Modulation of Ca2+ Signals by Epigallocatechin-3-gallate(EGCG) in Cultured Rat Hippocampal Neurons

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
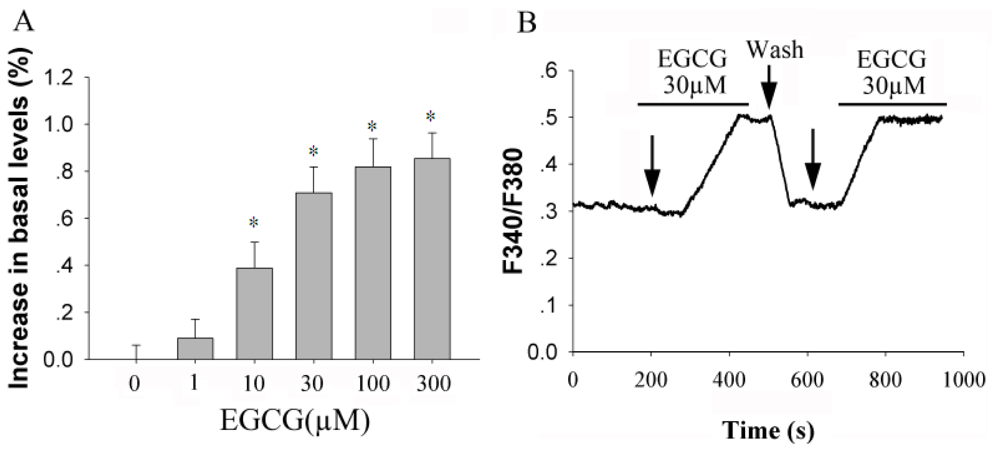
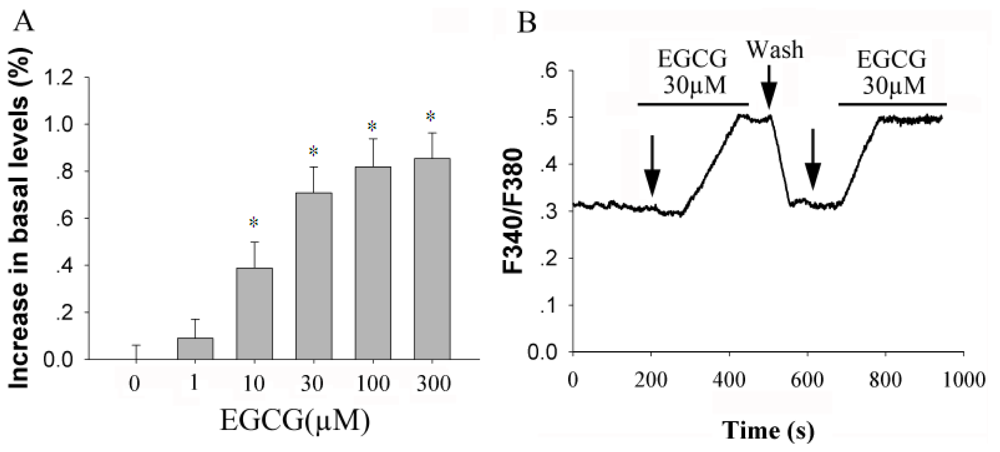
2.1. EGCG Elevates [Ca2+]i in Rat Hippocampal Neurons in a Dose-Dependent Manner
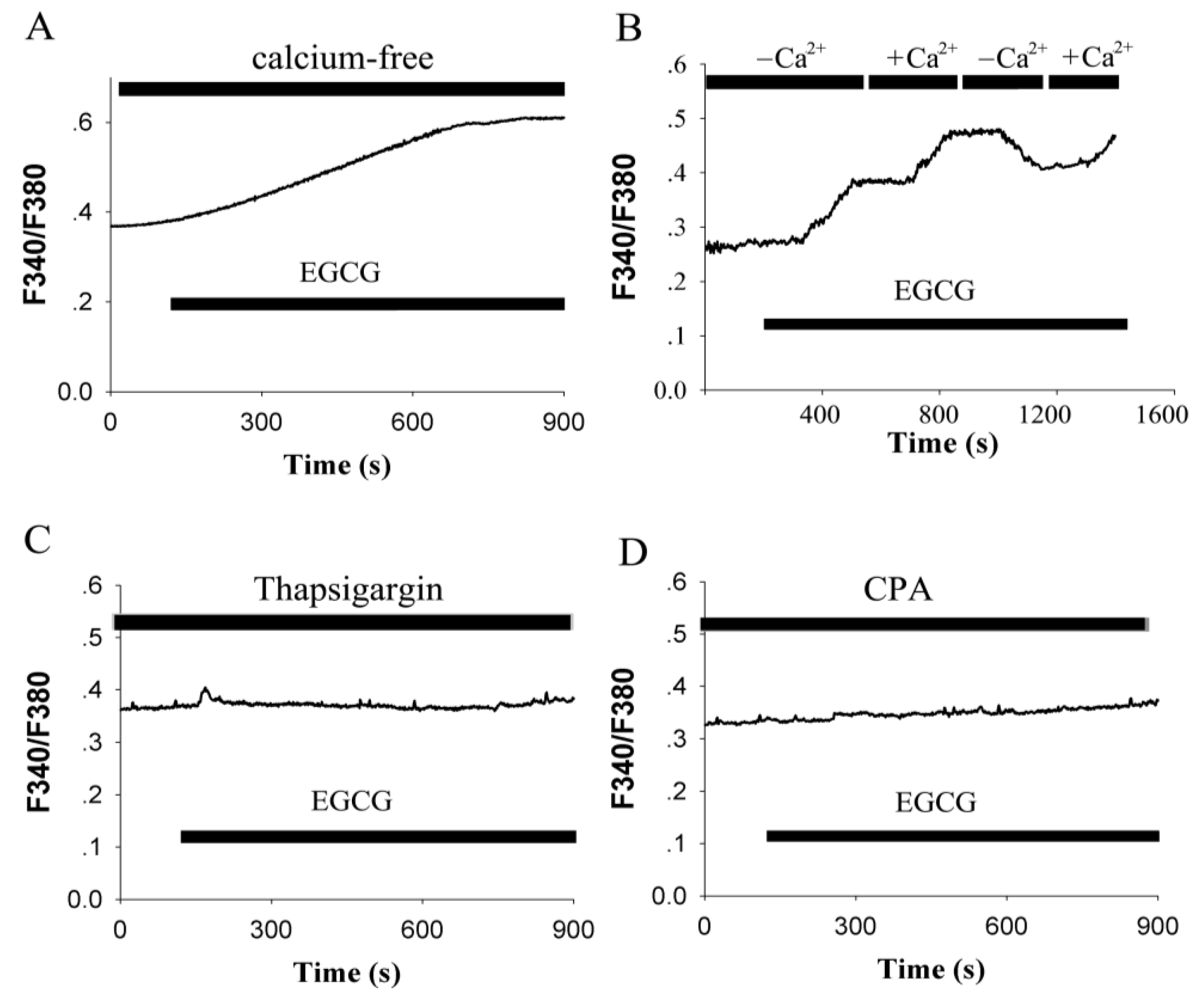
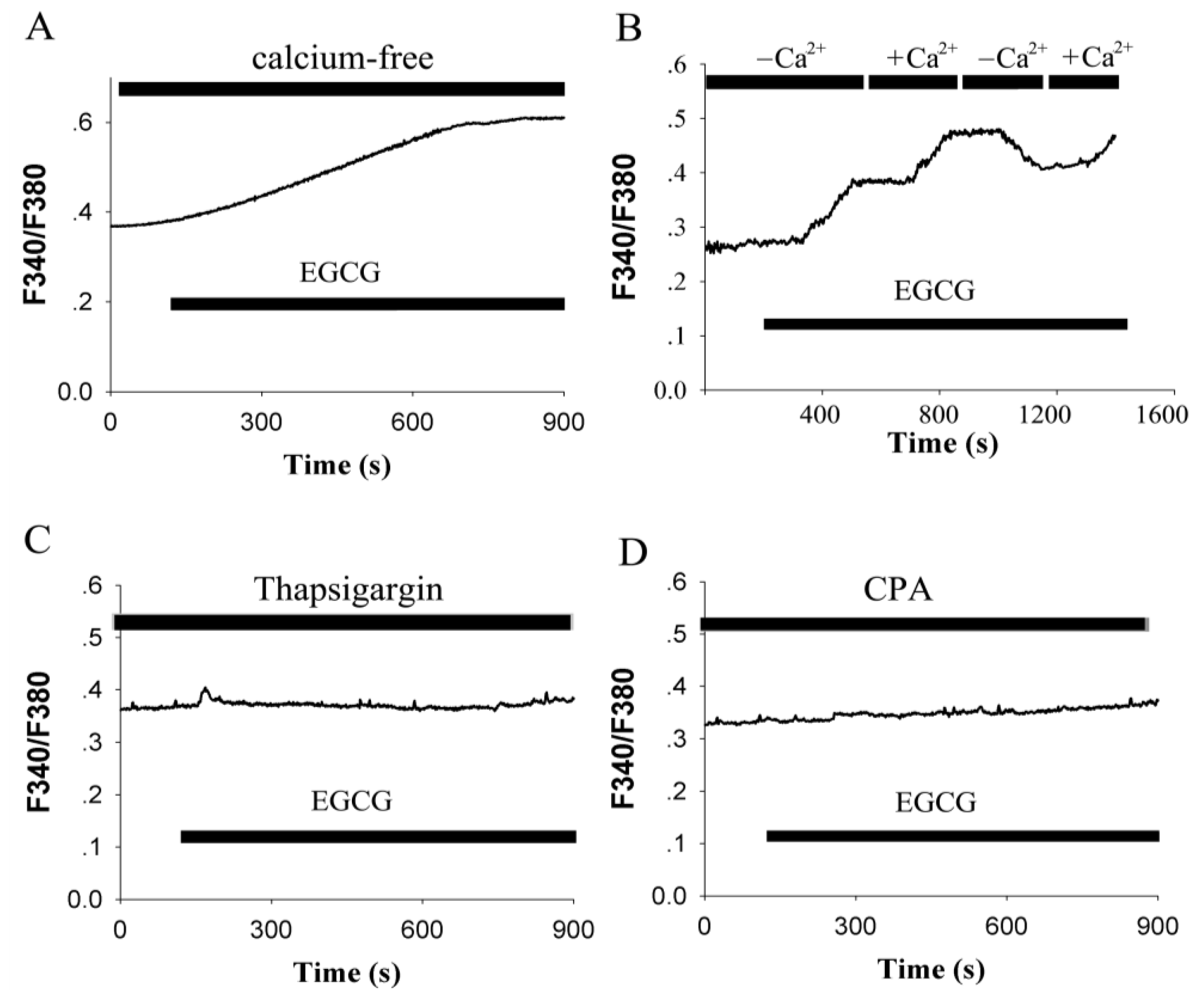
2.2. EGCG-Evoked Enhancement of [Ca2+]i Depends on the Release from Intracellular Calcium Store and Ca2+ Influx
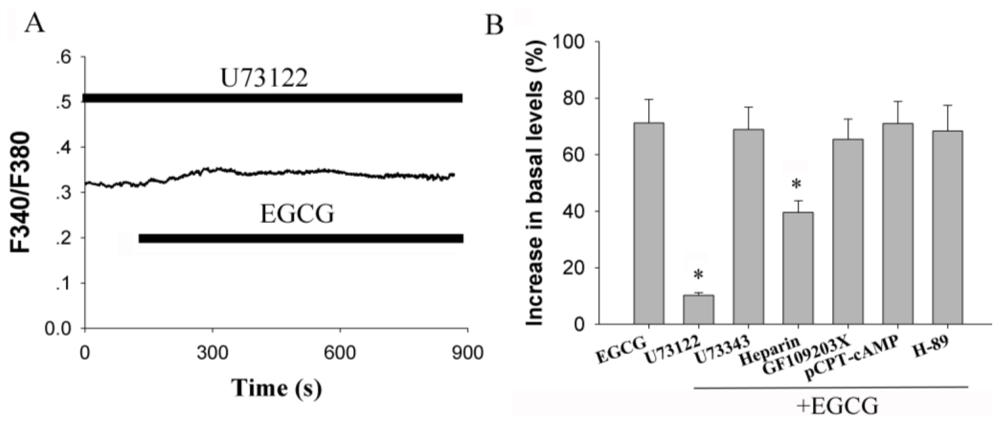
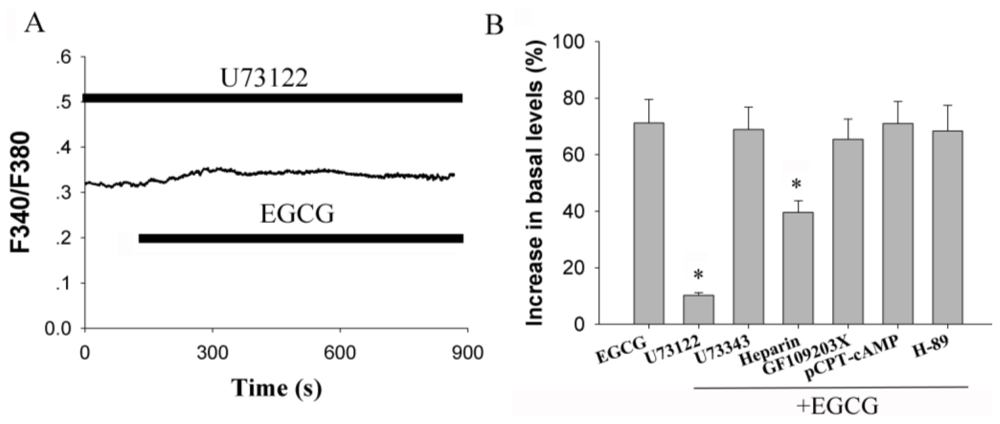
2.3. Activation of Phospholipase C (PLC) Signaling Pathways is Essential for EGCG-Stimulated [Ca2+]i Elevation
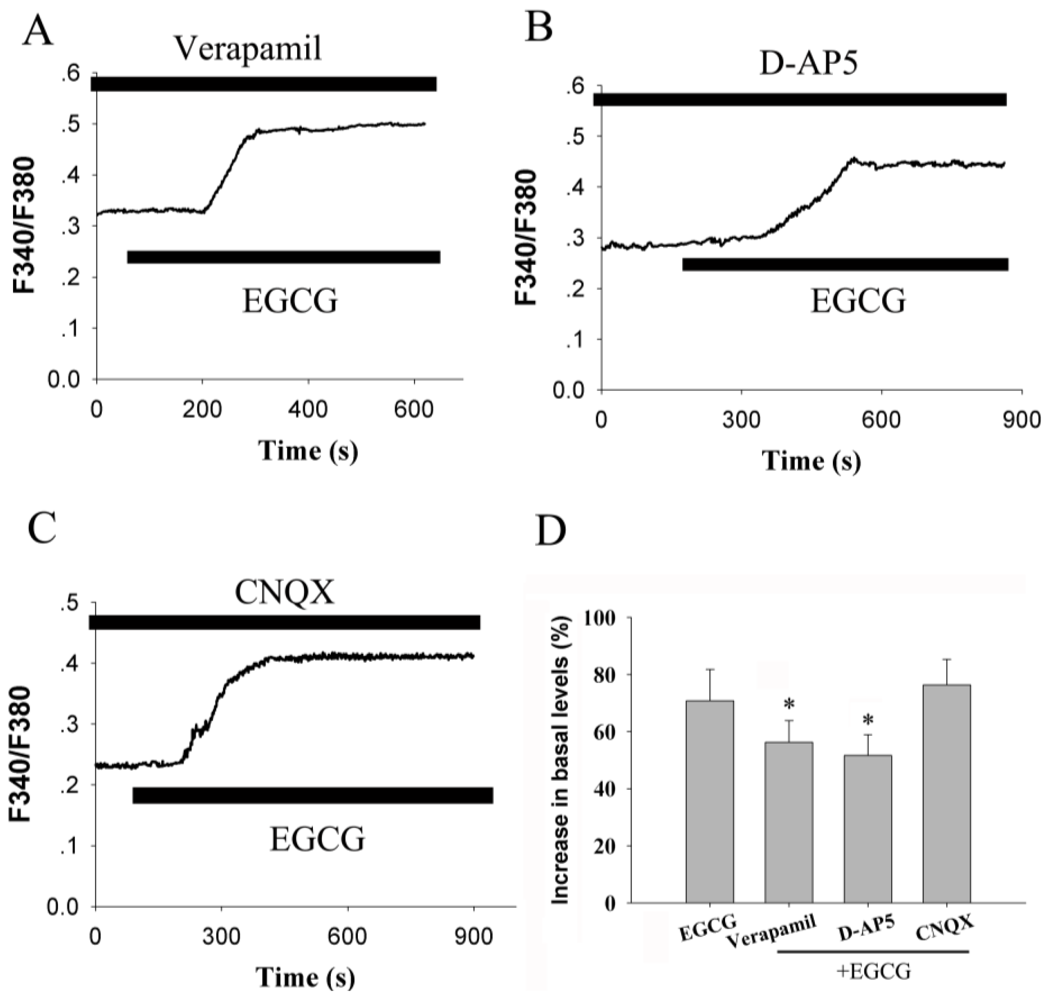
2.4. Voltage-Gated Calcium Channel and Receptor-Operated Calcium Channel Contribute to the Increase of [Ca2+]i
2.5. EGCG Suppresses IHVA and INMDA via Elevating Intracellular Ca2+ Concentration
2.6. Discussion
3. Experimental Section
3.1. Materials
3.2. Cell Culture
3.3. Calcium Imaging Experiment
3.4. Whole Cell Patch Clamp Recording
3.5. Statistical Analysis
4. Conclusions
Acknowledgements
References
- Shi, X; Ye, J; Leonard, S; Ding, M; Vallyathan, V; Castranova, V; Rojanasakul, Y; Dong, Z. Antioxidant properties of (–)-epicatechin-3-gallate and its inhibition of Cr (VI)-induced DNA damage and Cr (IV)- or TPA-stimulated NF-κB activation. Mol. Cell. Biochem 2000, 206, 125–132. [Google Scholar]
- Valcic, S; Burr, JA; Timmermann, BN; Liebler, DC. Antioxidant chemistry of green tea catechins. New oxidation products of (–)-epicatechin-3-gallate from their reactions with perosyl radicals. Chem. Res. Toxicol 2000, 13, 801–810. [Google Scholar]
- Komori, A; Yatsunami, J; Okabe, S; Abe, S; Hara, K; Suganuma, M; Kim, SJ; Fujiki, H. Anticarcinogenic activity of green tea polyphenols. Jpn. J. Clin. Oncol 1993, 23, 186–190. [Google Scholar]
- Kuroda, Y; Hara, Y. Antimutagenic and anticarcinogenic activity of tea polyphenols. Mutat. Res 1999, 436, 69–97. [Google Scholar]
- Wand, SL; Mukhtar, H. Gene expression profile in human prostate LNCaP cancer cells by (–)-epicatechin-3-gallate. Cancer Lett 2002, 182, 43–51. [Google Scholar]
- Dona, M; Dell’Aica, I; Calabrese, F; Benelli, R; Morini, M; Albini, A; Garbisa, S. Neutrophil restraint by green tea: inhibiton of inflammation, associated angiogenesis, and pulmonary fibrosis. J. Immunol 2003, 170, 4335–4341. [Google Scholar]
- Nakagawa, K; Miyazawa, T. Absorption and distribution of tea catechin, (–)-epicatechin-3-gallate, in the rat. J. Nutr. Sci. Vitaminol 1997, 43, 679–684. [Google Scholar]
- Berridge, MJ; Lipp, P; Bootman, MD. The versatility and universality of calcium signaling. Nat. Rev. Mol. Cell Biol 2000, 1, 11–21. [Google Scholar]
- Shieh, PB; Hu, SC; Bobb, K; Timmusk, T; Ghosh, A. Identification of a signaling pathway involved in calcium regulation of BDNF expression. Neuron 1998, 20, 727–740. [Google Scholar]
- Shoop, RD; Esquenazi, E; Yamada, N; Ellisman, MH; Berg, DK. Ultra-structure of a somatic spine mat for nicotinic signaling in neurons. J. Neurosci 2002, 22, 748–756. [Google Scholar]
- Tao, X; Finkbeiner, S; Arnold, DB; Shaywitz, AJ; Greenberg, ME. Ca2+ influx regulates BDNF transcription by a CREB family transcription factor-dependent mechanism. Neuron 1998, 20, 709–726. [Google Scholar]
- Bae, JH; Mun, KC; Park, WK; Lee, SR; Suh, SI; Baek, WK; Yim, MB; Kwon, TK; Song, DK. EGCG attenuates AMPA-induced intracellular calcium increase in hippocampal neurons. Biochem. Biophys. Res. Commun 2002, 290, 1506–1512. [Google Scholar]
- Lee, H; Bae, JH; Lee, SR. Protective effect of green tea polyphenol EGCG against neuronal damage and brain edema after unilateral cerebral ischemia in gerbils. J. Neurosci. Res 2004, 77, 892–900. [Google Scholar]
- Yin, ST; Tang, ML; Deng, HM; Xing, TR; Chen, JT; Wang, HL; Ruan, DY. Epigallocatechin-3-gallate induced primary cultures of rat hippocampal neurons death linked to calcium overload and oxidative stress. Naunyn-Schmiedeberg’s Arch. Pharmacol 2009, 379, 551–564. [Google Scholar]
- Tedford, HW; Zamponi, GW. Direct G protein modulation of Cav2 calcium channels. Pharmacol. Rev 2006, 58, 837–862. [Google Scholar]
- Levites, Y; Amit, T; Youdim, MBH; Mandel, S. Involvement of protein kinase C activation and cell survival/cell cycle genes in green tea polyphenol (–)-epigallocatechin-3-gallate neuroprotective action. J. Biol. Chem 2002, 277, 30574–30580. [Google Scholar]
- Choi, YB; Kim, YI; Lee, KS; Kim, BS; Kim, DJ. Protective effect of epigallocatechin gallate on brain damage after transient middle cerebral artery occlusion in rats. Brain Res 2004, 1019, 47–54. [Google Scholar]
- Levites, Y; Amit, T; Mandel, S; Youdim, MB. Neuroprotection and neurorescue against Abeta toxicity and PKC-dependent release of non-amyloidogenic soluble precursor protein by green tea polyphenol (–)-epigallocatechin-3-gallate. FASEB J 2003, 17, 952–954. [Google Scholar]
- Weinreb, O; Mandel, S; Youdim, MBH. cDNA gene expression profile homology of antioxidants and their anti-apoptotic and pro-apoptotic activities in human neuroblastoma cells. FASEB J 2003, 17, 935–937. [Google Scholar]
- Schroeter, H; Williams, RJ; Matin, R; Iversen, L; Rice-Evans, CA. Phenolic antioxidants attenuate neuronal cell death following uptake of oxidized low-density lipoprotein. Free Radic Biol Med 2000, 29, 1222–1233. [Google Scholar]
- Isbrucer, RA; Bausch, J; Edwards, JA; Wolz, E. Safety studies on epigallocatechin gallate (EGCG) preparations. Part 1. Genotoxicity. Food Chem. Toxicol 2006, 44, 626–635. [Google Scholar]
- Isbruker, RA; Edwards, JA; Wolz, E; Davidocich, A; Bausch, J. Safety studyies on epigallocatechin gallate (EGCG) preparations. Part 3. Teratogenicity and reproductive toxicity studies in rats. Food Chem. Toxicol 2006, 44, 651–661. [Google Scholar]
- Kanadzu, M; Lu, Y; Morimoto, K. Dual function of (–)-epigallocatechin gallate (EGCG) in healthy human lymphocytes. Cancer Lett 2006, 241, 250–255. [Google Scholar]
- Jeong, HS; Kim, YS; Park, JS. Modulation of neuronal activity by EGCG. Brain Res 2005, 1047, 267–270. [Google Scholar]
- Deng, HM; Yin, ST; Yan, D; Tang, ML; Li, CC; Chen, JT; Wang, M; Ruan, DY. Effects of EGCG on voltage-gated sodium channels in primary cultures of rat hippocampal CA1 neurons. Toxicology 2008, 252, 1–8. [Google Scholar]
- Bootman, MD; Collins, TJ; Peppiatt, CM; Prothero, LS; Mac-Kenzaie, L; de Smet, P; Travers, M; Tovey, SC; Seo, JT; Berridge, MJ; Ciccolini, F; Lipp, P. Calcium signaling—An overview. Semin. Cell Dev. Biol 2001, 12, 3–10. [Google Scholar]
- Putney, JW, Jr. Capacitative calcium entry in the nervous system. Cell Calcium 2003, 34, 339–344. [Google Scholar]
- Marchenko, SM. Mechanism of modulation of GABA-activated current by internal calcium in rat central neurons. Brain Res 1991, 546, 355–357. [Google Scholar]
- Lukyanetz, EA; Piper, TP; Sihra, T. Calcineurin involvement in the regulation of high-threshold Ca2+ channels in NG108-15 (rodent neuroblastoma × glioma hybrid) cells. J. Physiol 1998, 510, 371–385. [Google Scholar]
- Brehm, P; Eckert, R. Calcium entry leads to inactivation of calcium channel in paramecium. Science 1978, 202, 1203–1206. [Google Scholar]
- Stevens, TR; Krueger, SR; Fitzsimonds, RM; Picciotto, MR. Neuroprotection by nicotine in mouse primary cortical cultures involves activation of calcineurin and L-type calcium channel inactivation. J. Neurosci 2003, 23, 10093–10099. [Google Scholar]
- Mandel, SA; Avramovich-Tirosh, Y; Reznichenko, L; Zheng, H; Weinreb, O; Amit, T; Youdim, MBH. Multifunctional Activities of Green Tea Catechins in Neuroprotection. Neurosignals 2005, 14, 46–60. [Google Scholar]
- Williams, RJ; Spencer, JPE; Rice-Evans, C. Flavonoids: Antioxidants or signaling molecules? Free Radic. Biol. Med 2004, 36, 838–849. [Google Scholar]
- Ming, Y; Zhang, H; Long, L; Wang, F; Chen, J; Zhen, X. Modulation of Ca2+ signals by phosphatidylinositol-linked novel D1 dopamine receptor in hippocampal neurons. J. Neurochem 2006, 98, 1316–1323. [Google Scholar]
- Wang, JH; Wang, F; Yang, MJ; Yu, DF; Wu, WN; Liu, J; Ma, LQ; Cai, F; Chen, JG. Leptin regulated calcium channels of neuropeptide Y and proopiomelanocortin neurons by activation of different signal pathways. Neuroscience 2008, 156, 89–98. [Google Scholar]


© 2011 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, J.-H.; Cheng, J.; Li, C.-R.; Ye, M.; Ma, Z.; Cai, F. Modulation of Ca2+ Signals by Epigallocatechin-3-gallate(EGCG) in Cultured Rat Hippocampal Neurons. Int. J. Mol. Sci. 2011, 12, 742-754. https://doi.org/10.3390/ijms12010742
Wang J-H, Cheng J, Li C-R, Ye M, Ma Z, Cai F. Modulation of Ca2+ Signals by Epigallocatechin-3-gallate(EGCG) in Cultured Rat Hippocampal Neurons. International Journal of Molecular Sciences. 2011; 12(1):742-754. https://doi.org/10.3390/ijms12010742
Chicago/Turabian StyleWang, Jiang-Hua, Jin Cheng, Cai-Rong Li, Mao Ye, Zhe Ma, and Fei Cai. 2011. "Modulation of Ca2+ Signals by Epigallocatechin-3-gallate(EGCG) in Cultured Rat Hippocampal Neurons" International Journal of Molecular Sciences 12, no. 1: 742-754. https://doi.org/10.3390/ijms12010742
APA StyleWang, J.-H., Cheng, J., Li, C.-R., Ye, M., Ma, Z., & Cai, F. (2011). Modulation of Ca2+ Signals by Epigallocatechin-3-gallate(EGCG) in Cultured Rat Hippocampal Neurons. International Journal of Molecular Sciences, 12(1), 742-754. https://doi.org/10.3390/ijms12010742