Eco-Contribution for the Production of N-Arylnitrones: Solvent-Free and Assisted by Microwaves

Abstract
:1. Introduction
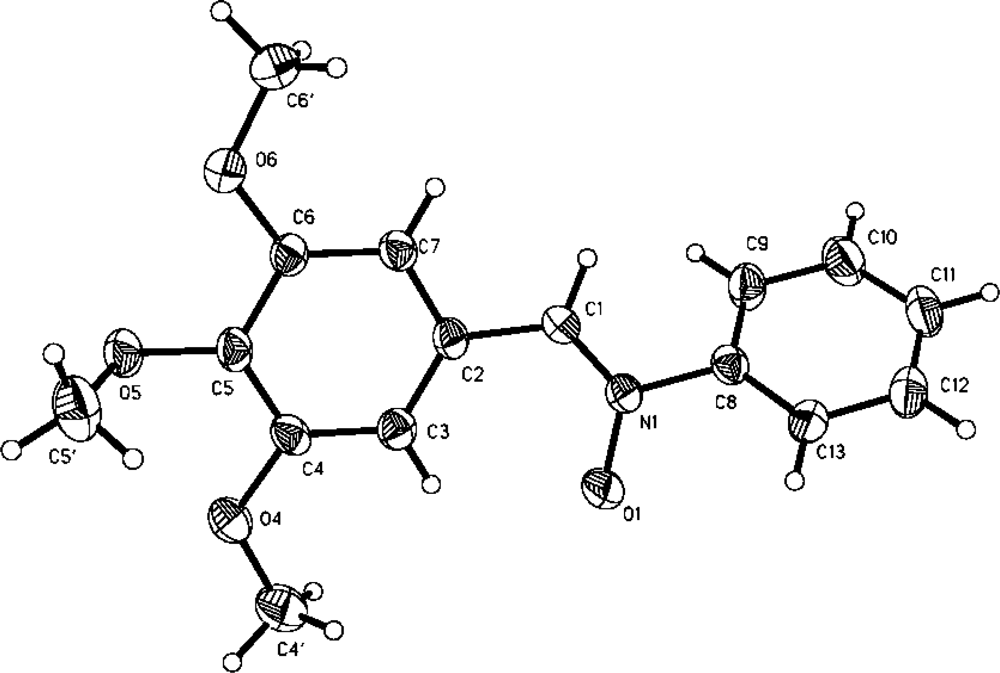
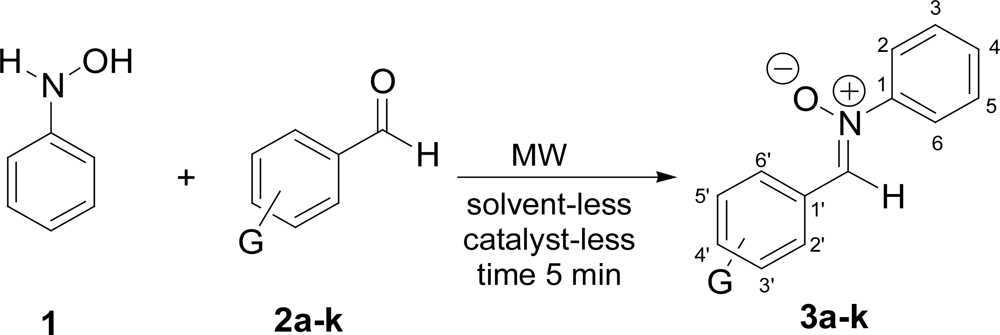
2. Results and Discussion
3. Experimental Section
4. Conclusions
Acknowledgments
References and Notes
- Nand, KS; Kamar, SS. Microwave-assisted, one-pot multicomponent synthesis of highly substituted pyridines of medicinal utility using KF/alumina. ARKIVOC 2009, XIII, 153–160. [Google Scholar]
- Torssell, KBG. Nitrile Oxides, Nitrones, and Nitronates in Organic Synthesis; VCH Publishers: New York, NY, USA, 1988. [Google Scholar]
- Janzen, EG. Spin trapping. Acc. Chem. Res 1971, 4, 31–40. [Google Scholar]
- Evans, CA. Spin trapping. Aldrichim. Acta 1979, 12, 23–29. [Google Scholar]
- Hyeon, KL; Jong, SC; Chwang, SP. Facile transformation of 3,4-disubstituted 2-azetidines to chiral 5,6-dihydro-2-pyridones. Tetrahedron Lett 2001, 42, 3483–3486. [Google Scholar]
- Martyn, F. Optically active isoxazolidines via asymmetric cycloaddition reactions of nitrones with alkenes: Applications inorganic synthesis. Tetrahedron 1997, 53, 403–425. [Google Scholar]
- Gothelf, KV; Jorgensen, KA. Asymmetric 1,3-dipolar cycloaddition reactions. Chem. Rev 1998, 98, 863–909. [Google Scholar]
- Torssell, K; Zeuthen, O. Reactions of t-butyl nitrones and trimethylsilyl nitronates. Synthesis and reactions of isoxazolidines and 2-isoxazolines. Acta Chem. Scand. Ser 1978, 32b, 118–124. [Google Scholar]
- Merino, P; Padwa, A. Nitrones and cyclic analogues. In Science of Synthesis; Thieme: Stuttgart, Germany, 2004. [Google Scholar]
- Breuer, EI; Patai, S. The Chemistry of Amino, Nitrosos and Nitro Compounds and their Derivates; Wiley: New York, NY, USA, 1982; p. 459. [Google Scholar]
- Murahashi, SI; Shiota, T; Imada, Y. Oxidation of secondary amines to nitrones: 6-methyl-2,3,4,5-tetrahydropyridine N-oxide. Org. Synth 1991, 70, 265–268. [Google Scholar]
- Keana, JF; Lee, TD. Versatile synthesis of doxyl spin labels by passing the usual ketone precursors. J. Am. Chem. Soc 1976, 97, 1273–1274. [Google Scholar]
- Ashburn, SP; Coates, RM. Generation and [3 + 2] cycloaddition reactions of oxazoline N-oxides. J. Org. Chem 1984, 49, 3127–3133. [Google Scholar]
- Ashburn, SP; Coates, RM. Preparation of oxazoline N-oxides and imidate N-oxides by amide acetal condensation and their [3 + 2] cycloaddition reactions. J. Org. Chem 1985, 50, 3076–3081. [Google Scholar]
- Soldaini, G; Cardona, F; Goti, A. Catalytic oxidation of imines based on methyltrioxorhenium/urea hydrogenperoxide: A mild and easy chemo- and regioselective entry to nitrones. Org. Lett 2007, 9, 473–476. [Google Scholar]
- Colasino, E; Nun, P; Colacino, FM; Martinéz, J; Lamaty, F. Solvent-free synthesis of nitrones in a ball-mill. Tetrahedron 2008, 64, 5569–5576. [Google Scholar]
- Andrade, M; Barros, M; Pinto, R. Exploiting microwave-assisted neat procedures: Synthesis of N-aryl and N-alkylnitrones and their cycloaddition en route for isoxazolidines. Tetrahedron 2008, 64, 10521–10530. [Google Scholar]
- Gómez, PR; Ramírez-San Juan, E; Miranda, R; Villalobos-Molina, R; Delgado, F; Osnaya, R; Trujillo, JF. Vasodilator effects of bis-dihydropyridines structurally related to nifedipine. Med. Chem 2006, 2, 527–534. [Google Scholar]
- Noguez, MO; García, A; Ibarra, C; Cabrera, A; Aceves, JM; Miranda, R. Green synthesis of bis-biginelli esters, with vasodilatory effects, their mass spectrometric and physical studies. Trends Inorg Chem, 2009; in press. [Google Scholar]
- Miranda, R. El mechero Bunsen del siglo XXI un acercamiento al protocolo de la química verde. In Aplicaciones de microondas en química y biología; El Colegio Nacional: México Distrito Federal, México, 2009; pp. 57–82. [Google Scholar]
- Ballesteros, L; Noguez, O; Arroyo, G; Velasco, B; Delgado, F; Miranda, R. Eco-friendly conditions for the production of 1,3-dithianes using microwaves irradiation. J. Mex. Chem. Soc 2005, 49, 302–306. [Google Scholar]
- Miranda, R; Noguez, O; Velasco, B; Arroyo, G; Penieres, G; Martínez, J; Delgado, F. Irradiación infrarroja una alternativa para la activación de reacciones y su contribución a la química verde. Educ. Quim 2009, 20, 421–425. [Google Scholar]
- Gilman, H; Blatt, AH. Organic Synthesis, 2nd ed; John Wiley & Sons, Inc: New York, NY, USA, 1941; pp. 445–446. [Google Scholar]
- Borch, RF; Berstein, MD; Durst, HD. The Cyanohydridoborate anion as a selective reducing agent. J. Am. Chem. Soc 1971, 93, 2897–2904. [Google Scholar]
- SHELX97-Programs for Crystal Structure Analysis, Release 97-2; Institüt für Anorganische Chemie der Universität; D-3400 Göttingen, Germany, 1998.



{kind=link}
{kind=link}
| Product | G | Melting point (°C) | Yield (%) |
|---|---|---|---|
| 3a | m-OH | 95 | 76 |
| 3b | o-OH | 147 | 85 |
| 3c | p-NO2 | 180 | 83 |
| 3d | o-NO2 | 85 | 77 |
| 3e | p-OCH3 | 108 | 86 |
| 3f | H | 130 | 81 |
| 3g | m-NO2 | 110 | 68 |
| 3h | p-N(CH3)2 | 126 | 75 |
| 3i | 3,4,5-triOCH3 | 187 | 78 |
| 3j | o-OCH3 | 115 | 80 |
| 3k | 2,3-diOH | 210 | 79 |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Reyes, L.; Corona, S.; Arroyo, G.; Delgado, F.; Miranda, R. Eco-Contribution for the Production of N-Arylnitrones: Solvent-Free and Assisted by Microwaves. Int. J. Mol. Sci. 2010, 11, 2576-2583. https://doi.org/10.3390/ijms11062576
Reyes L, Corona S, Arroyo G, Delgado F, Miranda R. Eco-Contribution for the Production of N-Arylnitrones: Solvent-Free and Assisted by Microwaves. International Journal of Molecular Sciences. 2010; 11(6):2576-2583. https://doi.org/10.3390/ijms11062576
Chicago/Turabian StyleReyes, Leonor, Sandra Corona, Gabriel Arroyo, Francisco Delgado, and René Miranda. 2010. "Eco-Contribution for the Production of N-Arylnitrones: Solvent-Free and Assisted by Microwaves" International Journal of Molecular Sciences 11, no. 6: 2576-2583. https://doi.org/10.3390/ijms11062576
APA StyleReyes, L., Corona, S., Arroyo, G., Delgado, F., & Miranda, R. (2010). Eco-Contribution for the Production of N-Arylnitrones: Solvent-Free and Assisted by Microwaves. International Journal of Molecular Sciences, 11(6), 2576-2583. https://doi.org/10.3390/ijms11062576