Scientific Evidence and Rationale for the Development of Curcumin and Resveratrol as Nutraceutricals for Joint Health


Abstract
:1. Introduction
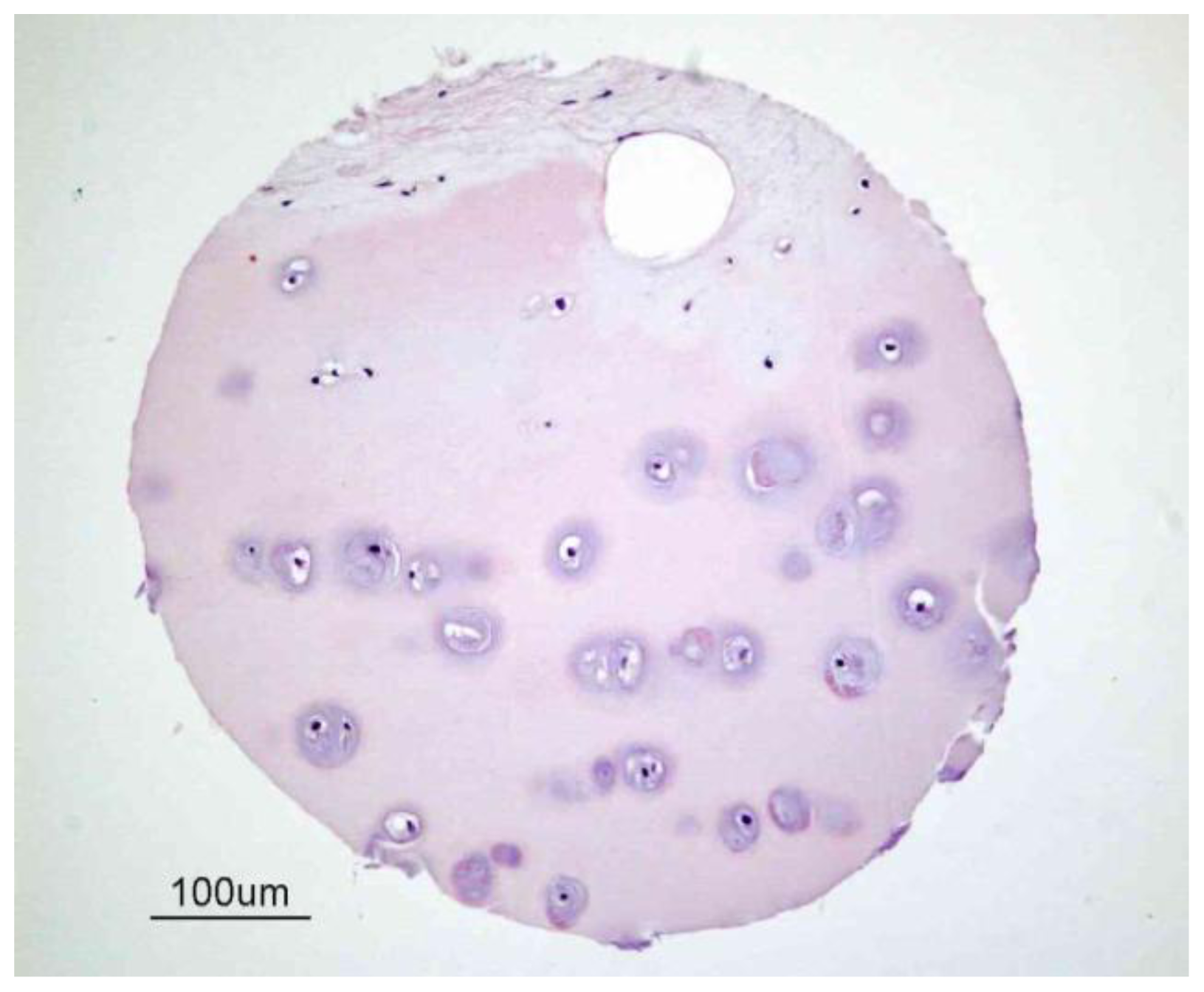
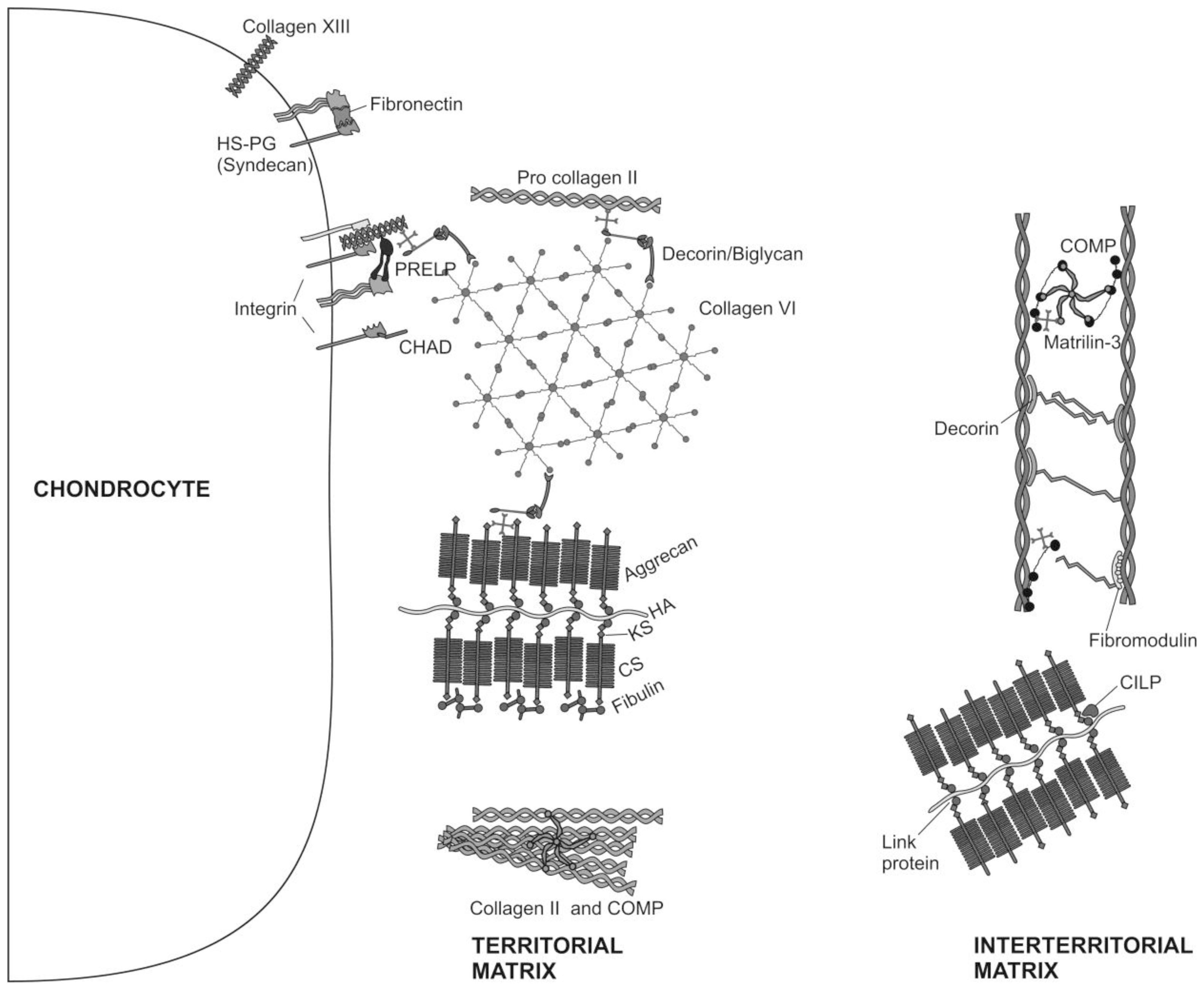
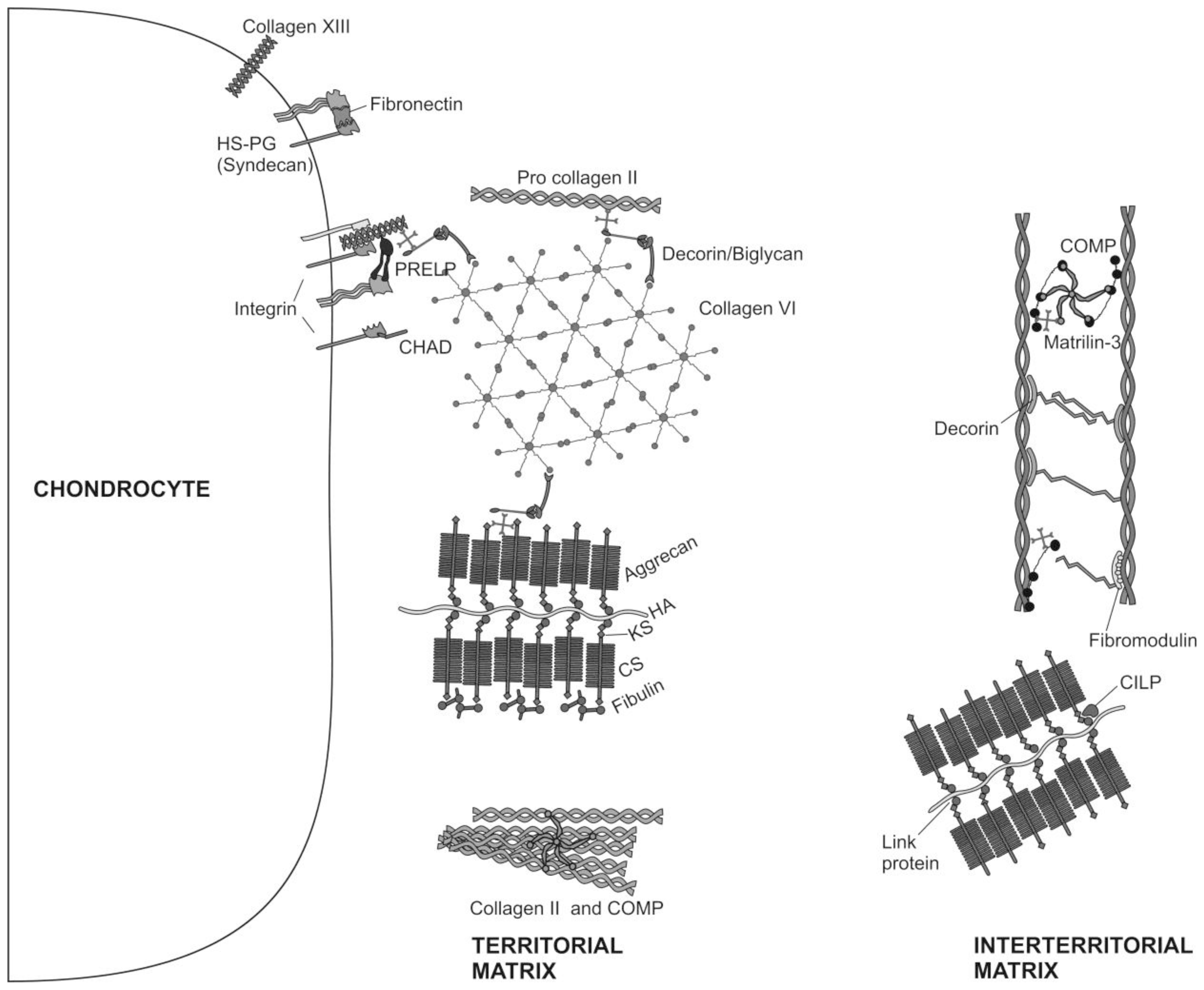
2. Articular Cartilage-Structure and Function
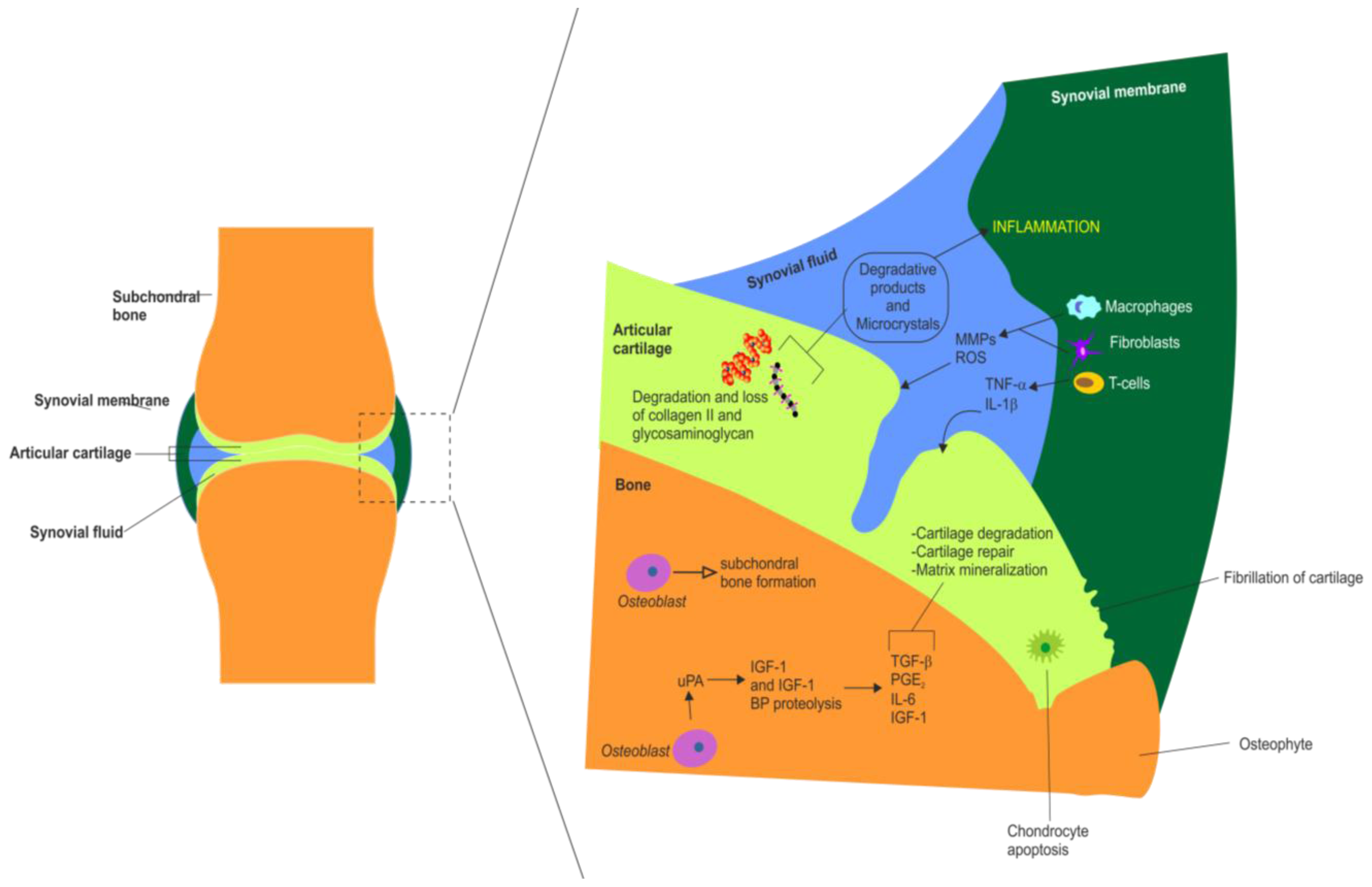
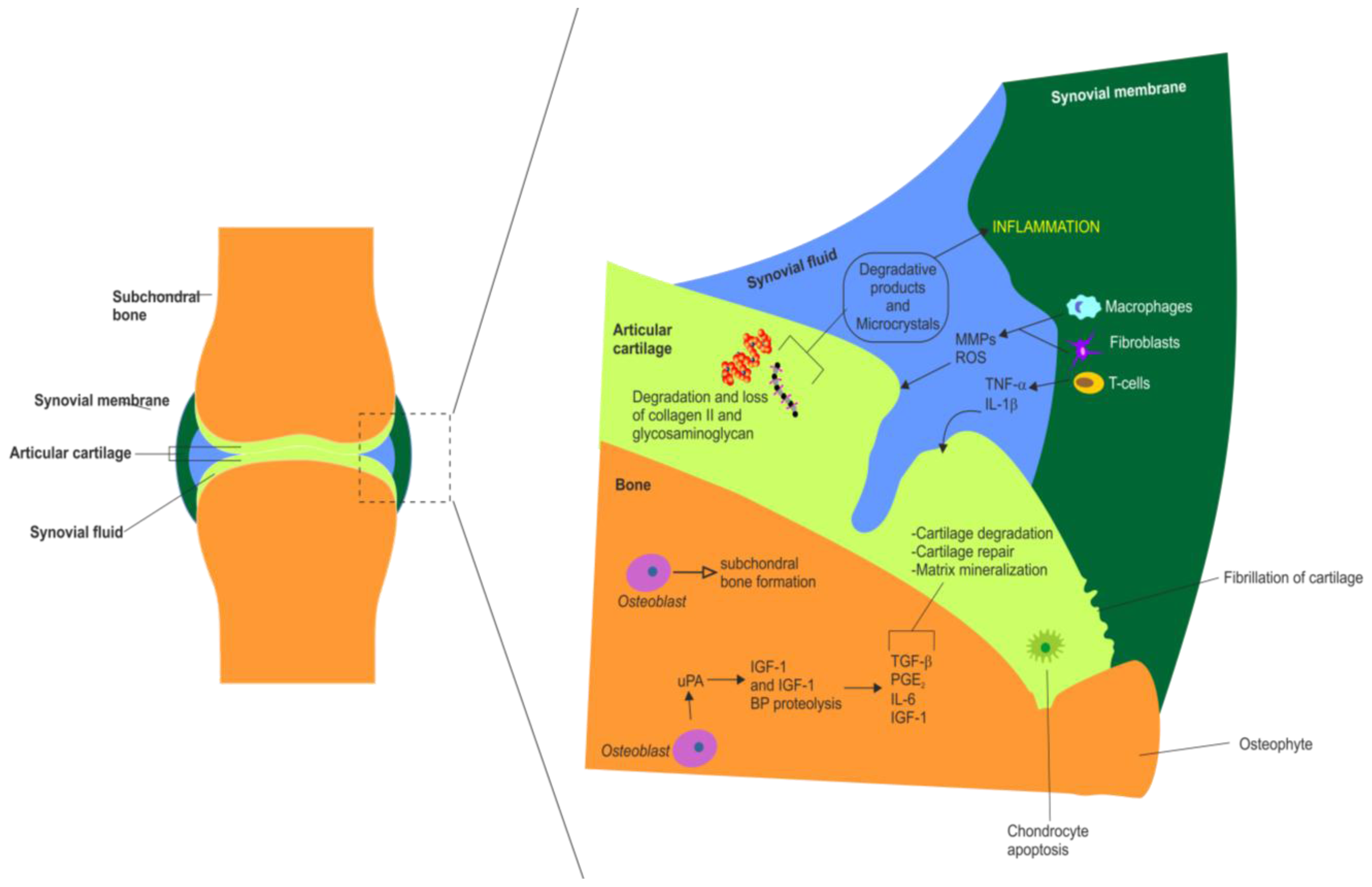
3. Articular Cartilage Degradation in OA
4. Cytokines and OA
5. The Role of Cytokines in Arthritis
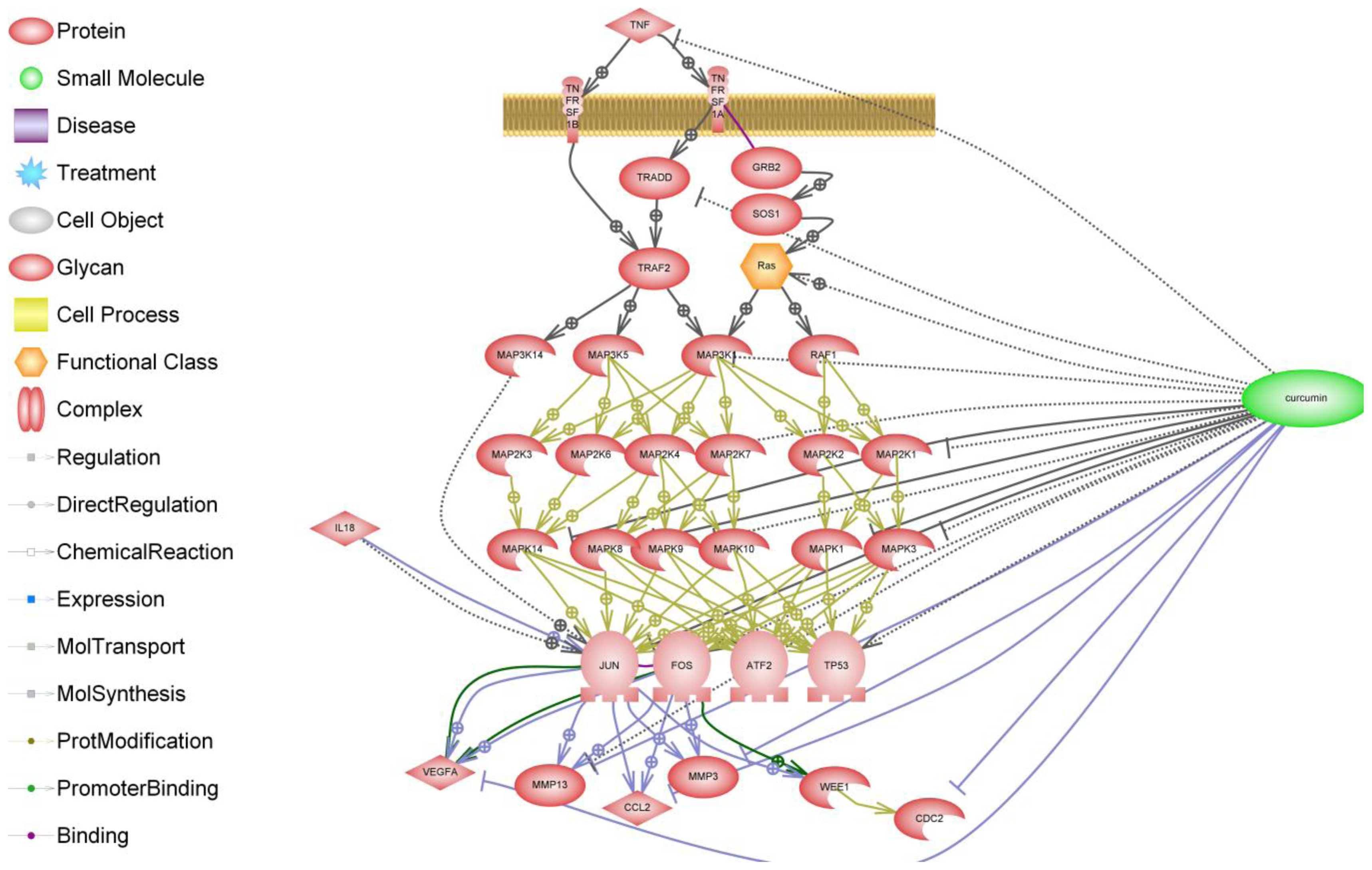
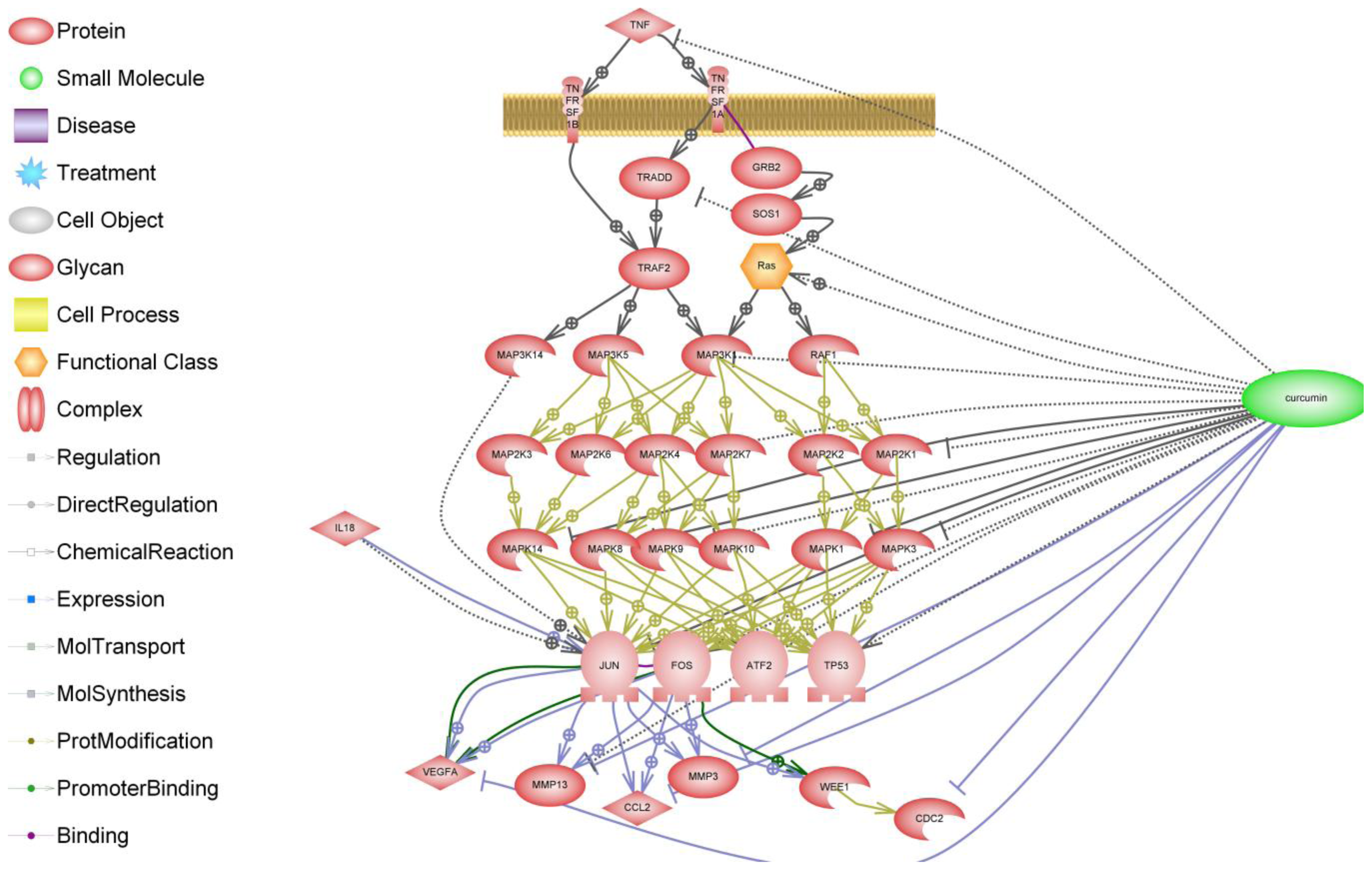
6. NF-κB Signaling in Arthritis
7. Curcumin and Resveratrol—Naturally Occurring NF-κB Inhibitors
8. Curcumin
9. Clinical Trials of Curcumin
10. Bioavailability and Topical Delivery of Curcumin
11. Synergistic Effects of Curcumin and NSAIDs
12. Resveratrol
13. Resveratrol and Transcription factor NF-κB
14. Resveratrol and OA
15. Clinical Trials of Other Phytochemical Based Products Approved as Medical Foods
16. Concluding Remarks
- For function claims: To maintain or to improve a function
- For reduction of disease risk claims: To reduce a risk factor for the development of a human disease (not reduction of the risk of the disease)—a risk factor that may serve as a predictor of development of that disease
- Joint space width on radiographs
- Mobility
- Stiffness
- Joint discomfort (i.e., pain)
References
- Oeppen, J.; Vaupel, J.W. The Disability Study Expert Group Members. Demography. Broken limits to life expectancy. Science 2002, 296, 1029–1031. [Google Scholar]
- Lafortune, G.; Balestat, G. Trends in Severe Disability Among Elderly People: Assessing the Evidence in 12 OECD Countries and the Future Implications. Available online: http://www.oecd.org/dataoecd/13/8/38343783.pdf accessed on 20 March 2012.
- The World Health Organization Home Page. Available online: http://www.who.int/en/ accessed on 20 March 2012.
- Weigl, M.; Cieza, A.; Cantista, P.; Reinhardt, J.D.; Stucki, G. Determinants of disability in chronic musculoskeletal health conditions: A literature review. Eur. J. Phys. Rehabil. Med 2008, 44, 67–79. [Google Scholar]
- Woolf, A.D.; Pfleger, B. Burden of major musculoskeletal conditions. Bull. World Health Organ 2003, 81, 646–656. [Google Scholar]
- McGowan, J.A. Perspectives on the future of bone and joint diseases. J. Rheumatol. Suppl 2003, 67, 62–64. [Google Scholar]
- The Bone and Joint Decade. Available online: http://www.boneandjointdecade.org/ accessed on 20 March 2012.
- Brooks, P.M. Impact of osteoarthritis on individuals and society: how much disability? Social consequences and health economic implications. Curr. Opin. Rheumatol 2002, 14, 573–577. [Google Scholar]
- Brooks, P.M. The burden of musculoskeletal disease—A global perspective. Clin. Rheumatol 2006, 25, 778–781. [Google Scholar]
- Buckwalter, J.A.; Mankin, H.J. Articular cartilage: Degeneration and osteoarthritis, repair, regeneration, and transplantation. Instr. Course Lect 1998, 47, 487–504. [Google Scholar]
- Buckwalter, J.A.; Mankin, H.J.; Grodzinsky, A.J. Articular cartilage and osteoarthritis. Instr. Course Lect 2005, 54, 465–480. [Google Scholar]
- Buckwalter, J.A.; Martin, J.; Mankin, H.J. Synovial joint degeneration and the syndrome of osteoarthritis. Instr. Course Lect 2000, 49, 481–489. [Google Scholar]
- Sutton, S.; Clutterbuck, A.; Harris, P.; Gent, T.; Freeman, S.; Foster, N.; Barrett-Jolley, R.; Mobasheri, A. The contribution of the synovium, synovial derived inflammatory cytokines and neuropeptides to the pathogenesis of osteoarthritis. Vet. J 2009, 179, 10–24. [Google Scholar]
- Shane Anderson, A.; Loeser, R.F. Why is osteoarthritis an age-related disease? Best Pract. Res. Clin. Rheumatol 2010, 24, 15–26. [Google Scholar]
- Sharma, L.; Kapoor, D.; Issa, S. Epidemiology of osteoarthritis: An update. Curr. Opin. Rheumatol 2006, 18, 147–156. [Google Scholar]
- Cushnaghan, J.; Dieppe, P. Study of 500 patients with limb joint osteoarthritis. I. Analysis by age, sex, and distribution of symptomatic joint sites. Ann. Rheum. Dis 1991, 50, 8–13. [Google Scholar]
- Felson, D.T. Risk factors for osteoarthritis: Understanding joint vulnerability. Clin. Orthop. Relat. Res 2004, 427, S16–S21. [Google Scholar]
- Lees, P. Pharmacology of drugs used to treat osteoarthritis in veterinary practice. Inflammopharmacology 2003, 11, 385–399. [Google Scholar]
- Ilyin, S.E.; Belkowski, S.M.; Plata-Salaman, C.R. Biomarker discovery and validation: Technologies and integrative approaches. Trends Biotechnol 2004, 22, 411–416. [Google Scholar]
- Mobasheri, A.; Airley, R.; Foster, C.S.; Schulze-Tanzil, G.; Shakibaei, M. Post-genomic applications of tissue microarrays: Basic research, prognostic oncology, clinical genomics and drug discovery. Histol. Histopathol 2004, 19, 325–335. [Google Scholar]
- Garnero, P.; Delmas, P.D. Biomarkers in osteoarthritis. Curr. Opin. Rheumatol 2003, 15, 641–646. [Google Scholar]
- The Cooperative Human Tissue Network (CHTN). Available online: http://www.chtn.nci.nih.gov/ accessed on 20 March 2012.
- The National Cancer Institute (NCI). Available online: http://www.cancer.gov/ accessed on 20 March 2012.
- Buckwalter, J.A.; Mankin, H.J. Articular cartilage: Tissue design and chondrocyte-matrix interactions. Instr. Course Lect 1998, 47, 477–486. [Google Scholar]
- Muir, H. The chondrocyte, architect of cartilage. Biomechanics, structure, function and molecular biology of cartilage matrix macromolecules. Bioessays 1995, 17, 1039–1048. [Google Scholar]
- Eyre, D.R. Collagens and cartilage matrix homeostasis. Clin. Orthop. Relat. Res 2004, S118–122. [Google Scholar]
- Kuettner, K.E.; Aydelotte, M.B.; Thonar, E.J. Articular cartilage matrix and structure: A minireview. J. Rheumatol. Suppl 1991, 27, 46–48. [Google Scholar]
- Guilak, F.; Alexopoulos, L.G.; Upton, M.L.; Youn, I.; Choi, J.B.; Cao, L.; Setton, L.A.; Haider, M.A. The pericellular matrix as a transducer of biomechanical and biochemical signals in articular cartilage. Ann. N. Y. Acad. Sci 2006, 1068, 498–512. [Google Scholar]
- Roughley, P.J.; Lee, E.R. Cartilage proteoglycans: Structure and potential functions. Microsc. Res. Tech 1994, 28, 385–397. [Google Scholar]
- Dudhia, J. Aggrecan, aging and assembly in articular cartilage. Cell Mol. Life Sci 2005, 62, 2241–2256. [Google Scholar]
- Kiani, C.; Chen, L.; Wu, Y.J.; Yee, A.J.; Yang, B.B. Structure and function of aggrecan. Cell Res 2002, 12, 19–32. [Google Scholar]
- Luo, W.; Guo, C.; Zheng, J.; Chen, T.L.; Wang, P.Y.; Vertel, B.M.; Tanzer, M.L. Aggrecan from start to finish. J. Bone Miner. Metab 2000, 18, 51–56. [Google Scholar]
- Kosher, R.A.; Lash, J.W.; Minor, R.R. Environmental enhancement of in vitro chondrogenesis. IV. Stimulation of somite chondrogenesis by exogenous chondromucoprotein. Dev. Biol 1973, 35, 210–220. [Google Scholar]
- Kosher, R.A.; Church, R.L. Stimulation of in vitro somite chondrogenesis by procollagen and collagen. Nature 1975, 258, 327–330. [Google Scholar]
- Von der Mark, K.; Gauss, V.; von der Mark, H.; Muller, P. Relationship between cell shape and type of collagen synthesised as chondrocytes lose their cartilage phenotype in culture. Nature 1977, 267, 531–532. [Google Scholar]
- Hewitt, A.T.; Varner, H.H.; Silver, M.H.; Martin, G.R. The role of chondronectin and cartilage proteoglycan in the attachment of chondrocytes to collagen. Prog. Clin. Biol. Res 1982, 110, 25–33. [Google Scholar]
- Sommarin, Y.; Larsson, T.; Heinegard, D. Chondrocyte-matrix interactions. Attachment to proteins isolated from cartilage. Exp. Cell Res 1989, 184, 181–192. [Google Scholar]
- Ramachandrula, A.; Tiku, K.; Tiku, M.L. Tripeptide RGD-dependent adhesion of articular chondrocytes to synovial fibroblasts. J. Cell Sci 1992, 101, 859–871. [Google Scholar]
- Ruoslahti, E.; Reed, J.C. Anchorage dependence, integrins, and apoptosis. Cell 1994, 77, 477–478. [Google Scholar]
- Enomoto-Iwamoto, M.; Iwamoto, M.; Nakashima, K.; Mukudai, Y.; Boettiger, D.; Pacifici, M.; Kurisu, K.; Suzuki, F. Involvement of α5β1 integrin in matrix interactions and proliferation of chondrocytes. J. Bone Miner. Res 1997, 12, 1124–1132. [Google Scholar]
- Gonzalez, F.A.; Seth, A.; Raden, D.L.; Bowman, D.S.; Fay, F.S.; Davis, R.J. Serum-induced translocation of mitogen-activated protein kinase to the cell surface ruffling membrane and the nucleus. J. Cell Biol 1993, 122, 1089–1101. [Google Scholar]
- Jenniskens, Y.M.; Koevoet, W.; de Bart, A.C.; Weinans, H.; Jahr, H.; Verhaar, J.A.; DeGroot, J.; van Osch, G.J. Biochemical and functional modulation of the cartilage collagen network by IGF1, TGFβ2 and FGF2. Osteoarthr. Cartil 2006, 14, 1136–1146. [Google Scholar]
- Trippel, S.B.; Corvol, M.T.; Dumontier, M.F.; Rappaport, R.; Hung, H.H.; Mankin, H.J. Effect of somatomedin-C/insulin-like growth factor I and growth hormone on cultured growth plate and articular chondrocytes. Pediatr. Res 1989, 25, 76–82. [Google Scholar]
- Isgaard, J. Expression and regulation of IGF-I in cartilage and skeletal muscle. Growth Regul 1992, 2, 16–22. [Google Scholar]
- Hunziker, E.B.; Wagner, J.; Zapf, J. Differential effects of insulin-like growth factor I and growth hormone on developmental stages of rat growth plate chondrocytes in vivo. J. Clin. Investig 1994, 93, 1078–1086. [Google Scholar]
- Sah, R.L.; Chen, A.C.; Grodzinsky, A.J.; Trippel, S.B. Differential effects of bFGF and IGF-I on matrix metabolism in calf and adult bovine cartilage explants. Arch. Biochem. Biophys 1994, 308, 137–147. [Google Scholar]
- Loeser, R.F. Growth factor regulation of chondrocyte integrins. Differential effects of insulin-like growth factor 1 and transforming growth factor β on α 1 β 1 integrin expression and chondrocyte adhesion to type VI collagen. Arthritis Rheum 1997, 40, 270–276. [Google Scholar]
- Di Cesare, P.E.; Carlson, C.S.; Stolerman, E.S.; Hauser, N.; Tulli, H.; Paulsson, M. Increased degradation and altered tissue distribution of cartilage oligomeric matrix protein in human rheumatoid and osteoarthritic cartilage. J. Orthop. Res 1996, 14, 946–955. [Google Scholar]
- Burton-Wurster, N.; Lust, G.; Macleod, J.N. Cartilage fibronectin isoforms: In search of functions for a special population of matrix glycoproteins. Matrix Biol 1997, 15, 441–454. [Google Scholar]
- Mobasheri, A.; Carter, S.D.; Martin-Vasallo, P.; Shakibaei, M. Integrins and stretch activated ion channels; putative components of functional cell surface mechanoreceptors in articular chondrocytes. Cell Biol. Int 2002, 26, 1–18. [Google Scholar]
- Millward-Sadler, S.J.; Salter, D.M. Integrin-dependent signal cascades in chondrocyte mechanotransduction. Ann. Biomed. Eng 2004, 32, 435–446. [Google Scholar]
- Buckwalter, J.A.; Lane, N.E. Athletics and osteoarthritis. Am. J. Sports Med 1997, 25, 873–881. [Google Scholar]
- Maffulli, N.; King, J.B. Effects of physical activity on some components of the skeletal system. Sports Med 1992, 13, 393–407. [Google Scholar]
- Martin, J.A.; Brown, T.; Heiner, A.; Buckwalter, J.A. Post-traumatic osteoarthritis: The role of accelerated chondrocyte senescence. Biorheology 2004, 41, 479–491. [Google Scholar]
- Buckwalter, J.A. Sports, joint injury, and posttraumatic osteoarthritis. J. Orthop. Sports Phys. Ther 2003, 33, 578–588. [Google Scholar]
- Newman, A.P. Articular cartilage repair. Am. J. Sports Med 1998, 26, 309–324. [Google Scholar]
- Solursh, M. Formation of cartilage tissue in vitro. J. Cell Biochem 1991, 45, 258–260. [Google Scholar]
- Hudelmaier, M.; Glaser, C.; Hohe, J.; Englmeier, K.H.; Reiser, M.; Putz, R.; Eckstein, F. Age-related changes in the morphology and deformational behavior of knee joint cartilage. Arthritis Rheum 2001, 44, 2556–2561. [Google Scholar]
- Eckstein, F.; Reiser, M.; Englmeier, K.H.; Putz, R. In vivo morphometry and functional analysis of human articular cartilage with quantitative magnetic resonance imaging—from image to data, from data to theory. Anat. Embryol. (Berl.) 2001, 203, 147–173. [Google Scholar]
- Ralphs, J.R.; Benjamin, M. The joint capsule: Structure, composition, ageing and disease. J. Anat 1994, 184, 503–509. [Google Scholar]
- Sarzi-Puttini, P.; Cimmino, M.A.; Scarpa, R.; Caporali, R.; Parazzini, F.; Zaninelli, A.; Atzeni, F.; Canesi, B. Osteoarthritis: An overview of the disease and its treatment strategies. Semin. Arthritis Rheum 2005, 35, 1–10. [Google Scholar]
- Poole, A.R. An introduction to the pathophysiology of osteoarthritis. Front. Biosci 1999, 4, D662–D670. [Google Scholar]
- Setton, L.A.; Elliott, D.M.; Mow, V.C. Altered mechanics of cartilage with osteoarthritis: Human osteoarthritis and an experimental model of joint degeneration. Osteoarthr.Cartil 1999, 7, 2–14. [Google Scholar]
- Shakibaei, M.; John, T.; de Souza, P.; Rahmanzadeh, R.; Merker, H.J. Signal transduction by β1 integrin receptors in human chondrocytes in vitro: Collaboration with the insulin-like growth factor-I receptor. Biochem. J 1999, 342, 615–623. [Google Scholar]
- Aigner, T.; Rose, J.; Martin, J.; Buckwalter, J. Aging theories of primary osteoarthritis: From epidemiology to molecular biology. Rejuvenation Res 2004, 7, 134–145. [Google Scholar]
- Abramson, S.B.; Attur, M. Developments in the scientific understanding of osteoarthritis. Arthritis Res. Ther 2009, 11. [Google Scholar] [CrossRef]
- Goldring, M.B.; Goldring, S.R. Osteoarthritis. J. Cell Physiol 2007, 213, 626–634. [Google Scholar]
- The National Institute of Arthritis and Musculoskeletal and Skin Diseases Home Page. Available online: http://www.niams.nih.gov/ accessed on 20 March 2012.
- Freedman, V.A.; Crimmins, E.; Schoeni, R.F.; Spillman, B.C.; Aykan, H.; Kramarow, E.; Land, K.; Lubitz, J.; Manton, K.; Martin, L.G.; et al. Resolving inconsistencies in trends in old-age disability: Report from a technical working group. Demography 2004, 41, 417–441. [Google Scholar]
- Australian Bureau of Statistics. Disability, Ageing and Carers, Australia: Summary of Findings. 2003. Available online: http://www.abs.gov.au/AUSSTATS/abs@.nsf/Lookup/4430.0Main+Features12003 accessed on 20 March 2012.
- Sellam, J.; Berenbaum, F. The role of synovitis in pathophysiology and clinical symptoms of osteoarthritis. Nat. Rev. Rheumatol 2010, 6, 625–635. [Google Scholar]
- Liles, W.C.; van Voorhis, W.C. Review: Nomenclature and biologic significance of cytokines involved in inflammation and the host immune response. J. Infect. Dis 1995, 172, 1573–1580. [Google Scholar]
- Feldmann, M.; Brennan, F.M.; Maini, R.N. Role of cytokines in rheumatoid arthritis. Annu. Rev. Immunol 1996, 14, 397–440. [Google Scholar]
- Maini, R.N.; Taylor, P.C. Anti-cytokine therapy for rheumatoid arthritis. Annu. Rev. Med 2000, 51, 207–229. [Google Scholar]
- Loeser, R.F. Aging and osteoarthritis. Curr. Opin. Rheumatol 2011, 23, 492–496. [Google Scholar]
- Bondeson, J.; Wainwright, S.D.; Lauder, S.; Amos, N.; Hughes, C.E. The role of synovial macrophages and macrophage-produced cytokines in driving aggrecanases, matrix metalloproteinases, and other destructive and inflammatory responses in osteoarthritis. Arthritis Res. Ther 2006, 8. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Alaaeddine, N.; Pelletier, J.P. Cytokines and their role in the pathophysiology of osteoarthritis. Front. Biosci 1999, 4, D694–D703. [Google Scholar]
- Sadouk, M.B.; Pelletier, J.P.; Tardif, G.; Kiansa, K.; Cloutier, J.M.; Martel-Pelletier, J. Human synovial fibroblasts coexpress IL-1 receptor type I and type II mRNA. The increased level of the IL-1 receptor in osteoarthritic cells is related to an increased level of the type I receptor. Lab. Investig 1995, 73, 347–355. [Google Scholar]
- Alaaeddine, N.; DiBattista, J.A.; Pelletier, J.P.; Cloutier, J.M.; Kiansa, K.; Dupuis, M.; Martel-Pelletier, J. Osteoarthritic synovial fibroblasts possess an increased level of tumor necrosis factor-receptor 55 (TNF-R55) that mediates biological activation by TNF-α. J. Rheumatol 1997, 24, 1985–1994. [Google Scholar]
- Bertolini, D.R.; Nedwin, G.E.; Bringman, T.S.; Smith, D.D.; Mundy, G.R. Stimulation of bone resorption and inhibition of bone formation in vitro by human tumour necrosis factors. Nature 1986, 319, 516–518. [Google Scholar]
- Kumar, S.; Votta, B.J.; Rieman, D.J.; Badger, A.M.; Gowen, M.; Lee, J.C. IL-1- and TNF-induced bone resorption is mediated by p38 mitogen activated protein kinase. J. Cell Physiol 2001, 187, 294–303. [Google Scholar]
- Seguin, C.A.; Bernier, S.M. TNFα suppresses link protein and type II collagen expression in chondrocytes: Role of MEK1/2 and NF-κB signaling pathways. J. Cell Physiol 2003, 197, 356–369. [Google Scholar]
- Sakai, T.; Kambe, F.; Mitsuyama, H.; Ishiguro, N.; Kurokouchi, K.; Takigawa, M.; Iwata, H.; Seo, H. Tumor necrosis factor α induces expression of genes for matrix degradation in human chondrocyte-like HCS-2/8 cells through activation of NF-κB: Abrogation of the tumor necrosis factor α effect by proteasome inhibitors. J. Bone Miner. Res 2001, 16, 1272–1280. [Google Scholar]
- Beutler, B.A. The role of tumor necrosis factor in health and disease. J. Rheumatol. Suppl 1999, 57, 16–21. [Google Scholar]
- Honorati, M.C.; Cattini, L.; Facchini, A. IL-17, IL-1β and TNF-α stimulate VEGF production by dedifferentiated chondrocytes. Osteoarthr. Cartil 2004, 12, 683–691. [Google Scholar]
- Seitz, M.; Loetscher, P.; Dewald, B.; Towbin, H.; Ceska, M.; Baggiolini, M. Production of interleukin-1 receptor antagonist, inflammatory chemotactic proteins, and prostaglandin E by rheumatoid and osteoarthritic synoviocytes—Regulation by IFN-γ and IL-4. J. Immunol 1994, 152, 2060–2065. [Google Scholar]
- Lisignoli, G.; Toneguzzi, S.; Pozzi, C.; Piacentini, A.; Riccio, M.; Ferruzzi, A.; Gualtieri, G.; Facchini, A. Proinflammatory cytokines and chemokine production and expression by human osteoblasts isolated from patients with rheumatoid arthritis and osteoarthritis. J. Rheumatol 1999, 26, 791–799. [Google Scholar]
- Goodstone, N.J.; Hardingham, T.E. Tumour necrosis factor α stimulates nitric oxide production more potently than interleukin-1β in porcine articular chondrocytes. Rheumatol. (Oxf.) 2002, 41, 883–891. [Google Scholar]
- Schuerwegh, A.J.; Dombrecht, E.J.; Stevens, W.J.; van Offel, J.F.; Bridts, C.H.; de Clerck, L.S. Influence of pro-inflammatory (IL-1 α, IL-6, TNF-α, IFN-γ) and anti-inflammatory (IL-4) cytokines on chondrocyte function. Osteoarthr. Cartil 2003, 11, 681–687. [Google Scholar]
- Arend, W.P.; Dayer, J.M. Cytokines and cytokine inhibitors or antagonists in rheumatoid arthritis. Arthritis Rheum 1990, 33, 305–315. [Google Scholar]
- Van Beuningen, H.M.; Arntz, O.J.; van den Berg, W.B. In vivo effects of interleukin-1 on articular cartilage. Prolongation of proteoglycan metabolic disturbances in old mice. Arthritis Rheum 1991, 34, 606–615. [Google Scholar]
- Takafuji, V.A.; McIlwraith, C.W.; Howard, R.D. Effects of equine recombinant interleukin-1α and interleukin-1β on proteoglycan metabolism and prostaglandin E2 synthesis in equine articular cartilage explants. Am. J. Vet. Res 2002, 63, 551–558. [Google Scholar]
- Tung, J.T.; Fenton, J.I.; Arnold, C.; Alexander, L.; Yuzbasiyan-Gurkan, V.; Venta, P.J.; Peters, T.L.; Orth, M.W.; Richardson, D.W.; Caron, J.P. Recombinant equine interleukin-1β induces putative mediators of articular cartilage degradation in equine chondrocytes. Can. J. Vet. Res 2002, 66, 19–25. [Google Scholar]
- Spiers, S.; May, S.A.; Bennett, D.; Edwards, G.B. Cellular sources of proteolytic enzymes in equine joints. Equine Vet. J 1994, 26, 43–47. [Google Scholar]
- Palmer, R.M.; Hickery, M.S.; Charles, I.G.; Moncada, S.; Bayliss, M.T. Induction of nitric oxide synthase in human chondrocytes. Biochem. Biophys.Res. Commun 1993, 193, 398–405. [Google Scholar]
- Jikko, A.; Wakisaka, T.; Iwamoto, M.; Hiranuma, H.; Kato, Y.; Maeda, T.; Fujishita, M.; Fuchihata, H. Effects of interleukin-6 on proliferation and proteoglycan metabolism in articular chondrocyte cultures. Cell Biol. Int 1998, 22, 615–621. [Google Scholar]
- Damiens, C.; Fortun, Y.; Charrier, C.; Heymann, D.; Padrines, M. Modulation by soluble factors of gelatinase activities released by osteoblastic cells. Cytokine 2000, 12, 1727–1731. [Google Scholar]
- Flannery, C.R.; Little, C.B.; Hughes, C.E.; Curtis, C.L.; Caterson, B.; Jones, S.A. IL-6 and its soluble receptor augment aggrecanase-mediated proteoglycan catabolism in articular cartilage. Matrix Biol 2000, 19, 549–553. [Google Scholar]
- Endo, H.; Akahoshi, T.; Nishimura, A.; Tonegawa, M.; Takagishi, K.; Kashiwazaki, S.; Matsushima, K.; Kondo, H. Experimental arthritis induced by continuous infusion of IL-8 into rabbit knee joints. Clin. Exp. Immunol 1994, 96, 31–35. [Google Scholar]
- Leonard, E.J.; Yoshimura, T. Neutrophil attractant/activation protein-1 (NAP-1 [interleukin-8]). Am. J. Respir. Cell Mol. Biol 1990, 2, 479–486. [Google Scholar]
- Yu, C.L.; Sun, K.H.; Shei, S.C.; Tsai, C.Y.; Tsai, S.T.; Wang, J.C.; Liao, T.S.; Lin, W.M.; Chen, H.L.; Yu, H.S.; et al. Interleukin 8 modulates interleukin-1 β, interleukin-6 and tumor necrosis factor-α release from normal human mononuclear cells. Immunopharmacology 1994, 27, 207–214. [Google Scholar]
- Merz, D.; Liu, R.; Johnson, K.; Terkeltaub, R. IL-8/CXCL8 and growth-related oncogene α/CXCL1 induce chondrocyte hypertrophic differentiation. J. Immunol 2003, 171, 4406–4415. [Google Scholar]
- Shalom-Barak, T.; Quach, J.; Lotz, M. Interleukin-17-induced gene expression in articular chondrocytes is associated with activation of mitogen-activated protein kinases and NF-κB. J. Biol. Chem 1998, 273, 27467–27473. [Google Scholar]
- Fahmi, H.; di Battista, J.A.; Pelletier, J.P.; Mineau, F.; Ranger, P.; Martel-Pelletier, J. Peroxisome proliferator—Activated receptor γ activators inhibit interleukin-1β-induced nitric oxide and matrix metalloproteinase 13 production in human chondrocytes. Arthritis Rheum 2001, 44, 595–607. [Google Scholar]
- Yao, Z.; Painter, S.L.; Fanslow, W.C.; Ulrich, D.; Macduff, B.M.; Spriggs, M.K.; Armitage, R.J. Human IL-17: A novel cytokine derived from T cells. J. Immunol 1995, 155, 5483–5486. [Google Scholar]
- Honorati, M.C.; Neri, S.; Cattini, L.; Facchini, A. Interleukin-17, a regulator of angiogenic factor release by synovial fibroblasts. Osteoarthr. Cartil 2006, 14, 345–352. [Google Scholar]
- Puren, A.J.; Fantuzzi, G.; Gu, Y.; Su, M.S.; Dinarello, C.A. Interleukin-18 (IFNγ-inducing factor) induces IL-8 and IL-1β via TNFα production from non-CD14+ human blood mononuclear cells. J. Clin. Investig 1998, 101, 711–721. [Google Scholar]
- Gracie, J.A.; Forsey, R.J.; Chan, W.L.; Gilmour, A.; Leung, B.P.; Greer, M.R.; Kennedy, K.; Carter, R.; Wei, X.Q.; Xu, D.; et al. A proinflammatory role for IL-18 in rheumatoid arthritis. J. Clin. Investig 1999, 104, 1393–1401. [Google Scholar]
- Cho, M.L.; Jung, Y.O.; Moon, Y.M.; Min, S.Y.; Yoon, C.H.; Lee, S.H.; Park, S.H.; Cho, C.S.; Jue, D.M.; Kim, H.Y. Interleukin-18 induces the production of vascular endothelial growth factor (VEGF) in rheumatoid arthritis synovial fibroblasts via AP-1-dependent pathways. Immunol. Lett 2006, 103, 159–166. [Google Scholar]
- Leung, B.P.; McInnes, I.B.; Esfandiari, E.; Wei, X.Q.; Liew, F.Y. Combined effects of IL-12 and IL-18 on the induction of collagen-induced arthritis. J. Immunol 2000, 164, 6495–6502. [Google Scholar]
- John, T.; Kohl, B.; Mobasheri, A.; Ertel, W.; Shakibaei, M. Interleukin-18 induces apoptosis in human articular chondrocytes. Histol. Histopathol 2007, 22, 469–482. [Google Scholar]
- Villiger, P.M.; Geng, Y.; Lotz, M. Induction of cytokine expression by leukemia inhibitory factor. J. Clin. Investig 1993, 91, 1575–1581. [Google Scholar]
- Henrotin, Y.E.; de Groote, D.D.; Labasse, A.H.; Gaspar, S.E.; Zheng, S.X.; Geenen, V.G.; Reginster, J.Y. Effects of exogenous IL-1 β, TNF α, IL-6, IL-8 and LIF on cytokine production by human articular chondrocytes. Osteoarthr. Cartil 1996, 4, 163–173. [Google Scholar]
- Varghese, S.; Yu, K.; Canalis, E. Leukemia inhibitory factor and oncostatin M stimulate collagenase-3 expression in osteoblasts. Am. J. Physiol 1999, 276, E465–E471. [Google Scholar]
- Carroll, G.J.; Bell, M.C. Leukaemia inhibitory factor stimulates proteoglycan resorption in porcine articular cartilage. Rheumatol. Intern 1993, 13, 5–8. [Google Scholar]
- Bell, M.C.; Carroll, G.J. Leukaemia inhibitory factor (LIF) suppresses proteoglycan synthesis in porcine and caprine cartilage explants. Cytokine 1995, 7, 137–141. [Google Scholar]
- Carroll, G.J.; Bell, M.C.; Chapman, H.M.; Mills, J.N.; Robinson, W.F. Leukemia inhibitory factor induces leukocyte infiltration and cartilage proteoglycan degradation in goat joints. J. Interferon Cytokine Res 1995, 15, 567–573. [Google Scholar]
- Aggarwal, B.B.; Shishodia, S. Suppression of the nuclear factor-κB activation pathway by spice-derived phytochemicals: Reasoning for seasoning. Ann. N. Y. Acad. Sci 2004, 1030, 434–441. [Google Scholar]
- Aggarwal, B.B.; Sundaram, C.; Malani, N.; Ichikawa, H. Curcumin: The Indian solid gold. Adv. Exp. Med. Biol 2007, 595, 1–75. [Google Scholar]
- Bremner, P.; Heinrich, M. Natural products as targeted modulators of the nuclear factor-κB pathway. J. Pharm. Pharmacol 2002, 54, 453–472. [Google Scholar]
- Marcu, K.B.; Otero, M.; Olivotto, E.; Borzi, R.M.; Goldring, M.B. NF-κB signaling: Multiple angles to target OA. Curr. Drug. Targets 2010, 11, 599–613. [Google Scholar]
- Hak, A.E.; Choi, H.K. Lifestyle and gout. Curr. Opin. Rheumatol 2008, 20, 179–186. [Google Scholar]
- Sale, J.E.; Gignac, M.; Hawker, G. The relationship between disease symptoms, life events, coping and treatment, and depression among older adults with osteoarthritis. J. Rheumatol 2008, 35, 335–342. [Google Scholar]
- Corson, T.W.; Crews, C.M. Molecular understanding and modern application of traditional medicines: Triumphs and trials. Cell 2007, 130, 769–774. [Google Scholar]
- Curtis, C.L.; Rees, S.G.; Little, C.B.; Flannery, C.R.; Hughes, C.E.; Wilson, C.; Dent, C.M.; Otterness, I.G.; Harwood, J.L.; Caterson, B. Pathologic indicators of degradation and inflammation in human osteoarthritic cartilage are abrogated by exposure to n-3 fatty acids. Arthritis Rheum 2002, 46, 1544–1553. [Google Scholar]
- Henrotin, Y.; Clutterbuck, A.L.; Allaway, D.; Lodwig, E.M.; Harris, P.; Mathy-Hartert, M.; Shakibaei, M.; Mobasheri, A. Biological actions of curcumin on articular chondrocytes. Osteoarthr.Cartil 2010, 18, 141–149. [Google Scholar]
- Lee, B.; Moon, S.K. Resveratrol inhibits TNF-α-induced proliferation and matrix metalloproteinase expression in human vascular smooth muscle cells. J. Nutr 2005, 135, 2767–2773. [Google Scholar]
- Annabi, B.; Currie, J.C.; Moghrabi, A.; Beliveau, R. Inhibition of HuR and MMP-9 expression in macrophage-differentiated HL-60 myeloid leukemia cells by green tea polyphenol EGCg. Leuk. Res 2007, 31, 1277–1284. [Google Scholar]
- Kim, M.; Murakami, A.; Ohigashi, H. Modifying effects of dietary factors on (−)-epigallocatechin-3-gallate-induced pro-matrix metalloproteinase-7 production in HT-29 human colorectal cancer cells. Biosci. Biotechnol. Biochem 2007, 71, 2442–2450. [Google Scholar]
- Kong, C.S.; Kim, Y.A.; Kim, M.M.; Park, J.S.; Kim, J.A.; Kim, S.K.; Lee, B.J.; Nam, T.J.; Seo, Y. Flavonoid glycosides isolated from Salicornia herbacea inhibit matrix metalloproteinase in HT1080 cells. Toxicol. In Vitro 2008, 22, 1742–1748. [Google Scholar]
- Vijayababu, M.R.; Arunkumar, A.; Kanagaraj, P.; Venkataraman, P.; Krishnamoorthy, G.; Arunakaran, J. Quercetin downregulates matrix metalloproteinases 2 and 9 proteins expression in prostate cancer cells (PC-3). Mol. Cell. Biochem 2006, 287, 109–116. [Google Scholar]
- Shishodia, S.; Singh, T.; Chaturvedi, M.M. Modulation of transcription factors by curcumin. Adv. Exp. Med. Biol 2007, 595, 127–148. [Google Scholar]
- Aggarwal, B.B.; Kumar, A.; Bharti, A.C. Anticancer potential of curcumin: Preclinical and clinical studies. Anticancer Res 2003, 23, 363–398. [Google Scholar]
- Largo, R.; Alvarez-Soria, M.A.; Diez-Ortego, I.; Calvo, E.; Sanchez-Pernaute, O.; Egido, J.; Herrero-Beaumont, G. Glucosamine inhibits IL-1β-induced NFκB activation in human osteoarthritic chondrocytes. Osteoarthr.Cartil 2003, 11, 290–298. [Google Scholar]
- Liacini, A.; Sylvester, J.; Li, W.Q.; Huang, W.; Dehnade, F.; Ahmad, M.; Zafarullah, M. Induction of matrix metalloproteinase-13 gene expression by TNF-α is mediated by MAP kinases, AP-1, and NF-κB transcription factors in articular chondrocytes. Exp. Cell Res 2003, 288, 208–217. [Google Scholar]
- Singh, S. From exotic spice to modern drug? Cell 2007, 130, 765–768. [Google Scholar]
- Csaki, C.; Keshishzadeh, N.; Fischer, K.; Shakibaei, M. Regulation of inflammation signalling by resveratrol in human chondrocytes in vitro. Biochem. Pharmacol 2008, 75, 677–687. [Google Scholar]
- Csaki, C.; Mobasheri, A.; Shakibaei, M. Synergistic chondroprotective effects of curcumin and resveratrol in human articular chondrocytes: Inhibition of IL-1β-induced NF-κB-mediated inflammation and apoptosis. Arthritis Res. Ther 2009, 11. [Google Scholar] [CrossRef]
- Deodhar, S.D.; Sethi, R.; Srimal, R.C. Preliminary study on antirheumatic activity of curcumin (diferuloyl methane). Indian J. Med. Res 1980, 71, 632–634. [Google Scholar]
- Joe, B.; Rao, U.J.; Lokesh, B.R. Presence of an acidic glycoprotein in the serum of arthritic rats: Modulation by capsaicin and curcumin. Mol. Cell. Biochem 1997, 169, 125–134. [Google Scholar]
- Onodera, S.; Kaneda, K.; Mizue, Y.; Koyama, Y.; Fujinaga, M.; Nishihira, J. Macrophage migration inhibitory factor up-regulates expression of matrix metalloproteinases in synovial fibroblasts of rheumatoid arthritis. J. Biol. Chem 2000, 275, 444–450. [Google Scholar]
- Jackson, J.K.; Higo, T.; Hunter, W.L.; Burt, H.M. The antioxidants curcumin and quercetin inhibit inflammatory processes associated with arthritis. Inflamm. Res 2006, 55, 168–175. [Google Scholar]
- Molnar, V.; Garai, J. Plant-derived anti-inflammatory compounds affect MIF tautomerase activity. Int. Immunopharmacol 2005, 5, 849–856. [Google Scholar]
- Mathy-Hartert, M.; Jacquemond-Collet, I.; Priem, F.; Sanchez, C.; Lambert, C.; Henrotin, Y. Curcumin inhibits pro-inflammatory mediators and metalloproteinase-3 production by chondrocytes. Inflamm. Res 2009, 58, 899–908. [Google Scholar]
- Park, C.; Moon, D.O.; Choi, I.W.; Choi, B.T.; Nam, T.J.; Rhu, C.H.; Kwon, T.K.; Lee, W.H.; Kim, G.Y.; Choi, Y.H. Curcumin induces apoptosis and inhibits prostaglandin E(2) production in synovial fibroblasts of patients with rheumatoid arthritis. Int. J. Mol. Med 2007, 20, 365–372. [Google Scholar]
- Neff, L.; Zeisel, M.; Sibilia, J.; Scholler-Guinard, M.; Klein, J.P.; Wachsmann, D. NF-κB and the MAP kinases/AP-1 pathways are both involved in interleukin-6 and interleukin-8 expression in fibroblast-like synoviocytes stimulated by protein I/II, a modulin from oral streptococci. Cell Microbiol 2001, 3, 703–712. [Google Scholar]
- Liacini, A.; Sylvester, J.; Li, W.Q.; Zafarullah, M. Inhibition of interleukin-1-stimulated MAP kinases, activating protein-1 (AP-1) and nuclear factor κ B (NF-κ B) transcription factors down-regulates matrix metalloproteinase gene expression in articular chondrocytes. Matrix Biol 2002, 21, 251–262. [Google Scholar]
- Shakibaei, M.; Schulze-Tanzil, G.; John, T.; Mobasheri, A. Curcumin protects human chondrocytes from IL-l1β-induced inhibition of collagen type II and β1-integrin expression and activation of caspase-3: An immunomorphological study. Ann. Anat 2005, 187, 487–497. [Google Scholar]
- Cho, M.L.; Jung, Y.O.; Moon, Y.M.; Min, S.Y.; Yoon, C.H.; Lee, S.H.; Park, S.H.; Cho, C.S.; Jue, D.M.; Kim, H.Y. Interleukin-18 induces the production of vascular endothelial growth factor (VEGF) in rheumatoid arthritis synovial fibroblasts via AP-1-dependent pathways. Immunol. Lett 2006, 103, 159–166. [Google Scholar]
- Funk, J.L.; Oyarzo, J.N.; Frye, J.B.; Chen, G.; Lantz, R.C.; Jolad, S.D.; Solyom, A.M.; Timmermann, B.N. Turmeric extracts containing curcuminoids prevent experimental rheumatoid arthritis. J. Nat. Prod 2006, 69, 351–355. [Google Scholar]
- Tohda, C.; Nakayama, N.; Hatanaka, F.; Komatsu, K. Comparison of anti-inflammatory activities of six curcuma rhizomes: A possible curcuminoid-independent pathway mediated by curcuma phaeocaulis extract. Evid. Based Complement. Alternat. Med 2006, 3, 255–260. [Google Scholar]
- Shakibaei, M.; John, T.; Schulze-Tanzil, G.; Lehmann, I.; Mobasheri, A. Suppression of NF-κB activation by curcumin leads to inhibition of expression of cyclo-oxygenase-2 and matrix metalloproteinase-9 in human articular chondrocytes: Implications for the treatment of osteoarthritis. Biochem. Pharmacol 2007, 73, 1434–1445. [Google Scholar]
- Anand, P.; Kunnumakkara, A.B.; Newman, R.A.; Aggarwal, B.B. Bioavailability of curcumin: Problems and promises. Mol. Pharm 2007, 4, 807–818. [Google Scholar]
- Toegel, S.; Wu, S.Q.; Piana, C.; Unger, F.M.; Wirth, M.; Goldring, M.B.; Gabor, F.; Viernstein, H. Comparison between chondroprotective effects of glucosamine, curcumin, and diacerein in IL-1β-stimulated C-28/I2 chondrocytes. Osteoarthr.Cartil 2008, 16, 1205–1212. [Google Scholar]
- Bright, J.J. Curcumin and autoimmune disease. Adv. Exp. Med. Biol 2007, 595, 425–451. [Google Scholar]
- ClinicalTrials.gov Home Page. A service of the National Institutes of Health (NIH). Available online: http://clinicaltrials.gov/ accessed on 20 March 2012.
- University of California. Curcumin in Rheumatoid Arthritis. Available online: http://clinicaltrials.gov/ct2/show/NCT00752154 accessed on 20 March 2012.
- Gupta, N.K.; Dixit, V.K. Bioavailability enhancement of curcumin by complexation with phosphatidyl choline. J. Pharm. Sci 2011, 100, 1987–1995. [Google Scholar]
- Hegge, A.B.; Masson, M.; Kristensen, S.; Tonnesen, H.H. Investigation of curcumin-cyclodextrin inclusion complexation in aqueous solutions containing various alcoholic co-solvents and alginates using an UV-VIS titration method. Pharmaze 2009, 64, 382–389. [Google Scholar]
- Hegge, A.B.; Schuller, R.B.; Kristensen, S.; Tonnesen, H.H. In vitro release of curcumin from vehicles containing alginate and cyclodextrin. Pharmaze 2008, 63, 585–592. [Google Scholar]
- Banerjee, M.; Tripathi, L.M.; Srivastava, V.M.; Puri, A.; Shukla, R. Modulation of inflammatory mediators by ibuprofen and curcumin treatment during chronic inflammation in rat. Immunopharmacol. Immunotoxicol 2003, 25, 213–224. [Google Scholar]
- Lev-Ari, S.; Strier, L.; Kazanov, D.; Elkayam, O.; Lichtenberg, D.; Caspi, D.; Arber, N. Curcumin synergistically potentiates the growth-inhibitory and pro-apoptotic effects of celecoxib in osteoarthritis synovial adherent cells. Rheumatol. (Oxf.) 2006, 45, 171–177. [Google Scholar]
- Lev-Ari, S.; Lichtenberg, D.; Arber, N. Compositions for treatment of cancer and inflammation. Recent Pat. Anticancer Drug Discov 2008, 3, 55–62. [Google Scholar]
- Jurenka, J.S. Anti-inflammatory properties of curcumin, a major constituent of Curcuma longa: A review of preclinical and clinical research. Altern. Med. Rev 2009, 14, 141–153. [Google Scholar]
- Rahman, I.; Biswas, S.K.; Kirkham, P.A. Regulation of inflammation and redox signaling by dietary polyphenols. Biochem. Pharmacol 2006, 72, 1439–1452. [Google Scholar]
- Bertelli, A.A.; Ferrara, F.; Diana, G.; Fulgenzi, A.; Corsi, M.; Ponti, W.; Ferrero, M.E.; Bertelli, A. Resveratrol, a natural stilbene in grapes and wine, enhances intraphagocytosis in human promonocytes: A co-factor in antiinflammatory and anticancer chemopreventive activity. Int. J. Tissue React 1999, 21, 93–104. [Google Scholar]
- Elliott, P.J.; Jirousek, M. Sirtuins: Novel targets for metabolic disease. Curr. Opin. Investig. Drugs 2008, 9, 371–378. [Google Scholar]
- Soleas, G.J.; Diamandis, E.P.; Goldberg, D.M. Wine as a biological fluid: History, production, and role in disease prevention. J. Clin. Lab. Anal 1997, 11, 287–313. [Google Scholar]
- Elmali, N.; Esenkaya, I.; Harma, A.; Ertem, K.; Turkoz, Y.; Mizrak, B. Effect of resveratrol in experimental osteoarthritis in rabbits. Inflamm. Res 2005, 54, 158–162. [Google Scholar]
- Penberthy, W.T. Pharmacological targeting of IDO-mediated tolerance for treating autoimmune disease. Curr. Drug. Metab 2007, 8, 245–266. [Google Scholar]
- Andlauer, W.; Kolb, J.; Siebert, K.; Furst, P. Assessment of resveratrol bioavailability in the perfused small intestine of the rat. Drugs. Exp. Clin. Res 2000, 26, 47–55. [Google Scholar]
- Aggarwal, B.B.; Bhardwaj, A.; Aggarwal, R.S.; Seeram, N.P.; Shishodia, S.; Takada, Y. Role of resveratrol in prevention and therapy of cancer: Preclinical and clinical studies. Anticancer Res 2004, 24, 2783–2840. [Google Scholar]
- Yu, C.; Shin, Y.G.; Chow, A.; Li, Y.; Kosmeder, J.W.; Lee, Y.S.; Hirschelman, W.H.; Pezzuto, J.M.; Mehta, R.G.; van Breemen, R.B. Human, rat, and mouse metabolism of resveratrol. Pharm. Res 2002, 19, 1907–1914. [Google Scholar]
- Kumar, A.; Takada, Y.; Boriek, A.M.; Aggarwal, B.B. Nuclear factor-κB: Its role in health and disease. J. Mol. Med 2004, 82, 434–448. [Google Scholar]
- Tang, X.; Liu, D.; Shishodia, S.; Ozburn, N.; Behrens, C.; Lee, J.J.; Hong, W.K.; Aggarwal, B.B.; Wistuba, II. Nuclear factor-κB (NF-κB) is frequently expressed in lung cancer and preneoplastic lesions. Cancer 2006, 107, 2637–2646. [Google Scholar]
- Sarkar, F.H.; Li, Y. NF-κB: A potential target for cancer chemoprevention and therapy. Front. Biosci 2008, 13, 2950–2959. [Google Scholar]
- Aggarwal, B.B.; Takada, Y.; Shishodia, S.; Gutierrez, A.M.; Oommen, O.V.; Ichikawa, H.; Baba, Y.; Kumar, A. Nuclear transcription factor NF-κ B: Role in biology and medicine. Indian J. Exp. Biol 2004, 42, 341–353. [Google Scholar]
- Manna, S.K.; Mukhopadhyay, A.; Aggarwal, B.B. Resveratrol suppresses TNF-induced activation of nuclear transcription factors NF-κ B, activator protein-1, and apoptosis: Potential role of reactive oxygen intermediates and lipid peroxidation. J. Immunol 2000, 164, 6509–6519. [Google Scholar]
- Pinto, M.C.; Garcia-Barrado, J.A.; Macias, P. Resveratrol is a potent inhibitor of the dioxygenase activity of lipoxygenase. J. Agric. Food Chem 1999, 4842–4846. [Google Scholar]
- Xie, W.L.; Chipman, J.G.; Robertson, D.L.; Erikson, R.L.; Simmons, D.L. Expression of a mitogen-responsive gene encoding prostaglandin synthase is regulated by mRNA splicing. Proc. Natl. Acad. Sci. USA 1991, 88, 2692–2696. [Google Scholar]
- Subbaramaiah, K.; Chung, W.J.; Michaluart, P.; Telang, N.; Tanabe, T.; Inoue, H.; Jang, M.; Pezzuto, J.M.; Dannenberg, A.J. Resveratrol inhibits cyclooxygenase-2 transcription and activity in phorbol ester-treated human mammary epithelial cells. J. Biol. Chem 1998, 273, 21875–21882. [Google Scholar]
- Elmali, N.; Baysal, O.; Harma, A.; Esenkaya, I.; Mizrak, B. Effects of resveratrol in inflammatory arthritis. Inflammation 2007, 30, 1–6. [Google Scholar]
- Shakibaei, M.; John, T.; Seifarth, C.; Mobasheri, A. Resveratrol inhibits IL-1 β-induced stimulation of caspase-3 and cleavage of PARP in human articular chondrocytes in vitro. Ann. N. Y. Acad. Sci 2007, 1095, 554–563. [Google Scholar]
- Shakibaei, M.; Csaki, C.; Nebrich, S.; Mobasheri, A. Resveratrol suppresses interleukin-1β-induced inflammatory signaling and apoptosis in human articular chondrocytes: Potential for use as a novel nutraceutical for the treatment of osteoarthritis. Biochem. Pharmacol 2008, 76, 1426–1439. [Google Scholar]
- Kelly, G.S. A review of the sirtuin system, its clinical implications, and the potential role of dietary activators like resveratrol: Part 2. Altern. Med. Rev 2010, 15, 313–328. [Google Scholar]
- Limbrel. Available online: http://www.limbrel.com/ accessed on 20 March 2012.
- Study of Flavocoxid (Limbrel) Versus Naproxen in Subjects With Moderate-Severe Osteoarthritis of the Knee. Available online: http://clinicaltrials.gov/ct2/show/NCT00928837 accessed on 20 March 2012.
- The European Food Safety Authority (EFSA). Available online: http://www.efsa.europa.eu/ accessed on 20 March 2012.
- EFSA Panel on Dietetic Products, N.A.A.N. Scientific Opinion on the substantiation of health claims related to glucosamine alone or in combination with chondroitin sulphate and maintenance of joints (ID 1561, 1562, 1563, 1564, 1565) and reduction of inflammation (ID 1869) pursuant to Article 13(1) of Regulation (EC) No 1924/2006. EFSA J 2009, 7. [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cytokine | Expression | Functions | References |
|---|---|---|---|
| TNF-α | Synoviocytes Chondrocytes | Increase cartilage degradation and bone resorption | [80,81] |
| Inhibit glycoprotein and collagen synthesis. | [82] | ||
| Upregulate MMP expression | [83] | ||
| Stimulate other cells to produce proinflammatory cytokines and growth factors | [84] | ||
| Stimulate proangiogenic factor release | [85] | ||
| Stimulate other cells to produce chemotactic cytokines | [86,87] | ||
| Stimulate Nitric Oxide (NO) production | [88] | ||
| Induce chondrocyte apoptosis | [89] | ||
| IL-1β | Synoviocytes Chondrocytes Macrophages | Increase cartilage degradation and bone resorption | [80,81,90] |
| Inhibit proteoglycan synthesis | [91,92] | ||
| Upregulate MMP expression | [93] | ||
| Production of proteolytic enzymes | [94] | ||
| Stimulate other cells to produce proinflammatory cytokines | [77] | ||
| Stimulate other cells to produce chemotactic cytokines | [86,87] | ||
| Stimulate proangiogenic factor release | [85] | ||
| Stimulate NO production | [95] | ||
| Induce chondrocyte apoptosis | [89] | ||
| IL-6 | Synoviocytes Chondrocytes osteoblasts | Inhibit proteoglycan synthesis | [96] |
| Reduce chondrocyte proliferation | [96] | ||
| Increase MMP-2 activity | [97] | ||
| Increase aggrecanase-mediated proteoglycan catabolism | [98] | ||
| IL-8 | Monocytes Synoviocytes Chondrocytes Osteoblasts | Recruits leucocytes | [99] |
| Neutrophil chemoattractant | [100] | ||
| Stimulates release of proinflammatory cytokines | [101] | ||
| Hypertrophic differentiation and calcification of chondrocytes | [102] | ||
| IL-17 | Activated T-lymphocytes | Induce NO synthesis | [103,104] |
| Induce MMP synthesis | [103,104] | ||
| Increase production of IL-1β, Il-6 and IL-8 | [103,105] | ||
| Stimulate release of proangiogenic factors | [106] | ||
| IL-18 | Macrophages Synovial fibroblasts | Stimulate release of proinflammatory cytokines | [107,108] |
| Stimulate angiogenesis | [109] | ||
| Induce NO synthesis | [108] | ||
| Synovial hyperplasia and inflammatory cell recruitment | [110] | ||
| Induce chondrocyte apoptosis | [111] | ||
| Reduce expression of cartilage matrix components | [111] | ||
| Up-regulate fibronectin- a mediator of cartilage destruction | [111] | ||
| Leukaemia Inhibitory Factor (LIF) | Synovial fibroblasts Chondrocytes | Stimulate proinflammatory cytokine expression | [112,113] |
| Increase pro-MMP-2 synthesis | [97] | ||
| Increase MMP-13 synthesis and activity | [114] | ||
| Increase cartilage resorption | [115] | ||
| Decrease proteoglycan synthesis | [116] | ||
| Leukocyte infiltration into synovial fluid | [117] | ||
| Increase cartilage degradation when in combination with IL-1β and TNF-α | [115] | ||
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Mobasheri, A.; Henrotin, Y.; Biesalski, H.-K.; Shakibaei, M. Scientific Evidence and Rationale for the Development of Curcumin and Resveratrol as Nutraceutricals for Joint Health. Int. J. Mol. Sci. 2012, 13, 4202-4232. https://doi.org/10.3390/ijms13044202
Mobasheri A, Henrotin Y, Biesalski H-K, Shakibaei M. Scientific Evidence and Rationale for the Development of Curcumin and Resveratrol as Nutraceutricals for Joint Health. International Journal of Molecular Sciences. 2012; 13(4):4202-4232. https://doi.org/10.3390/ijms13044202
Chicago/Turabian StyleMobasheri, Ali, Yves Henrotin, Hans-Konrad Biesalski, and Mehdi Shakibaei. 2012. "Scientific Evidence and Rationale for the Development of Curcumin and Resveratrol as Nutraceutricals for Joint Health" International Journal of Molecular Sciences 13, no. 4: 4202-4232. https://doi.org/10.3390/ijms13044202
APA StyleMobasheri, A., Henrotin, Y., Biesalski, H.-K., & Shakibaei, M. (2012). Scientific Evidence and Rationale for the Development of Curcumin and Resveratrol as Nutraceutricals for Joint Health. International Journal of Molecular Sciences, 13(4), 4202-4232. https://doi.org/10.3390/ijms13044202