Abstract
Ornithine cyclodeaminase (OCD) is an NAD+-dependent deaminase that is found in bacterial species such as Pseudomonas putida. Importantly, it catalyzes the direct conversion of the amino acid L-ornithine to L-proline. Using molecular dynamics (MD) and a hybrid quantum mechanics/molecular mechanics (QM/MM) method in the ONIOM formalism, the catalytic mechanism of OCD has been examined. The rate limiting step is calculated to be the initial step in the overall mechanism: hydride transfer from the L-ornithine’s Cα–H group to the NAD+ cofactor with concomitant formation of a Cα=NH2 + Schiff base with a barrier of 90.6 kJ mol−1. Importantly, no water is observed within the active site during the MD simulations suitably positioned to hydrolyze the Cα=NH2 + intermediate to form the corresponding carbonyl. Instead, the reaction proceeds via a non-hydrolytic mechanism involving direct nucleophilic attack of the δ-amine at the Cα-position. This is then followed by cleavage and loss of the α-NH2 group to give the Δ1-pyrroline-2-carboxylate that is subsequently reduced to L-proline.
1. Introduction
l-proline (Pro) is one of the naturally occurring 20 genetically encoded amino acids and is unique amongst them in being the only one with a secondary α-amine. The effect of its structure and functionality upon the proteins in which it is found has long been studied. In addition, however, it is also known to have a number of key physiologically important roles. For example, it has been shown to be an important antioxidant needed by microorganisms, plants, and animals [1–8]. Furthermore, in certain pathogenic bacteria, it has been suggested that proline metabolism plays a role in enabling the pathogen to survive under harsh conditions [9–11]. Plants also depend on proline production for cell wall biosynthesis, while mediating abiotic and biotic cell stresses [12,13]. In mammals, the interconversion between proline and Δ1-pyrroline-5-carboxylate (P5C), an intermediate in its biosynthesis and catabolism, is believed to be involved in cell apoptosis [1,14–18]. Importantly, this cycle provides a redox shuttle between the cytosol and mitochondria, controlling the formation of reactive oxygen species [14,19,20]. Consequently, it is important to understand the functions of the enzymes that metabolize proline.
In general, Pro is synthesized within cells and organisms via multi-enzymatic pathways from either glutamate (Glu) or arginine (Arg) [9,14,21–24]. In the former, the enzymes involved require the use of γ-glutamyl kinase and glutamate-γ-semialdehyde dehydrogenase, which require the cofactors ATP and NADH or NADPH, respectively. In contrast, the latter pathway utilizes the metalloenzyme arginase and the pyridoxal-5′-phosphate (PLP)-dependent enzyme, ornithine δ-minotransferase. Both pathways, however, lead to the formation of a common intermediate, glutamic-semialdehyde. This then undergoes a non-enzymatic cyclization via an intramolecular condensation reaction to give the imine, P5C [14,25]. Then, the common enzyme P5C reductase (P5CR) reduces the latter cyclic intermediate to form L-Pro [9,26]. In mammals, the former pathway in which glutamate is converted to proline is believed to be the major metabolic route.
More recently, however, several bacteria have been shown to be able to utilize an unusual enzymatic route for proline biosynthesis [27,28]. More specifically, they use the NAD+-dependent non-metalloenzyme ornithine cyclodeaminase (OCD) to directly convert the amino acid L-ornithine (L-Orn), itself also an intermediate along the above “Arg-pathway”, to proline [9]. Thus, in contrast to the two general pathways discussed above in which the actual cyclization step is done without enzymatic participation, the mechanism of OCD includes the cyclization of a linear intermediate to a cyclic product (Scheme I) [9,29,30]. Furthermore, it not only produces L-proline stereospecifically and without additional enzymes being involved, but does so in an irreversible fashion [9,31].
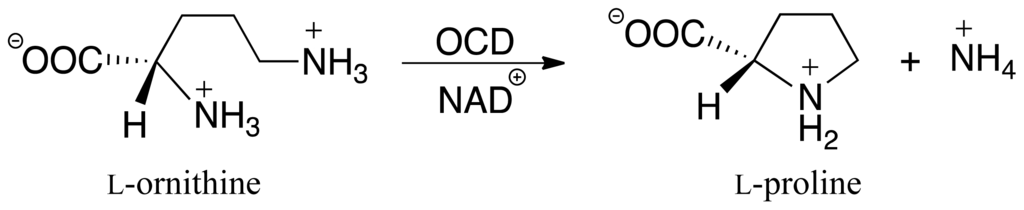
Scheme I.
Overall reaction for conversion of L-ornithine to L-proline as catalyzed by ornithine cyclodeaminase (OCD).
It is generally believed that the overall mechanism of OCD begins with an initial hydride transfer from the L-Orn substrate’s Cα–H moiety onto the C4 center of the nicotinamide ring of the NAD+ cofactor. This results in the formation of an iminium (Cα=NH2 + containing) intermediate complex [32–37]. Unfortunately, however, it is then unclear how the mechanism proceeds. Based on the results of experimental studies including X-ray crystallographic structures and mass spectroscopic data, two possible pathways have been proposed (Scheme II). While they share some common features such as involving formation of a Schiff base, a chemically and biochemically important reaction process that has been previously studied in detail both experimentally and computationally, they also have some very important differences [38–47]. In particular, in the hydrolytic pathway, an active site water nucleophilically attacks the Cα center of the iminium intermediate, resulting in loss of ammonium with formation of the keto acid, 5-amino-2-oxopentanoate (Scheme IIA) [28,34]. This then undergoes a cyclization reaction in which the δ-amine nucleophilically attacks the carbonyl carbon to form a cyclic Δ1-pyrroline-2-carboxylate (P2C) species. This latter intermediate has previously been shown to be involved in other biochemical processes including, for example, lysine catabolism. It is further noted that this ring formation step can be considered analogous to the cyclization of glutamic γ-semialdehyde in which an amine (α-amine) reacts with a carbonyl carbon center. This step is then followed by a hydride transfer from the NADH moiety onto the Cα-position (C2) of P2C to give L-proline.
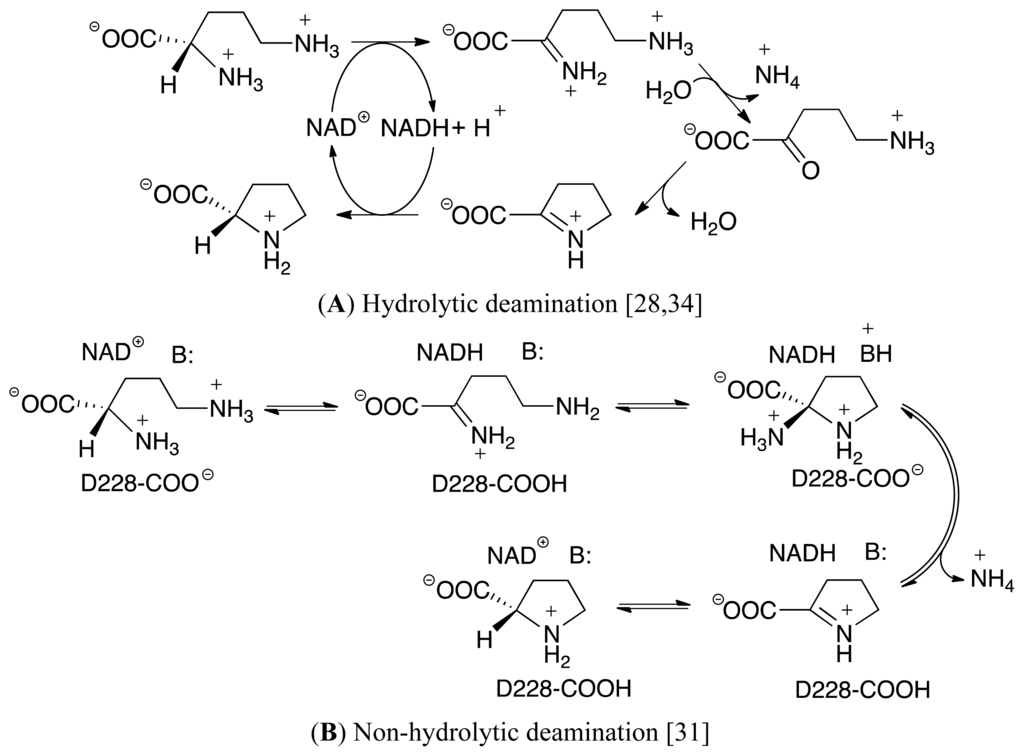
Scheme II.
Proposed (A) hydrolytic and (B) non-hydrolytic pathways for the conversion of L-ornithine to L-proline as catalyzed by the enzyme ornithine cyclodeaminase.
In contrast, in the alternate proposed mechanism known as the non-hydrolytic pathway, and shown above in Scheme IIB [31], the δ-amino tail of the iminium intermediate is neutralized by a suitable active site residue. Consequently, it is now able to nucleophilically attack the Cα-center of the iminium intermediate to form a cyclic 2-aminoproline species. This then undergoes loss of the α-amino group as ammonia or ammonium to give P2C. The final step is as in the hydrolytic pathway; hydride transfer from the NADH moiety onto the Cα-position (C2) of P2C to give the final product.
As noted above, the enzyme OCD itself has only been found in a few select bacteria [27]. However, it has been noted that it shows close phylogenetic resemblance to some crystallin enzymes, in particular those found in mammals. These proteins and enzymes are primarily known for their role in lens and cornea structure and transparency [48,49]. However, their malfunctioning has been linked to a number of diseases including cataract formation and cancer. Indeed, OCD is a member of the μ-crystallin family of enzymes which have been found to be abundant in the eye lens of marsupials [50] and are believed to have a similar chemical mechanism to that of OCD [31,51]. Thus, a clearer elucidation of the mechanism of OCD can provide invaluable insights not only into its catalytic abilities and proline biosynthesis, but also into related physiologically important enzymes.
Computational chemistry is a proven invaluable tool for the study of enzymatic mechanisms [52]. Thus, we have complementarily applied molecular dynamics (MD) simulations and an ONIOM QM/MM approach to investigate the catalytic mechanism of OCD. In particular, we have examined the feasibility of both the proposed hydrolytic and non-hydrolytic pathways for conversion of L-ornithine to L-proline.
2. Computational Methods
2.1. Molecular Dynamics (MD) Equilibration
The Molecular Operating Environment (MOE) [53] program was used for model preparation and the molecular dynamics (MD) simulations. For the model, the structure of OCD used was taken from an X-ray crystallographic structure of an OCD homo-dimer; each active site was complexed with L-ornithine (L-Orn) and NADH (PDB ID: 1X7D) [31]. While the protein is homo-dimeric, we have chosen to use a single monomer for the MD simulations. This model simplification is reasonable since the catalytic site does not include interface residues. Thus, a single protomeric enzyme-substrate-cofactor complex was selected while the other was removed. In addition, all selenomethionine (Se-Met) residues were mutated to the native methionines. The bound NADH cofactor was oxidized to give the catalytically required NAD+ (i.e., a hydride was removed from the C4 center of NADH). The coordinates of missing hydrogens were added using the MOE default method. A 7-Å spherical layer of water molecules was then added to solvate the enzyme-substrate complex. In order to force the system to lie within the volume of space defined by the surrounding shell of waters, an ellipsoidal potential wall with a scaling constant of 2 was placed around the solvated enzyme-substrate complex. To allow the electrostatic and van der Waals potentials to decay smoothly, a damping functional factor was included. The geometry of each solvated complex was then optimized using the CHARMM22 force field until the root mean square gradient of the total energy fell below 0.21 kJ mol−1 Å−1. The MD simulations were performed under constrained pressure and temperature. The equations of motion were coupled with the Nosé-Poincaré thermostat [54] and the time step for numerical integration was set to 2 fs. Initially, the system was heated from 150 to 300 K for a period of 50 ps, followed by an equilibration period of 100 ps at the constant temperature of 300 K and pressure of 1 atm. Based on rmsd and cluster analyses, a representative structure was chosen from the trajectory. This structure was then optimized with the CHARMM22 force field and used to obtain a suitable enzyme complex for further investigation (see below). We have successfully applied this MD protocol in the study of other enzymatic systems [55].
2.2. QM/MM Computations
The Gaussian 09 suite of programs [56] was used for all QM/MM calculations using the ONIOM formalism [57–65]. Optimized structures were obtained at the ONIOM (B3LYP/6-31G(d):AMBER96) level of theory [66–69]. Specifically, the AMBER96 molecular mechanics force field was used to describe the low-layer (i.e., protein environment surrounding the active center) while the high-layer (i.e., the active center) was described using the density functional theory-based method B3LYP in conjunction with the 6-31G(d) basis set. Harmonic vibrational frequencies were calculated at this same level of theory in order to obtain the corresponding Gibbs free energy corrections at SATP (ΔEGibbs) in addition to characterize the nature of the stationary points on the potential energy surface (PES) (i.e., as energy minima or first-order transition states (TSs)). Single point energy calculations on the above optimized structures were performed at the ONIOM (B3LYP/6-311 + G(2df,p):AMBER96//B3LYP/6-31G(d):AMBER96) + ΔEGibbs level of theory within a mechanical embedding (ME) formalism.
The final CHARMM22 optimized MD structure (see above) was used to obtain a suitable chemical model for the fully bound active site for use in the QM/MM-based mechanistic studies. More specifically, all residues and waters up to 15 Å from the L-ornithine substrate were extracted to be used as the enzyme-substrate-cofactor model. This distance was chosen as it has been previously shown that the steric and electrostatic effects arising from the protein environment surrounding the active site within this distance can have important effects on the mechanism and its intermediates and transition states [70].
The QM-region included the L-ornithine substrate and the active site groups with which it directly interacts or are experimentally known or proposed to be involved in the mechanism. Namely, the R-groups of Arg112, Lys69, Glu56 and Asp228 were included along with two active site waters observed within the MD simulations. The NAD+ cofactor was also included in part, the asterisk (*) indicating that only its nicotinamide and ribose ring was within the QM-region. The rest was replaced and modelled by a methyl group as shown in Figure 1. All those residues or waters shown in the outer-circle in Figure 1b were included within the MM-layer in their entirety. The only exceptions being those in which the R-group was included within the QM region. To ensure the integrity of the active site model during calculations, the α-carbons of each residue as well as that of the capping methyl in NAD+ were held fixed at their final CHARMM22 optimized positions (see above). This computational approach has been widely used previously, and successfully applied in studies on related enzymatic systems [55].
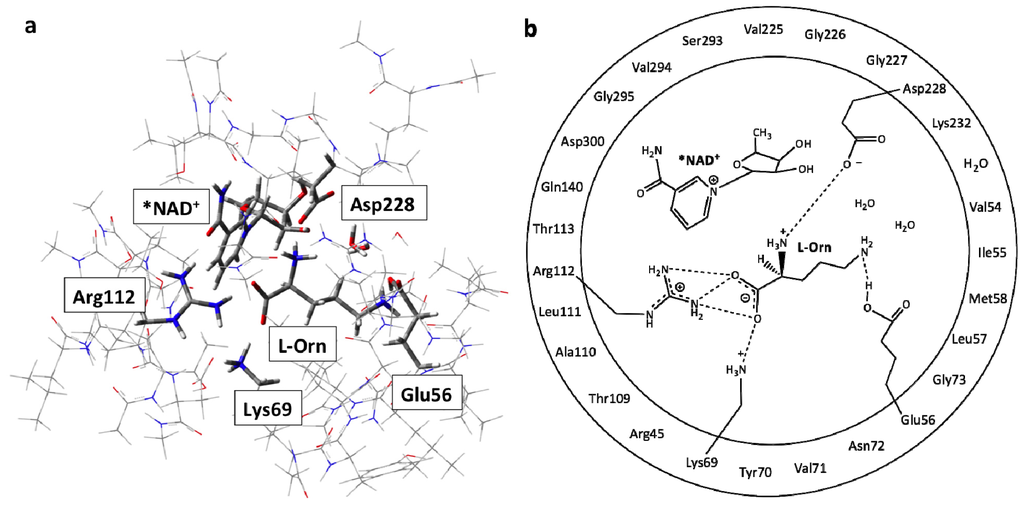
Figure 1.
Illustration of the ONIOM (QM/MM) fully bound active site model used: (a) the low or molecular mechanics (MM)-layer is shown in wire while the high or quantum mechanics (QM)-layer is shown in tube format; (b) those moieties within the inner circle were included within the QM-region, while those within the outer circle represent the residues contained in the MM-layer of the QM/MM model.
3. Results and Discussion
3.1. Structure of the Active Site with the Substrate L-Ornithine and NAD+ Cofactor Bound
We began this study by obtaining and examining the optimized structure of the fully bound active site complex (RC). That is, the complex in which both the NAD+ cofactor and L-ornithine (L-Orn) substrate are bound and which is shown in Figure 2a. As it can be seen, the L-Orn is bound electrostatically via interactions of each of its charged groups with active site residues. In particular, the α-COO− forms short, strong hydrogen bonds with the protonated R-groups of both Arg112 and Lys69 with distances of 1.73 and 1.53 Å, respectively. Meanwhile, its α-NH3 + group is strongly hydrogen bonded with the nearby R-group carboxylate of the active site aspartyl (Asp228) with an r(α-NH···−OOC-Asp228) distance of 1.62 Å. Notably, this results in a modest lengthening in the participating Nα–H bond to 1.08 Å. For the R-group, δ-NH3 +, however, it was found that it transferred a proton to the R-group carboxylate of the active site glutamyl residue (Glu56) with which it hydrogen bonds. Although they now form a neutral Nδ···HOOC–Glu56 hydrogen bond, its length is still decidedly short with a length of only 1.68 Å.
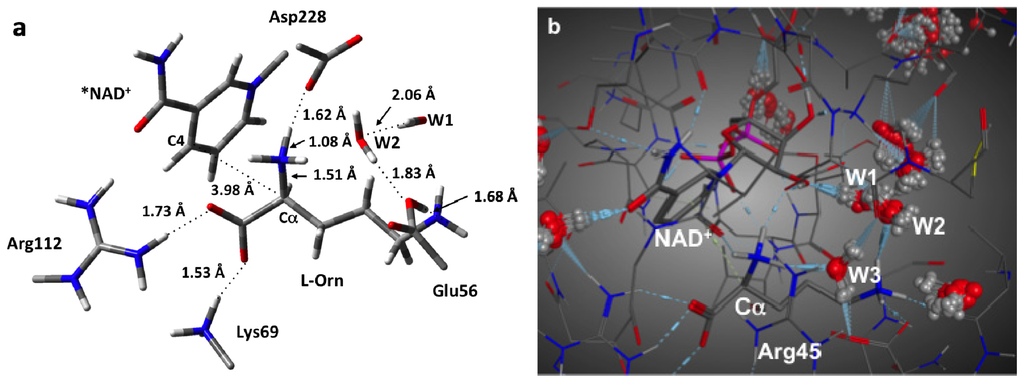
Figure 2.
(a) Optimized structure (see Computational Methods) of the initial fully bound active site complex RC with selected bond lengths shown (in Angstroms). (b) Solvated MD structure of the active site.
Importantly, in this bound conformation, the distance between the substrate’s Cα and the NAD+ cofactor’s C4 center to which a hydride would be transferred, i.e., r(Cα···C4(NAD+)), is 3.98 Å. It is noted that while in the X-ray crystal structure (PDB ID: 1X7D), the enzyme was co-crystallized with the substrate and NADH, the above calculated distance is in reasonable agreement with the experimentally measured distance of 3.8 Å. It is noted that the mechanistically important C4···H–Cα distance is 3.51 Å and that the Cα–H bond has lengthened marginally to 1.10 Å.
In addition, it is noted that in the MD structure of the initial reactant complex RC and shown in Figure 2b, three waters (W1, W2 and W3) were observed to lie in the active site and interact with the substrate. More specifically, one (W1) forms a hydrogen bonding bridge between the δ-NH3 + group and a sugar hydroxyl of the NAD+ cofactor. A second (W2) is simply hydrogen bonded to the substrates δ-NH3 + group while a third (W3) forms a hydrogen bonding bridge between the anionic R-group carboxylate of Glu56 and the substrate’s protonated α-NH3 + moiety. It should be noted that the oxygen of this latter water, OW3, is 3.50 Å away from the Cα center. These same interactions were also in agreement with the subsequent QM/MM optimized structure (not shown).
3.2. L-Orn Oxidation and Formation of 2-Aminoproline (AP)
The first catalytic step of OCD involves a hydride transfer from L-Orn to NAD+ via TS1 (Figure 3). Specifically, the hydride is transferred from the Cα–H moiety of the L-Orn substrate onto the C4 center of the nicotinamide ring of NAD+. This process occurs via TS1 with a relatively high barrier of 90.6 kJ mol−1 with respect to RC (Figure 4). This may reflect in part the lack of any stabilizing interactions between the active site and the hydride entity as well as the large structural changes predicted. For example, as it can be seen in Figure 3, in TS1 a significant reduction in the Cα···C4(NAD+) distance is seen. Specifically, it has shortened by 1.24 Å to 2.74 Å. As a result, the hydride is almost equidistant between the Cα and C4 centers with Cα···H− and C4···H− distances of 1.48 and 1.28 Å, respectively. However, concomitant with the H− transfer, a proton from α-NH3 + group has transferred to the R-group carboxylate of Asp228 (Figure 3). Indeed, in TS1, the proton has fully transferred to the carboxylate as indicated by an r(H–O-Asp228) distance of 0.98 Å while the Cα–N bond length has already significantly shortened by 0.16 Å to 1.35 Å. This suggests that the proton transfer occurs early in this reaction step.
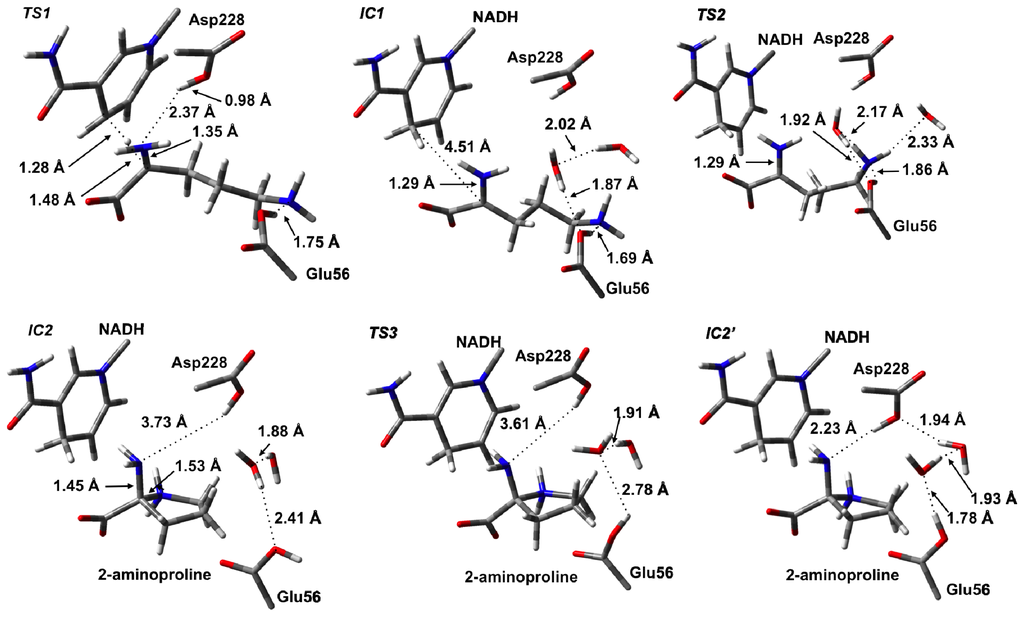
Figure 3.
Optimized structures (see Computational Methods) with selected bond lengths shown (in Angstroms) for the ornithine cyclodeaminase (OCD) catalytic mechanism prior to deamination as L-ornithine is converted to 2-aminoproline.
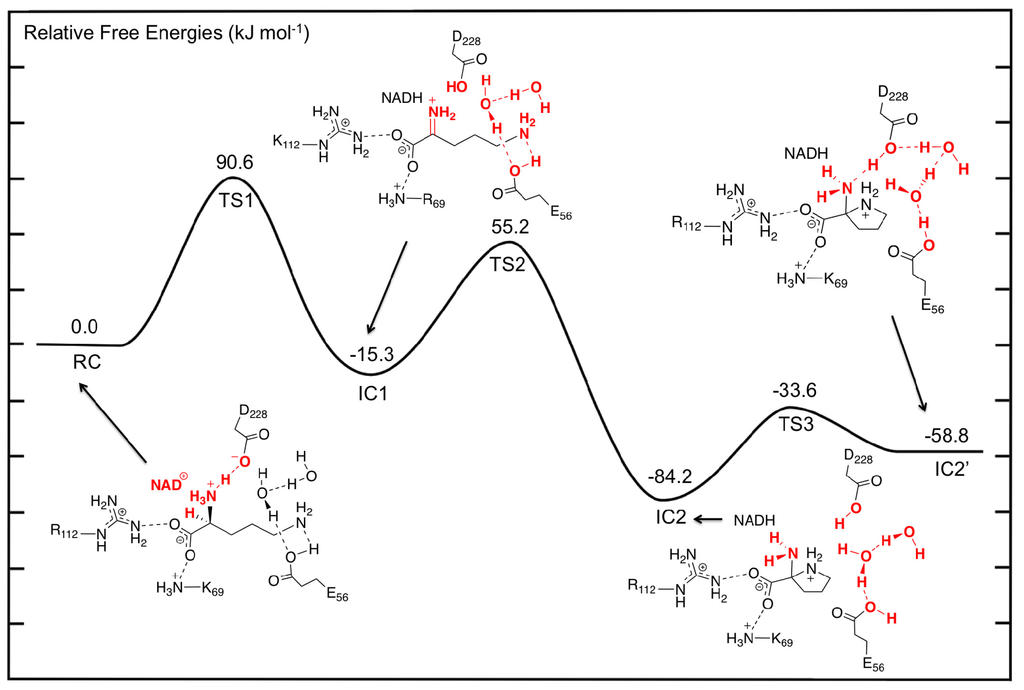
Figure 4.
Potential energy surface (PES) obtained (see Computational Methods) of the OCD catalytic mechanism prior to deamination as L-ornithine is converted to 2-aminoproline.
Complete H− transfer onto the NAD+ cofactor results in formation of the iminium ion intermediate (IC1) which lies lower in energy than RC by −15.3 kJ mol−1 (Figure 4). In IC1, the Cα···C4(NADH) distance has lengthened significantly to 4.51 Å and is in fact now even greater than that seen in RC (Figure 3). Furthermore, the Cα–N bond has shortened even further to just 1.29 Å indicating the formation of a Schiff base. In addition, the hydrogen bond interaction between the α-NH2 and now neutral Asp228-COOH group has been weakened with an r(Asp228-COOH···Nα) distance of 4.14 Å.
As detailed in the introduction section, it has been proposed that subsequent to hydride transfer and formation of the Schiff base intermediate, cyclization of L-Orn may occur via one of two possible pathways. Namely, it may either involve a first reaction with an active water to give the keto acid, 5-amino-2-oxopentanoate (A2O), or direct nucleophilic attack of the δ-NH2 nitrogen at the Cα center. Indeed, the former A2O intermediate is similar to the glutamate γ-semialdehyde, common in both Argand Glu-pathways for proline biosynthesis. In particular, the ketone functionality is exposed to nucleophilic attack by either the α-amine in the non-enzymatic reaction or by δ-amine in OCD. As noted previously for RC, the nearest active site water that may nucleophilically attack the Cα center lies approximately 3.5 Å from the Cα center. We did attempt to elucidate a possible reaction pathway for reaction of IC1 with a water molecule but were unable to do so. Analysis of the structure of IC1, however, suggests that in agreement with Goodman et al. [31], the steric presence of Arg45 packing against the substrate in IC1 prevents the water from approaching much closer. Indeed, the nearest distance between Arg45 and the Cα/Nα centers is only 3.40 Å. As a result, the hydrolytic pathway would seem unlikely to be physically feasible and hence is not being discussed further herein. Thus, the remainder of the discussion solely concerns the non-hydrolytic pathway unless otherwise mentioned. However, it is interesting to note that an iminium ion simulates a Lewis acid-activated carbonyl functional group [71]. In such Lewis acid activation, a lowering of the LUMO energy of the C=O π-system occurs promoting nucleophilic attack. Thus, it appears that OCD has optimized proline synthesis by maintaining the existence of a far more reactive intermediate such as an iminium ion rather than a ketone derivative.
With the iminium intermediate IC1 formed, it is now susceptible to nucleophilic attack by Nδ of L-Orn. However, presumably prior to attack, the “tail” is required to curl within the active site. It is noted that the δ-NH2 group in IC1, unlike say the α-COO− group, forms just one hydrogen bond interaction with an active site residue and that being with the now neutral R-group of Glu56. This would be expected to facilitate the required cleavage of this interaction and subsequent curling of the “tail” of IC1. Unfortunately, at the present level of theory we were unable to exactly optimize a TS for this process. Instead, a number of detailed systematic scans of the PES for such a curling was performed in order to determine an upper limit for the energy required for this step. In particular, it was found that a decrease in the ∠Cα–Cβ–Cγ–Cδ by 55.4° to 128.5° gave a structure (TS2) that corresponded to an energy maximum of 55.2 kJ mol−1 with respect to RC, which is 70.5 kJ mol−1 with respect to IC1. It is noted that in TS2, the Nδ···Cα distance has decreased from 4.51 Å in IC1 to 3.92 Å. Thus, clearly the desired Cα–Nδ bond has not yet formed. Interestingly, it should be noted that all attempts to optimize energy minima complexes with ∠Cα–Cβ–Cγ–Cδ less than 128.5° (without constraining the Cα–Cβ–Cγ–Cδ dihedral angle) led directly to the formation of the very low energy 2-aminoproline intermediate complex IC2 lying 84.2 kJ mol−1 lower in energy than RC. Importantly, in IC2, a Cα–Nδ bond has been formed with a length of 1.45 Å, typical for a single C–N bond. This suggests that in OCD, the active site structure allows for a reasonably low energy curling of the “tail” of the Schiff base intermediate and that once suitably positioned, the Nδ center appears able to nucleophilically attack at the Cα center readily. Thus, as stated above, given that OCD has protected the iminium intermediate from reacting with water, it has allowed for an apparently barrierless C–N bond formation without the need for activation by a Lewis acid. This is far different than what has been seen in various carbonyl analogues where significant barriers for initial C–N bond formation exist [38–47].
3.3. Deamination of 2-Aminoproline (AP)
In order to lose the α-amine of IC2 (i.e., deamination), it must first be protonated. This is analogous to the required protonation of the hydroxide to form water prior to Schiff base formation in the aldehyde analogues [38,40,41]. One possibility is that an intramolecular proton transfer may occur from the ring –NH2 +– group directly onto the leaving amine. However, this would necessarily involve a four-membered ring transition structure which is well-known to be higher in energy due to the inherent strain involved [72]. Alternatively, an active site residue may be able to act as a proton donor. Within the active site of OCD, however, the nearest acidic residue is the neutralized R-group carboxylic acid of Asp228. Within the present computational model in the formation of IC2 (i.e., during the preceding cyclization step), it was found that the Asp228-COOH group was no longer in a position to transfer a proton to the leaving α-amine (Figure 3). Thus, the Asp228-COOH···α-NH2 distance must first shorten allowing, for the formation of a hydrogen bond to the leaving α-amine. However, for this to occur IC2 must undergo a conformational or hydrogen bonding network change. In particular, the proton on Glu56 must rotate allowing for a rearrangement of the active site waters allowing for the Asp228-COOH···α-NH2 distance to be reduced.
The process occurs via TS3 with a barrier of 50.6 kJ mol−1 with respect to IC2 to give the alternate complex IC2′ (Figure 4). The latter complex, while lying 25.4 kJ mol−1 higher in energy than IC2, still lies markedly lower in energy than RC by 58.8 kJ mol−1. As expected, this rotation of the proton on Glu56-COOH group caused Asp228-COOH to hydrogen bond with the α-amino group of the substrate. Furthermore, the hydrogen bonding network between the Glu56-COOH group and Asp228-COOH involving the two waters also changed. Specifically, the Glu56-COOH moiety now acts as a hydrogen bond donor via the waters to the carboxylic group of Asp228.
However, within the present computational model we were unable to locate a transition structure for proton transfer from Asp228-COOH onto the leaving amine. This is likely due to the fact that this would result in several charged groups (Asp228-COO−, α-NH3 + and –NH2 +−), all being in close proximity and thus highly sensitive to the degree of inclusion of the electrostatics of the protein environment. While it is plausible that under experimental conditions Asp228-COOH may transfer its proton onto the α-NH2 group, previous computational investigations on Schiff base formation reactions have shown that such proton transfers generally occur via intramolecular proton transfer from the bridging amine; furthermore such proton transfers are facilitated by a water or other suitable group to prevent the formation of a four-membered TS [38,40,41].
Thus, we examined the possibility of a water molecule aiding proton transfer from either Asp228-COOH or the bridging –NH2 +– to the α-NH2 moiety. Indeed, once the cyclic pyrrolidine species is formed in IC2′ it is noted that there is now solvent-accessible space in the vicinity of the Asp228-COOH group. The resulting IC2′ complex in which a water was added is hereafter denoted as IC2′···H2O (Figure 5). The resulting PES obtained for the subsequent proton transfers is shown in Figure 6a. Importantly, in IC2′···H2O, a water simultaneously acts as a hydrogen bond acceptor via its oxygen with the Asp228-COOH group and pyrrolidine’s –NH2 +– moiety, while also acting as a hydrogen bond donor to the α-NH2 group of the pyrrolidine and the 2′-OH group of the NAD+ cofactor’s ribose. Indeed, in the optimized structure of IC2′···H2O r(Asp228-COOH···OH2) and r(αN···H2O) were found to be 1.74 and 1.77 Å, respectively, while for the ring amine r(NH2 +···OH2) is slightly longer at 1.95 Å (Figure 5).
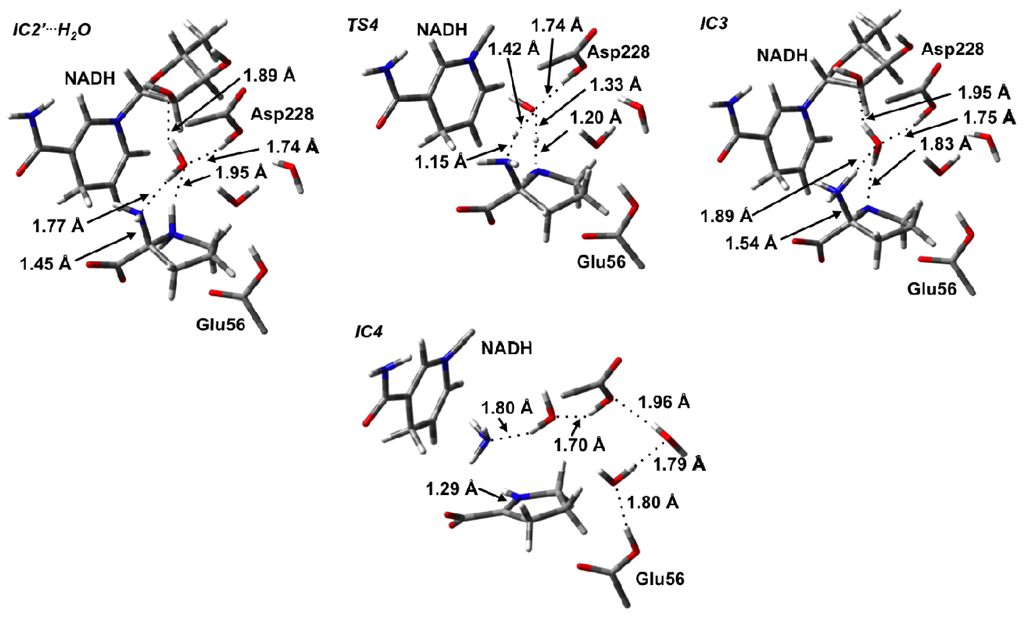
Figure 5.
Optimized structures (see Computational Methods) with selected bond lengths (in Angstroms) of the water-assisted intermediate complex (IC2′···H2O), TS4, cyclic intermediate IC3, and Schiff base intermediate IC4.
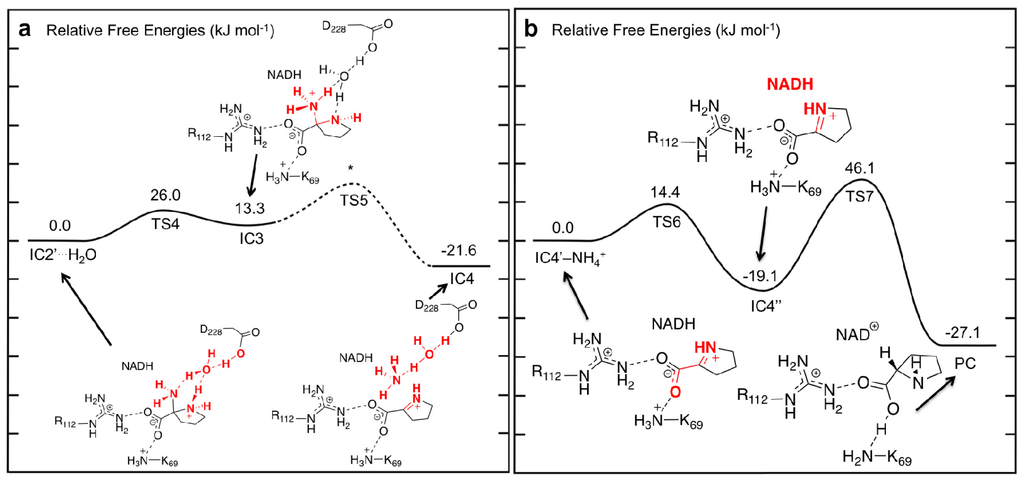
Figure 6.
PESs obtained (see Computational Methods) for (a) water-assisted deamination of 2-aminoproline to give Δ1-pyrroline-2-carboxylate (P2C) and, (b) reduction of P2C to give L-proline.
Interestingly, rather than a proton transfer from Aps228-COOH, it was found that the 2-aminoproline itself underwent an intramolecular proton transfer from its ring –NH2 +– moiety onto the α-NH2 group via the bridging water molecule. This process occurred via the six-membered ring transition structure, TS4, at a very low cost of only 26.0 kJ mol−1 with respect to IC2′···H2O (Figure 6a). The resulting intermediate IC3 formed lies only 13.3 kJ mol−1 higher in energy that IC2′···H2O (Figure 6a). Importantly, the protonation of the α-amine results in a significant lengthening in the Cα–Nα bond in IC3 by 0.09 Å to 1.54 Å and a shortening of the Cα–Nδ bond (0.10 Å) within the pyrrolidine. Such bond changes are likely to aid in deamination and Schiff base formation.
In previous computational studies on Schiff base formation involving a ketone/aldehyde, the loss of the leaving group –OH2 has been shown to occur in two steps [38]. First the adjacent bridging amine undergoes an inversion thus allowing for favorable overlap of its lone pair and the anti-bonding orbital of C–OH2 bond to be cleaved. The second step is then cleavage of the C–OH2 bond itself. For the 2-amino-2-carboxy-pyrrolidine, no stable intermediate corresponding to inversion of the ring –NH– was obtained nor was a concerted TS involving cleavage of the Cα–N bond with inversion of the bridging amine. However, it is noted that, while in previous studies, the barrier for loss of the water for the analogous carbonyl systems was found to be generally rate-limiting [38,39,41,42] cleavage of the Cα–NH3 + bond in OCD was found to be exothermic. In particular, with IC4 lying 34.9 kJ mol−1 lower in energy than IC3 or −21.6 kJ mol−1 relative to IC2′···H2O (Figure 6a). Thus, there is clearly a driving force for formation of this Schiff base intermediate. C–N bond cleavage with concomitant Schiff base formation results in the Δ1-pyrroline-2-carboxylate (P2C) containing complex IC4. It should also be noted that in IC4, the Asp228-COOH proton has also transferred onto the leaving NH3 to give an ammonium ion, NH4 + via H2O. The latter is then free to leave the active site.
3.4. Hydride Transfer from NADH onto P2C to Give the Final Product L-Proline
For the final stage, we considered formation of the L-proline product from P2C after loss of the cleaved NH4 + from the active site. Only minor structural changes were noticed in the resulting complex IC4′–NH4 +. In principle, L-proline can be formed via hydride transfer from the NADH moiety on the C2 (what will become Cα) of P2C. However, in IC4′–NH4 +, the key NADHC4···C2 is quite long at 4.00 Å. Furthermore, the P2C itself is not ideally positioned for the transfer. However, it is able to undergo an intramolecular rotation about its C2–COO− bond, i.e., a change in its ∠O1–C1–Cα–Nα dihedral angle, where the oxygen involved is the one hydrogen bonded to the Lys69. This process occurs via TS6 at a markedly low cost of only 14.4 kJ mol−1 with respect to IC4′–NH4 + suggesting that the rotation is likely reasonably unhindered (Figure 6b). It is noted that in TS6 the ∠O1–C1–Cα–Nα has decreased by 43.0° to 93.8°. This rotation is also exergonic with the resulting alternate conformer complex IC4″ being lower in energy than IC4′–NH4 + by 19.1 kJ mol−1. Importantly, as a result of this reorientation of P2C within the active site, its C2 center is now more suitably positioned for hydride transfer and the NADHC4···C2 distance has decreased significantly to 3.14 Å (Figure 7).
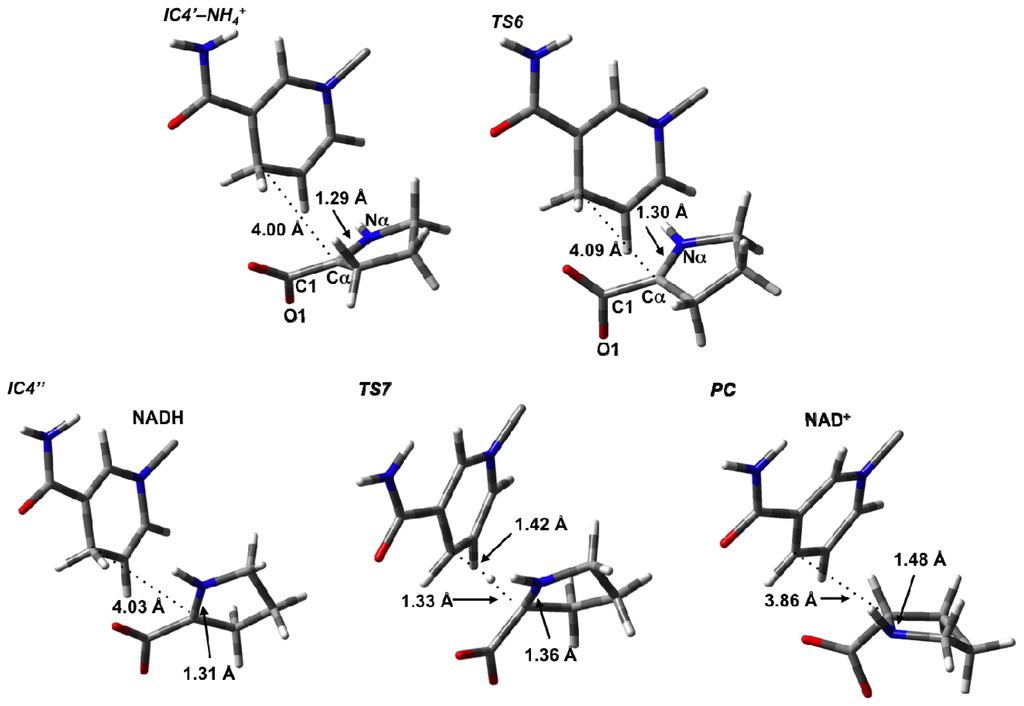
Figure 7.
Optimized structures (see Computational Methods) with selected bond lengths shown (in Angstroms) of the Δ1-pyrroline-2-carboxylate intermediate IC5, TS7, and L-proline product (PC).
Reduction of P2C via a hydride transfer from NADHC4–H onto its C2 center can then occur via TS7 with a barrier of 65.2 kJ mol−1 with respect to IC4″ (Figure 6b). This is notably lower than the cost of the initial hydride transfer from the L-Orn substrate to NAD+. This final step is also exergonic with the final product complex (PC) in which the L-proline is bound within the active site being a further 8.0 kJ mol−1 lower in energy than IC4″, and with a relative free energy of −27.1 kJ mol−1 with regards to IC4′–NH4 + (Figure 6b).
4. Conclusions
Using a combination of MD and QM/MM methods the catalytic mechanism of OCD has been investigated. From the results it was found that the initial hydride transfer from the Cα–H group of the L-ornithine substrate to the C4 center of the NAD+ cofactor with concomitant formation of a Schiff base, is the rate limiting step. In particular, this process occurred with a relative free energy barrier of 90.6 kJ mol−1. Experimentally, this hydride transfer step might be examined by deuterating H–(Cα) of L-Orn and conducting a kinetic isotope effect study.
For the remaining steps of the mechanism, while two pathways have been proposed, it appears that the enzyme most likely operates via a non-hydrolytic pathway. In particular, the MD and QM/MM results suggest that water is sterically hindered from attacking Cα after the initial Schiff base formation. Given that iminium ions are generally more reactive it seems that the active site of OCD evolved to guarantee such an intermediate exists by preventing its reaction with water. Indeed, following a conformational change of the substrate within the active site, a barrierless C–N bond formation occurred. This is considerably different than seen in previous investigations of various carbonyl analogues where significant barriers to C–N bond formation exists. While a transition state for deamination could not be found, the overall process was found to be thermodynamically favorable. Importantly, with deamination, a second Schiff base was formed. Like the initial C–N bond formation, this Schiff base would likely become more reactive. Indeed, the final step in the reaction was a H− transfer with a low barrier of 65.2 kJ mol−1. The resulting product, L-proline, being thermodynamically more favored than the preceding intermediate.
Thus, while the typical biosynthetic pathway for L-proline from arginine requires two enzymes and non-enzymatic cyclization of a glutamate γ-semialdehyde intermediate, this cyclic intermediate being common to the pathway involving glutamate, is later reduced to form L-Pro. In contrast, ornithine cyclodeaminase appears to first exploit the formation of a highly reactive Cα=NH2 +–containing iminium ion in order to enable cyclization and ultimately formation of a second iminium ion, Δ1-pyrroline-2-carboxylate, formed after loss of the α-NH3 group. The latter ion is readily reduced via a hydride transfer from the NADH cofactor onto its “Cα-center”, resulting in the formation of L-proline.
Acknowledgments
We thank the Natural Sciences and Engineering Research Council of Canada (NSERC) for funding, SHARCNET for additional computational resources, Eric A. C. Bushnell also gratefully acknowledges NSERC for CGS-M and PGS-D graduate scholarships, while Bogdan F. Ion thanks Ontario Graduate Scholarship (OGS) program for financial support.
References
- Meng, Z.H.; Lou, Z.Y.; Liu, Z.; Li, M.; Zhao, X.D.; Bartlam, M.; Rao, Z.H. Crystal structure of human pyrroline-5-carboxylate reductase. J. Mol. Biol 2006, 359, 1364–1377. [Google Scholar]
- Yura, T.; Vogel, H.J. Pyrroline-5-carboxylate reductase of Neurospora crassa; partial purification and some properties. J. Biol. Chem 1959, 234, 335–338. [Google Scholar]
- Valle, D.; Blaese, R.M.; Phang, J.M. Increased sensitivity of lymphocyte Δ1-pyrroline-5-carboxylate reductase to inhibition by proline with transformation. Nature 1975, 253, 214–216. [Google Scholar]
- Shiono, T.; Kador, P.F.; Kinoshita, J.H. Stimulation of the hexose-monophosphate pathway by pyrroline-5-carboxylate reductase in the lens. Exp. Eye Res 1985, 41, 767–775. [Google Scholar]
- Yeh, G.C.; Harris, S.C.; Phang, J.M. Pyrroline-5-carboxylate reductase in human-erythrocytes—A comparison of differential regulation. J. Clin. Invest 1981, 67, 1042–1046. [Google Scholar]
- Murahama, M.; Yoshida, T.; Hayashi, F.; Ichino, T.; Sanada, Y.; Wada, K. Purification and characterization of Δ1-pyrroline-5-carboxylate reductase isoenzymes, indicating differential distribution in spinach (Spinacia oleracea L.) leaves. Plant Cell Physiol 2001, 42, 742–750. [Google Scholar]
- Deutch, C.E.; Klarstrom, J.L.; Link, C.L.; Ricciardi, D.L. Oxidation of L-thiazolidine-4-carboxylate by Δ1-pyrroline-5-carboxylate reductase in Escherichia coli. Curr. Microbiol 2001, 42, 442–446. [Google Scholar]
- Basch, J.J.; Wickham, E.D.; Farrell, H.M. Pyrroline-5-carboxylate reductase in lactating bovine mammary glands. J. Dairy Sci 1996, 79, 1361–1368. [Google Scholar]
- Forlani, G.; Petrollino, D.; Fusetti, M.; Romanini, L.; Nocek, B.; Joachimiak, A.; Berlicki, L.; Kafarski, P. Δ1-pyrroline-5-carboxylate reductase as a new target for therapeutics: Inhibition of the enzyme from Streptococcus pyogenes and effects in vivo. Amino Acids 2012, 42, 2283–2291. [Google Scholar]
- Nakajima, K.; Natsu, S.; Mizote, T.; Nagata, Y.; Aoyania, K.; Fukuda, Y.; Nagata, K. Possible involvement of put A gene in Helicobacter pylori colonization in the stomach and motility. Biomed. Res 2008, 29, 9–18. [Google Scholar]
- Hoper, D.; Volker, U.; Hecker, M. Comprehensive characterization of the contribution of individual SigB-dependent general stress genes to stress resistance of Bacillus subtilis. J. Bacteriol 2005, 187, 2810–2826. [Google Scholar]
- Forlani, G.; Giberti, S.; Berlicki, L.; Petrollino, D.; Kafarski, P. Plant P5C reductase as a new target for aminomethylenebisphosphonates. J. Agric. Food Chem 2007, 55, 4340–4347. [Google Scholar]
- Hare, P.D.; Cress, W.A. Metabolic implications of stress-induced proline accumulation in plants. Plant Growth Regul 1997, 21, 79–102. [Google Scholar]
- Nocek, B.; Chang, C.; Li, H.; Lezondra, L.; Holzle, D.; Collart, F.; Joachimiak, A. Crystal structures of Δ1-pyrroline-5-carboxylate reductase from human pathogens Neisseria meningitides and Streptococcus pyogenes. J. Mol. Biol 2005, 354, 91–106. [Google Scholar]
- Polyak, K.; Xia, Y.; Zweier, J.L.; Kinzler, K.W.; Vogelstein, B. A model for p53-induced apoptosis. Nature 1997, 389, 300–305. [Google Scholar]
- Maxwell, S.A.; Davis, G.E. Differential gene expression in p53-mediated apoptosis-resistant vs. apoptosis-sensitive tumor cell lines. Proc. Natl. Acad. Sci. USA 2000, 97, 13009–13014. [Google Scholar]
- Donald, S.P.; Sun, X.Y.; Hu, C.A.A.; Yu, J.; Mei, J.M.; Valle, D.; Phang, J.M. Proline oxidase, encoded by p53-induced gene-6, catalyzes the generation of proline-dependent reactive oxygen species. Cancer Res 2001, 61, 1810–1815. [Google Scholar]
- Chen, C.B.; Dickman, M.B. Proline suppresses apoptosis in the fungal pathogen Colletotrichum trifolii. Proc. Natl. Acad. Sci. USA 2005, 102, 3459–3464. [Google Scholar]
- Hagedorn, C.H.; Phang, J.M. Catalytic transfer of hydride ions from NADPH to oxygen by the interconversions of proline and Δ1-pyrroline-5-carboxylate. Arch. Biochem. Biophys 1986, 248, 166–174. [Google Scholar]
- Hagedorn, C.H. Demonstration of a NADPH-linked Δ1-pyrroline-5-carboxylate proline shuttle in a cell-free rat-liver system. Biochim. Biophys. Acta 1986, 884, 11–17. [Google Scholar]
- Smith, R.J.; Downing, S.J.; Phang, J.M.; Lodato, R.F.; Aoki, T.T. Biosynthesis and metabolism of arginine in bacteria. Proc. Natl. Acad. Sci. USA 1980, 77, 5221–5225. [Google Scholar]
- Deutch, A.H.; Smith, C.J.; Rushlow, K.E.; Kretschmer, P.J. Escherichia-coli Δ1-pyrroline-5-carboxylate reductase—Gene sequence, protein overproduction and purification. Nucleic Acids Res 1982, 10, 7701–7714. [Google Scholar]
- Cunin, R.; Glansdorff, N.; Pierard, A.; Stalon, V. Biosynthesis and metabolism of arginine in bacteria. Microbiol. Rev 1986, 50, 314–352. [Google Scholar]
- Aral, B.; Kamoun, P. The proline biosynthesis in living organisms. Amino Acids 1997, 13, 189–217. [Google Scholar]
- Tanner, J.J. Structural biology of proline catabolism. Amino Acids 2008, 35, 719–730. [Google Scholar]
- Petrollino, D.; Forlani, G. Coenzyme preference of Streptococcus pyogenes Δ1-pyrroline-5-carboxylate reductase: Evidence supporting NADPH as the physiological electron donor. Amino Acids 2012, 43, 493–497. [Google Scholar]
- Soto, M.J.; Vandillewijn, P.; Olivares, J.; Toro, N. Ornithine cyclodeaminase activity in Rhizobium-meliloti. FEMS Microbiol. Lett 1994, 119, 209–213. [Google Scholar]
- Graupner, M.; White, R.H. Methanococcus jannaschii generates L-proline by cyclization of L-ornithine. J. Bacteriol 2001, 183, 5203–5205. [Google Scholar]
- Alam, S.; Wang, S.C.; Ruzicka, F.J.; Frey, P.A.; Wedekind, J.E. Crystallization and X-ray diffraction analysis of ornithine cyclodeaminase from Pseudomonas putida. Acta Crystallogr. Sect. D Biol. Crystallogr 2004, 60, 941–944. [Google Scholar]
- Gallagher, D.T.; Monbouquette, H.G.; Schroder, I.; Robinson, H.; Holden, M.J.; Smith, N.N. Structure of alanine dehydrogenase from Archaeoglobus: Active site analysis and relation to bacterial cyclodeaminases and mammalian μ-crystallin. J. Mol. Biol 2004, 342, 119–130. [Google Scholar]
- Goodman, J.L.; Wang, S.; Alam, S.; Ruzicka, F.J.; Frey, P.A.; Wedekind, J.E. Ornithine cyclodeaminase: Structure, mechanism of action, and implications for the μ-crystallin family. Biochemistry 2004, 43, 13883–13891. [Google Scholar]
- Carugo, O.; Argos, P. NADP-dependent enzymes. I. Conserved stereochemistry of cofactor binding. Proteins 1997, 28, 10–28. [Google Scholar]
- Andres, J.; Moliner, V.; Safont, V.S.; Domingo, L.R.; Picher, M.T. On transition structures for hydride transfer step in enzyme catalysis. A comparative study on models of glutathione reductase derived from semiempirical, HF, and DFT methods. J. Org. Chem 1996, 61, 7777–7783. [Google Scholar]
- Walsh, C. Enzymatic Reaction Mechanisms; W. H. Freeman: San Francisco, CA, USA, 1979. [Google Scholar]
- Klinman, J.P. Isotope-effects and structure-reactivity correlations in yeast alcohol-dehydrogenase reaction—Study of enzyme-catalyzed oxidation of aromatic alcohols. Biochemistry 1976, 15, 2018–2026. [Google Scholar]
- Lipscomb, W.N. Structure and catalysis of enzymes. Annu. Rev. Biochem 1983, 52, 17–34. [Google Scholar]
- Sans, N.; Schroder, G.; Schroder, J. The Noc region of Ti plasmid C58 codes for arginase and ornithine cyclodeaminase. Eur. J. Biochem 1987, 167, 81–87. [Google Scholar]
- Erdtman, E.; Bushnell, E.A.C.; Gauld, J.W.; Eriksson, L.A. Computational studies on Schiff-base formation: Implications for the catalytic mechanism of porphobilinogen synthase. Comput. Theor. Chem 2011, 963, 479–489. [Google Scholar]
- Almasi, J.N.; Bushnell, E.A.C.; Gauld, J.W. A QM/MM-based computational investigation on the catalytic mechanism of saccharopine reductase. Molecules 2011, 16, 8569–8589. [Google Scholar]
- Godoy-Alcantar, C.; Yatsimirsky, A.K.; Lehn, J.M. Structure-stability correlations for imine formation in aqueous solution. J. Phys. Org. Chem 2005, 18, 979–985. [Google Scholar]
- Hall, N.E.; Smith, B.J. High-level ab initio molecular orbital calculations of imine formation. J. Phys. Chem. A 1998, 102, 4930–4938. [Google Scholar]
- Szefczyk, B.; Kedzierski, P.; Sokalski, W.A.; Leszczynski, J. Theoretical insights into catalysis by phosphonoacetaldehyde hydrolase. Mol. Phys 2006, 104, 2203–2211. [Google Scholar]
- Conant, J.B.; Bartlett, P.D. A quantitative study of semicarbazone formation. J. Am. Chem. Soc 1932, 54, 2881–2899. [Google Scholar]
- Hill, R.L.; Crowell, T.I. Structural effects in the reactivity of primary amines with piperonal. J. Am. Chem. Soc 1956, 78, 2284–2286. [Google Scholar]
- Jencks, W.P. Reactions of nucleophilic reagents with phosphoramidate. J. Am. Chem. Soc 1959, 81, 475–481. [Google Scholar]
- Lowry, T.H.; Richardson, K.S. Mechanism and Theory in Organic Chemistry; Harper and Row Publishers: New York, NY, USA, 1976. [Google Scholar]
- Santerre, G.M.; Hansrote, C.J., Jr; Crowell, T.I. The reaction of aromatic aldehydes with n-butylamine. acid catalysis and substituent effects. J. Am. Chem. Soc. 1958, 80, 1254–1257. [Google Scholar]
- Hallen, A.; Cooper, A.J.L.; Jamie, J.F.; Haynes, P.A.; Willows, R.D. Mammalian forebrain ketimine reductase identified as μ-crystallin; potential regulation by thyroid hormones. J. Neurochem 2011, 118, 379–387. [Google Scholar]
- Kathiresan, T.; Krishnan, K.; Krishnakumar, V.; Agrawal, R.; Anand, A.; Muralidhar, D.; Mishra, A.K.; Dhople, V.M.; Aggrawal, R.K.; Sharma, Y. Triose phosphate isomerase, a novel enzyme-crystallin, and tau-crystallin in crocodile cornea—High accumulation of both proteins during late embryonic development. FEBS J 2006, 273, 3370–3380. [Google Scholar]
- Kim, R.Y.; Gasser, R.; Wistow, G.J. μ-crystallin is a mammalian homolog of Agrobacterium ornithine cyclodeaminase and is expressed in human retina. Proc. Natl. Acad. Sci. USA 1992, 89, 9292–9296. [Google Scholar]
- Jaenicke, R.; Slingsby, C. Lens crystallins and their microbial homologs: Structure, stability, and function. Crit. Rev. Biochem. Mol. Biol 2001, 36, 435–499. [Google Scholar]
- Llano, J.; Gauld, J.W. Mechanistics of Enzyme Catalysis: From Small to Large Active-Site Models. In Quantum Biochemistry: Electronic Structure and Biological Activity; Matta, C.F., Ed.; Wiley-VCH: Weinheim, Germany, 2010; Volume 2, pp. 643–666. [Google Scholar]
- MOE, version 2010.10; Chemical Computing Group: Montreal, QC, Canada, 2010.
- Bond, S.D.; Leimkuhler, B.J.; Laird, B.B. The Nosé-Poincaré method for constant temperature molecular dynamics. J. Comput. Phys 1999, 151, 114–134. [Google Scholar]
- Bushnell, E.A.C.; Erdtman, E.; Llano, J.; Eriksson, L.A.; Gauld, J.W. The first branching point in porphyrin biosynthesis: A systematic docking, molecular dynamics and quantum mechanical/molecular mechanical study of substrate binding and mechanism of uroporphyrinogen-III decarboxylase. J. Comput. Chem 2011, 32, 822–834. [Google Scholar]
- Gaussian 09, Revision B.01; Gaussian Inc: Wallingford, CT, USA, 2010.
- Bearpark, M.J.; Ogliaro, F.; Vreven, T.; Boggio-Pasqua, M.; Frisch, M.J.; Larkin, S.M.; Robb, M.A. CASSCF calculations for excited states of large molecules: Choosing when to use the RASSCF, ONIOM and MMVB approximations. Aip. Conf. Proc 2007, 2, 583–585. [Google Scholar]
- Dapprich, S.; Komaromi, I.; Byun, K.S.; Morokuma, K.; Frisch, M.J. A new ONIOM implementation in Gaussian98. Part I. The calculation of energies, gradients, vibrational frequencies and electric field derivatives. J. Mol. Struc. Theochem 1999, 461, 1–21. [Google Scholar]
- Humbel, S.; Sieber, S.; Morokuma, K. The IMOMO method: Integration of different levels of molecular orbital approximations for geometry optimization of large systems: Test for n-butane conformation and SN2 reaction: RCl + Cl−. J. Chem. Phys 1996, 105, 1959–1967. [Google Scholar]
- Maseras, F.; Morokuma, K. IMOMM—A new integrated ab-initio plus molecular mechanics geometry optimization scheme of equilibrium structures and transition-states. J. Comput. Chem 1995, 16, 1170–1179. [Google Scholar]
- Morokuma, K.; Musaev, D.G.; Vreven, T.; Basch, H.; Torrent, M.; Khoroshun, D.V. Model studies of the structures, reactivities, and reaction mechanisms of metalloenzymes. IBM J. Res. Dev 2001, 45, 367–395. [Google Scholar]
- Svensson, M.; Humbel, S.; Froese, R.D.J.; Matsubara, T.; Sieber, S.; Morokuma, K. ONIOM: A multilayered integrated MO + MM method for geometry optimizations and single point energy predictions. A test for Diels-Alder reactions and Pt(P(t-Bu)3)2 + H2 oxidative addition. J. Phys. Chem 1996, 100, 19357–19363. [Google Scholar]
- Vreven, T.; Byun, K.S.; Komaromi, I.; Dapprich, S.; Montgomery, J.A.; Morokuma, K.; Frisch, M.J. Combining quantum mechanics methods with molecular mechanics methods in ONIOM. J. Chem. Theory Comput 2006, 2, 815–826. [Google Scholar]
- Vreven, T.; Morokuma, K. On the application of the IMOMO (integrated molecular orbital plus molecular orbital) method. J. Comput. Chem 2000, 21, 1419–1432. [Google Scholar]
- Vreven, T.; Morokuma, K.; Farkas, O.; Schlegel, H.B.; Frisch, M.J. Geometry optimization with QM/MM, ONIOM, and other combined methods. I. Microiterations and constraints. J. Comput. Chem 2003, 24, 760–769. [Google Scholar]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys 1993, 98, 5648–5652. [Google Scholar]
- Becke, A.D. A new mixing of Hartree-Fock and local density-functional theories. J. Chem. Phys 1993, 98, 1372–1377. [Google Scholar]
- Lee, C.T.; Yang, W.T.; Parr, R.G. Development of the Colle-Salvetti correlation-energy formula into a functional of the electron-density. Phys. Rev. B 1988, 37, 785–789. [Google Scholar]
- Case, D.A.; Cheatham, T.E.; Darden, T.; Gohlke, H.; Luo, R.; Merz, K.M.; Onufriev, A.; Simmerling, C.; Wang, B.; Woods, R.J. The Amber biomolecular simulation programs. J. Comput. Chem 2005, 26, 1668–1688. [Google Scholar]
- Hu, L.H.; Soderhjelm, P.; Ryde, U. On the convergence of QM/MM energies. J. Chem. Theory Comput 2011, 7, 761–777. [Google Scholar]
- Brazier, J.B.; Tomkinson, N.C.O. Secondary and primary amine catalysts for iminium catalysis. Top. Curr. Chem 2009, 291, 281–347. [Google Scholar]
- Roy, D.; von Ragué Schleyer, P. Chemical Origin of Life: How do Five HCN Molecules Combine to form Adenine under Prebiotic and Interstellar Conditions. In Quantum Biochemistry: Electronic Structure and Biological Activity; Matta, C.F., Ed.; Wiley-VCH Verlag GmbH & Co. KGaA: Weinheim, Germany, 2010; Volume 1, pp. 199–217. [Google Scholar]
© 2012 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).