P.-C.; Yang, S.-H.; Cho, C.-L.; Weng, H.-C.; Tsai, Y.-D.; Wang, K.-W.; Chen, H.-J. Inhibition of extracellular signal-regulated kinases 1/2 provides neuroprotection in spinal cord ischemia/reperfusion injury in rats: Relationship with the nuclear factor-κB-regulated anti-apoptotic mechanisms. J. Neurochem 2010, 114, 237–246. [Google Scholar]
- Betti, M.; Ambrogini, P.; Minelli, A.; Floridi, A.; Lattanzi, D.; Ciuffoli, S.; Bucherelli, C.; Prospero, E.; Frontini, A.; Santarelli, L.; et al. Maternal dietary load of α-tocopherol depress protein kinase C signaling and synaptic plasticity in rat postnatal developing hppocampus and promotes permanent deficits in adult offspring. J. Nutr. Biochem 2011, 22, 60–70. [Google Scholar]
- Maltseva, E.L.; Palmina, N.P.; Burlakova, E.B. Natural (alpha-tocopherol) and synthetic (phenosan potassium salt) antioxidants regulate the protein kinase C activity in a broad concentration range (10−4–10−20 M). Membr. Cell Biol 1998, 12, 251–268. [Google Scholar]
- Martin-Nizard, F.; Boullier, A.; Fruchart, J.C.; Duriez, P. α-Tocopherol but not β-tocopherol inhibits thrombin-induced PKC activation and endothelin secretion in endothelial cells. J. Cardiovasc. Risk 1998, 5, 339–345. [Google Scholar]
- Zirrgiebel, U.; Ohga, Y.; Carter, B.; Behringer, B.; Inagaki, N.; Thoenen, H.; Lindholm, D. Characterization of TrkB receptor-mediated signaling pathways in rat cerebellar granule neurons: Involvement of protein kinase C in neuronal survival. J. Neurochem 1995, 65, 2241–2250. [Google Scholar]
- Wang, Y.; Hirai, K.; Ashraf, M. Activation of mitochondrial ATP-sensitive K+ channel for cardiac protection against ischemic injury is dependent on protein kinase C activity. Circ. Res 1999, 85, 731–741. [Google Scholar]
- Hongpaisan, J.; Sun, M.K.; Alkon, D.L. PKCɛ activation prevents synaptic loss, Aβ elevation, and cognitive deficits in Alzheimer’s disease transgenic mice. J. Neurosci 2011, 31, 630–643. [Google Scholar]
- Chan, J.Y.; Chang, A.Y.; Wang, L.L.; Ou, C.C.; Chan, S.H. Protein kinase C-dependent mitochondrial translocation of proapoptotic protein Bax on activation of inducible nitric oxide synthase in rostral ventrolateral medulla mediates cardiovascular depression during experimental endotoxemia. Mol. Pharmacol 2007, 71, 1129–1139. [Google Scholar]
- Sun, F.; Kanthasamy, A.; Song, C.; Yang, Y.; Anantharam, V.; Kanthasamy, A.G. Proteasome inhibitor-induced apoptosis is mediated by positive feedback amplification of PKCδ proteolytic activation and mitochondrial translocation. J. Cell. Mol. Med 2008, 12, 2467–2481. [Google Scholar]
- Vassault, A. Lactate dehydrogenase: UV-Method with Pyruvate and NADH. In Methods of Enzymatic Analysis; Bergmeyer, H.U., Ed.; Verlag Chemie: Weinheim, Germany, 1983; Volume 3, pp. 118–126. [Google Scholar]
- Dichter, M.A. Rat cortical neurons in cell culture: Culture methods, cell morphology, electrophysiology, and synapse formation. Brain Res 1978, 149, 279–293. [Google Scholar]
- Antonov, S.M.; Gmiro, V.E.; Johnson, J.W. Binding sites for permeant ions in the channel of NMDA receptors and their effects on channel block. Nat. Neurosci 1998, 1, 451–461. [Google Scholar]
- Gong, J.; Traganos, F.; Darzynkiewicz, Z. A selective procedure for DNA extraction from apoptotic cells applicable for gel electrophoresis and flow cytometry. Anal. Biochem 1994, 218, 314–319. [Google Scholar]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pre-incubation Time/Sample | 3 h | 6 h | 12 h | 18 h |
|---|---|---|---|---|
| Rescue Rates % | ||||
| 100 μM α-T | 67.7 ± 3.95 *,x | 72.6 ± 4.6 *,x | 67.1 ± 3.3 * | 66.2 ± 4.5 * |
| 100 nM α-T | 44.0 ± 6.9 *,x | 48.2 ± 5.8 *,x | 61.2 ± 3.6 *x | 63.9 ± 3.5 *,x |
| 10 nM α-T | 10.5 ± 2.4 * | 24.2 ± 1.2 * | 28.9 ± 5.4 *,# | 33.1 ± 2.4 *,# |
| 1 nM α-T | 2.90 ± 3.8 | 7.2 ± 4.1 | 10.5 ± 0.5 * | 15.8 ± 0.4 * |
| 0.1 nM α-T | 1.3 ± 3.2 | 2.3 ± 5.08 | 1.6 ± 7.1 | 6.9 ± 7.0 |
| Sample | Rescue Rates of α-T, % |
|---|---|
| 100 nM α-T | 53.3 ± 4.9 * |
| 100 nM α-T + 1 μM GF109203X | 20.7 ± 8.0 x |
| 100 nM α-T | 48.3 ± 6.0 * |
| 100 nM α-T + 10 μM SL327 | 34.5 ± 4.9 *,# |
| 100 nM α-T | 44.9 ± 6.6 * |
| 100 nM α-T + 50 μM LY294002 | 19.8 ± 10.1 # |
| 100 nM α-T | 53.7 ± 5.4 * |
| 100 nM α-T + 10 μM LY294002 | 48.2 ± 2.1 * |
| 100 nM α-T | 52.2 ± 6.3 * |
| 100 nM α-T + 10 μM SL327 + 10 μM LY294002 | 39.7 ± 8.3 *,x |
| 100 μM α-T | 55.9 ± 6.9 * |
| 100 μM α-T + 1 μM GF109203X | 19.7 ± 9.0 x |
| 100 μM α-T | 55.7 ± 6.2 * |
| 100 μM α-T + 10 μM SL327 | 36.5 ± 5.2 *,# |
| 100 μM α-T | 53.4 ± 9.5 * |
| 100 μM α-T + 50 μM LY294002 | 29.8 ± 6.9 *,# |
| 100 μM α-T | 52.6 ± 5.7 * |
| 100 μM α-T + 10 μM LY294002 | 48.3 ± 4.5 * |
| 100 μM α-T | 57.7 ± 7.6 * |
| 100 μM α-T + 10 μM SL327 + 10 μM LY294002 | 39.3 ± 7.4 *,x |
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Zakharova, I.O.; Sokolova, T.V.; Bayunova, L.V.; Vlasova, Y.A.; Rychkova, M.P.; Avrova, N.F. α-Tocopherol at Nanomolar Concentration Protects PC12 Cells from Hydrogen Peroxide-Induced Death and Modulates Protein Kinase Activities. Int. J. Mol. Sci. 2012, 13, 11543-11568. https://doi.org/10.3390/ijms130911543
Zakharova IO, Sokolova TV, Bayunova LV, Vlasova YA, Rychkova MP, Avrova NF. α-Tocopherol at Nanomolar Concentration Protects PC12 Cells from Hydrogen Peroxide-Induced Death and Modulates Protein Kinase Activities. International Journal of Molecular Sciences. 2012; 13(9):11543-11568. https://doi.org/10.3390/ijms130911543
Chicago/Turabian StyleZakharova, Irina O., Tatyana V. Sokolova, Liubov V. Bayunova, Yulia A. Vlasova, Maria P. Rychkova, and Natalia F. Avrova. 2012. "α-Tocopherol at Nanomolar Concentration Protects PC12 Cells from Hydrogen Peroxide-Induced Death and Modulates Protein Kinase Activities" International Journal of Molecular Sciences 13, no. 9: 11543-11568. https://doi.org/10.3390/ijms130911543
APA StyleZakharova, I. O., Sokolova, T. V., Bayunova, L. V., Vlasova, Y. A., Rychkova, M. P., & Avrova, N. F. (2012). α-Tocopherol at Nanomolar Concentration Protects PC12 Cells from Hydrogen Peroxide-Induced Death and Modulates Protein Kinase Activities. International Journal of Molecular Sciences, 13(9), 11543-11568. https://doi.org/10.3390/ijms130911543