Evolutionary Divergence of Duplicate Copies of the Growth Hormone Gene in Suckers (Actinopterygii: Catostomidae)

Abstract
:1. Introduction
2. Results and Discussion
2.1. Sequences of Catostomid GH
2.2. Phylogenetic Analysis of GHI and GHII
2.3. Selection Tests
3. Experimental Section
3.1. DNA Extraction, PCR, and Sequencing
3.2. Sequence Alignment,Variation, and Phylogenetic Analysis
3.3. Selection Tests
4. Conclusions
Acknowledgments
References and Notes
- Ohno, S. Evolution by Gene Duplication; Springer-Verlag: New York, NY, USA, 1970. [Google Scholar]
- Lynch, M; Conery, JS. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar]
- Vogel, G. Doubled genes may explain fish diversity. Science 1998, 281, 1119–1121. [Google Scholar]
- Meyer, A; Schartl, M. Gene and genome duplications in vertebrates: The one-to-four (-to-eight in fish) rule and the evolution of novel gene functions. Curr. Opin. Cell Biol 1999, 1, 699–704. [Google Scholar]
- Meyer, A; van de Peer, Y. From 2R to 3R: Evidence for a fish-specific genome duplication (FSGD). Bioessays 2005, 27, 937–945. [Google Scholar]
- Volff, JN. Genome evolution and biodiversity in teleost fish. Heredity 2005, 94, 280–294. [Google Scholar]
- Chen, TT; Marsh, A; Shamblott, M; Chan, K-M; Tang, Y-L; Cheng, CM; Yang, B-Y. Structure and evolution of fish growth hormone and insulinlike growth factor genes. In Fish Physiology: Molecular Endocrinology of Fish; Sherwood, NM, Hew, CL, Eds.; Academic Press, Inc: San Diego, CA, USA, 1994; Volume 13, pp. 179–209. [Google Scholar]
- Yowe, DL; Epping, RJ. Cloning of the barramundi growth hormone-encoding gene: a comparative analysis of higher and lower vertebrate GH genes. Gene 1995, 162, 255–259. [Google Scholar]
- Kawauchi, H; Suzuki, K; Yamazaki, T; Moiryama, S; Nozaki, M; Yamaguchi, K; Takahashi, A; Youson, J; Sower, SA. Identification of growth hormone in the sea lamprey, an extant representative of a group of the most ancient vertebrates. Endocrinology 2002, 143, 4916–4921. [Google Scholar]
- Yuri, T; Kimball, RT; Braun, EL; Braun, MJ. Duplication of accelerated evolution and growth hormone gene in passerine birds. Mol. Biol. Evol 2008, 25, 352–361. [Google Scholar]
- Forsyth, IA; Wallis, M. Growth hormone and prolactin—Molecular and functional evolution. J. Mam. Gland Biol. Neoplasia 2002, 7, 291–312. [Google Scholar]
- Rubin, DA; Dores, DM. Cloning of a growth hormone from a primitive bony fish and its phylogenetic relationships. Gen. Comp. Endo 1994, 95, 71–83. [Google Scholar]
- Rubin, DA; Youson, JH; Marra, LE; Dores, DM. Cloning of a gar (Lepisosteus osseus) GH cDNA: trends in actinopterygian GH structure. J. Mol. Endo 1996, 16, 73–80. [Google Scholar]
- Schneider, JF; Myster, SH; Hackett, PB; Guise, KS; Faras, AJ. Molecular cloning and sequence analysis of the cDNA for northern pike (Esox lucius) growth hormone. Mol. Mar. Biol. Biotech 1992, 1, 106–112. [Google Scholar]
- Venkatesh, B; Brenner, S. Genomic structure and sequence of the pufferfish (Fugu rubripes) growth hormone-encoding gene: A comparative analysis of teleost growth hormone genes. Gene 1997, 187, 211–215. [Google Scholar]
- Clements, MD; Bart, HL, Jr; Hurley, DL. Isolation and characterization of two distinct growth hormone cDNAs from the tetraploid smallmouth buffalofish (Ictiobus bubalus). Gen. Comp. Endo 2004, 136, 411–418. [Google Scholar]
- Bernardi, G; D'Onofrio, G; Caccio, S; Bernardi, G. Molecular phylogeny of bony fishes, based on the amino acid sequence of the growth hormone. J. Mol. Evo 1993, 37, 644–649. [Google Scholar]
- Almuly, R; Cavari, B; Ferstman, H; Kolodny, O; Funkenstein, B. Genomic structure and sequence of the gilthead seabream (Sparus aurata) growth hormone-encoding gene: identification of minisatellite polymorphism in intron I. Genome 2000, 43, 836–845. [Google Scholar]
- Rubin, DA; Dores, DM. Obtaining a more resolute teleost growth hormone phylogeny by introduction of gaps in sequence alignment. Mol. Phylo. Evol 1995, 4, 129–138. [Google Scholar]
- Oakley, TH; Phillips, RB. Phylogeny of salmonine fishes based on growth hormone introns: Atlantic (Salmo) and Pacific (Oncorhynchus) salmon are not sister taxa. Mol. Phylo. Evol 1999, 11, 381–393. [Google Scholar]
- Rajesh, R; Majumdar, KC. A comparative account of the structure of the growth hormone encoding gene and genetic interrelationship in six species of the genus Labeo. Fish Physiol. Biol 2007, 33, 311–333. [Google Scholar]
- Schlee, P; Fuchs, H; Blusch, J; Werner, T; Rotmann, O; Stein, H. Genetic polymorphism in the intron of the growth hormone gene of the bleak. J. Fish Biol 1996, 48, 1275–1277. [Google Scholar]
- Yue, G; Li, Y; Orban, L. Characterization of microsatellites in the IGF-2 and GH genes of Asian seabass (Lates calcarifer). Mar. Biotech 2001, 3, 1–3. [Google Scholar]
- Mayden, RL; Chen, W-J; Bart, HL, Jr; Doosey, MH; Simons, AM; Tang, KL; Wood, RM; Agnew, MK; Yang, L; Hirt, MV; Clements, MD; Saitoh, K; Sado, T; Miya, M; Nishida, M. Reconstructing the phylogenetic relationships of the Earth’s most diverse clade of freshwater fishes—Order Cypriniformes (Actinopterygii: Ostariophysi): A case study using multiple nuclear loci and the mitochondrial genome. Mol. Phylo. Evol 2009, 51, 500–514. [Google Scholar]
- Uyeno, T; Smith, GR. Tetraploid origin of the karyotype of catostomid fishes. Science 1972, 175, 644–646. [Google Scholar]
- Chiou, C-S; Chen, H-T; Chang, W-C. The complete nucleotide sequence of the growth-hormone gene from the common carp (Cyprinus carpio). Biochim. Biophys. Acta 1990, 1087, 91–94. [Google Scholar]
- Zhu, Z; He, L; Chen, TT. Primary-structural and evolutionary analyses of the growth-hormone gene from grass carp (Ctenopharyngodon idellus). Eur. J. Biochem 1992, 207, 643–648. [Google Scholar]
- Noh, JK; Cho, KN; Nam, YK; Kim, DS; Kim, CG. Genomic organization and sequence of the mud loach (Misgurnus mizolepis) growth hormone gene: A comparative analysis of teleost growth hormone genes. Mol. Cells 1999, 9, 638–645. [Google Scholar]
- Deeds, EJ; Shakhnavich, EI. A structure-centric view of protein evolution, design, and adaptation. Adv. Enzymol. Relat. Areas Mol. Biol 2007, 75, 133–191. [Google Scholar]
- Sawada, Y. Phylogeny and zoogeography of the superfamily Cobitoidea (Cyprinoidei, Cypriniformes). Mem. Fac. Fish. Hokkaido Univ 1982, 28, 65–223. [Google Scholar]
- Siebert, DJ. Interrelationships among Families of the ORDER CYPRINIFORMES (Teleostei) Unpubl. Ph.D. Thesis, City University of New York: New York, NY, USA, 1987.
- Harris, PM; Mayden, RL. Phylogenetic relationships of major clades of Catostomidae (Teleostei: Cypriniformes) as inferred from mitochondrial SSU and LSU rDNA sequences. Mol. Phylo. Evol 2001, 20, 225–237. [Google Scholar]
- Doosey, MH; Bart, HL, Jr; Saitoh, K; Miya, M. Phylogenetic relationships of catostomid fishes (Actinopterygii: Cypriniformes) based on mitochondrial ND4/ND5 gene sequences. Mol. Phylo. Evol 2010, 54, 1028–1034. [Google Scholar]
- Etherton, TD; Bauman, DE. Biology of somatotropin in growth and lactation of domestic animals. Physiol. Rev 1998, 78, 235–241. [Google Scholar]
- Waters, MJ; Shang, CA; Behnchen, SN; Tam, SP; Li, H; Shen, B; Lobie, PE. Growth hormone as a cytokine. Clin. Exp. Pharmacol. Physiol 1999, 26, 760–764. [Google Scholar]
- Sanders, ES; Harvey, S. Growth hormone as an early embryonic growth and differentiation factor. Anat. Embryol 2004, 209, 1–9. [Google Scholar]
- Thompson, JD; Higgins, DG; Gibson, TJ. CLUSTAL W: Improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position specific gap penalties and weight matrix choice. Nucleic Acids Res 1994, 22, 4673–4680. [Google Scholar]
- Hall, TA. BioEdit: a user-friendly biological sequence alignment editor and analysis program for Windows 95/98/NT. Nucleic Acids Symp 1999, 41, 95–98. [Google Scholar]
- Stamatakis, A; Hoover, P; Rougemont, J. A rapid bootstrap algorithm for the RAxML web servers. Systemat. Biol 2008, 57, 758–771. [Google Scholar]
- Swofford, DL. PAUP* Phylogenetic Analysis Using Parsimony (*and Other Methods), 4th ed; (40b10); Sinauer Associates: Sunderland, MA, USA, 2002. [Google Scholar]
- Nei, M; Gojobori, T. Simple methods for estimating the numbers of synonymous and nonsynonymous nucleotide substitutions. Mol. Biol. Evol 1986, 3, 418–426. [Google Scholar]
- Tamura, K; Dudley, J; Nei, M; Kumar, S. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0. Mol. Biol. Evol 2007, 24, 1596–1599. [Google Scholar]
- Nei, M; Kumar, S. Molecular Evolution and Phylogenetics; Oxford University Press: New York, NY, USA, 2000. [Google Scholar]
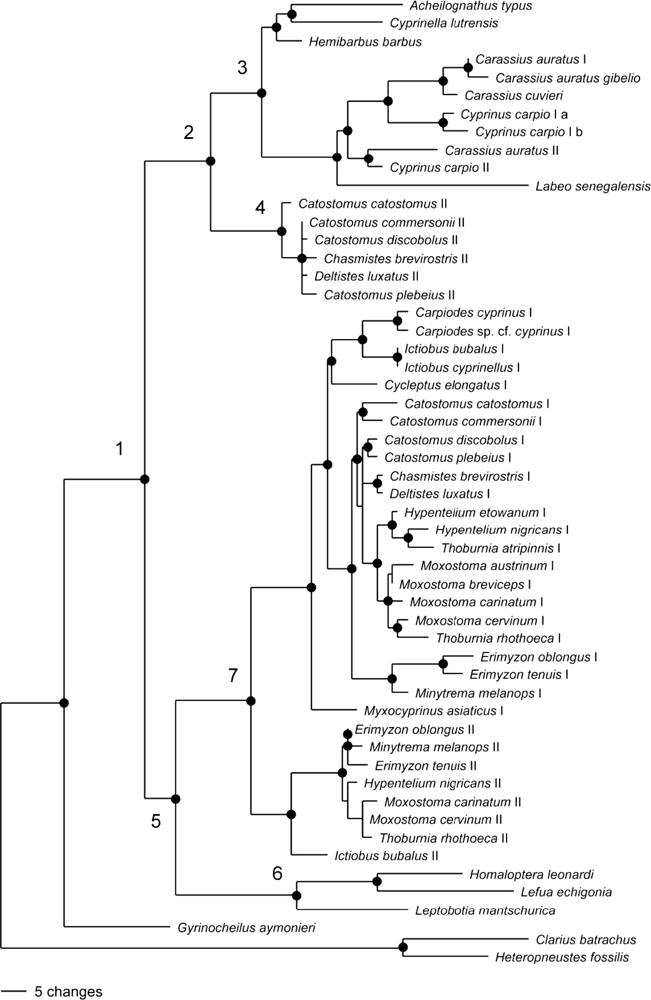

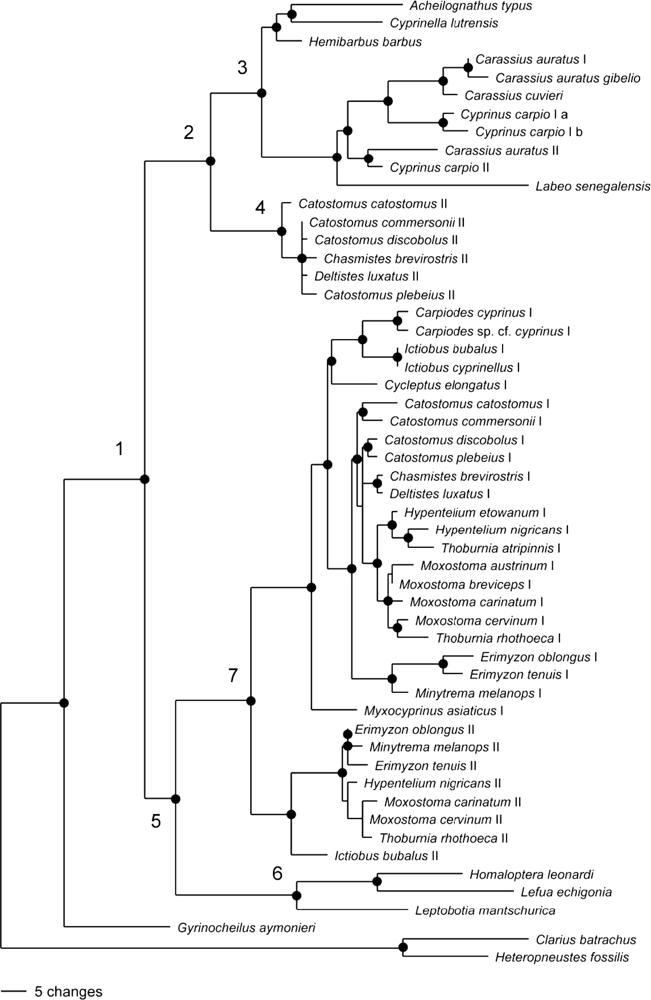{kind=link}
| Species | Copy | Source | GenBank number |
|---|---|---|---|
| Carpiodes cyprinus | I | this study | GU937834 |
| Carpiodes sp. cf. cyprinus | I | this study | GU937849 |
| Catostomus catostomus | I | this study | GU937824 |
| C. catostomus | II | this study | GU937826 |
| C. commersonii | I | Mayden et al. 2009 | FJ265027 |
| C. commersonii | II | this study | GU937823 |
| C. discobolus | I | this study | GU937830 |
| C. discobolus | II | this study | GU937832 |
| C. plebeius | I | this study | GU937833 |
| C. plebeius | II | this study | GU937829 |
| Chasmistes brevirostris | I | this study | GU937825 |
| C. brevirostris | II | this study | GU937827 |
| Cycleptus elongatus | I | Mayden et al. 2009 | FJ265028 |
| Deltistes luxatus | I | this study | GU937831 |
| D. luxatus | II | this study | GU937828 |
| Erimyzon oblongus | I | this study | GU937837 |
| E. oblongus | II | this study | GU945705 |
| E. tenuis | I | this study | GU937838 |
| E. tenuis | II | this study | GU937839 |
| Hypentelium etowanum | I | this study | GU937836 |
| H. nigricans | I | Mayden et al. 2009 | FJ265055 |
| Ictiobus bubalus | I | Clements et al. 2004 | AY375301 |
| I. bubalus | II | Clements et al. 2004 | AY375302 |
| I. cyprinellus | I | this study | GU937840 |
| Minytrema melanops | I | Mayden et al. 2009 | FJ265050 |
| M. melanops | II | this study | GU937822 |
| Moxostoma austrinum | I | this study | GU937841 |
| M. breviceps | I | this study | GU937842 |
| M. carinatum | I | this study | GU937843 |
| M. carinatum | II | this study | GU937835 |
| M. cervinum | I | this study | GU937844 |
| M. cervinum | II | this study | GU937845 |
| Myxocyprinus asiaticus | I | Mayden et al. 2009 | FJ265052 |
| Thoburnia atripinnis | I | this study | GU937846 |
| T. rhothoeca | I | this study | GU937847 |
| T. rhothoeca | II | this study | GU937848 |
| Acheilognathus typus | Mayden et al. 2009 | FJ265056 | |
| Carassius auratus | I | Law et al. 1996 | AF069398 |
| C. auratus | II | Law et al. 1996 | AF069399 |
| C. a. gibelio | unpublished | AY265352 | |
| Clarius batrachus | unpublished | AF416485 | |
| Cyprinella lutrensis | Mayden et al. 2009 | FJ265061 | |
| Cyprinus carpio | I a | Mayden et al. 2009 | FJ265047 |
| C. carpio | I b | unpublished | AJ640136 |
| C. carpio | II | unpublished | AJ640135 |
| Gyrinocheilus aymonieri | Mayden et al. 2009 | FJ265031 | |
| Hemibarbus barbus | Mayden et al. 2009 | FJ265032 | |
| Heteropneustus fossilis | unpublished | AF416489 | |
| Homaloptera leonardi | Mayden et al. 2009 | FJ265022 | |
| Labeo senegalensis | Mayden et al. 2009 | FJ265034 | |
| Lefua echigonia | Mayden et al. 2009 | FJ265023 | |
| Leptobotia mantschurica | Mayden et al. 2009 | FJ265035 |
| Species | Copy | Voucher | 5′ UTR | Intron 1 | Intron 2 | Intron 3 | Intron 4 | 3′ UTR | Gene | CDS |
|---|---|---|---|---|---|---|---|---|---|---|
| Carpiodes cyprinus | I | None | na | na | na | na | 607 | |||
| Carpiodes sp. cf. cyprinus | I | None | 207 | 985 | 154 | 1878 | 532 | |||
| Catostomus catostomus | I | UAIC 11218.05 | 228 | 194 | 949 | 154 | 2117 | 550 | ||
| C. catostomus | II | UAIC 11218.05 | 39 | 174 | 194 | 311 | 145 | 1470 | 607 | |
| C. commersonii | I | None | 47 | 235 | 175 | 592 | 102 | 1788 | 633 | |
| C. commersonii | II | None | 31 | 222 | 194 | 311 | 145 | 1513 | 610 | |
| C. discobolus | I | BYU 57986 | 36 | 235 | 198 | 599 | 102 | 1711 | 541 | |
| C. discobolus | II | BYU 57986 | 48 | 220 | 194 | 311 | 142 | 1531 | 613 | |
| C. plebeius | I | MSB 49632 | 235 | 198 | 599 | 102 | 1683 | 542 | ||
| C. plebeius | II | MSB 49632 | 220 | 194 | 311 | 145 | 1512 | 603 | ||
| Chasmistes brevirostris | I | OS 15963 | 35 | 235 | 198 | 596 | 102 | 2037 | 633 | |
| C. brevirostris | II | OS 15963 | 210 | 194 | 311 | 145 | 1492 | 593 | ||
| Cycleptus elongates | I | TU 192331 | 242 | 203 | 965 | 143 | 2125 | 571 | ||
| Deltistes luxatus | I | OS 15922 | 35 | 236 | 198 | 596 | 102 | 1925 | 633 | |
| D. luxatus | II | OS 15922 | 210 | 194 | 311 | 145 | 1517 | 611 | ||
| Erimyzon oblongus | I | NCSM 37439 | 185 | 955 | 154 | 1845 | 543 | |||
| E. oblongus | II | NCSM 37439 | 557 | 227 | 199 | 1157 | 150 | |||
| E. tenuis | I | None | 259 | 259 | 192 | 947 | 154 | 2395 | 633 | |
| E. tenuis | II | None | 139 | 225 | 199 | 826 | 263 | |||
| Hypentelium etowanum | I | None | 176 | 248 | 197 | 832 | 146 | 2293 | 633 | |
| H.nigricans | I | None | 147 | 252 | 197 | 901 | 146 | 2212 | 569 | |
| H. nigricans | II | None | 639 | 226 | 181 | 1287 | 225 | |||
| Ictiobus bubalus | I | TU 196158 | 56 | Na | na | na | na | 590 | 633 | |
| I.bubalus | II | TU 196158 | 56 | Na | na | na | na | 590 | 633 | |
| I. cyprinellus | I | None | 204 | 870 | 154 | 1787 | 559 | |||
| Minytrema melanops | I | TU 193988 | 147 | 254 | 193 | 1188 | 154 | 2569 | 633 | |
| M. melanops | II | TU 193988 | 627 | 225 | 182 | 320a | 154 | 2141a | 633 | |
| Moxostoma austrinus | I | None | 261 | 200 | 914 | 143 | 2074 | 548 | ||
| M. breviceps | I | None | 38 | 225 | 200 | 936 | 143 | 2084 | 542 | |
| M. carinatum | I | None | 211 | 259 | 200 | 936 | 143 | 2392 | 633 | |
| M. carinatum | II | None | 225 | 179 | 150 | |||||
| M. cervinum | I | None | 263 | 200 | 928 | 143 | 2092 | 542 | ||
| M. cervinum | II | None | 618 | 224 | 181 | 267 | ||||
| Myxocyprinus asiaticus | I | None | 31 | 269 | 215 | 969 | 154 | 2180 | 541 | |
| Thoburnia atripinnis | I | None | 211 | 252 | 195 | 936 | 152 | 2442 | 633 | |
| T. rhothoeca | I | None | 208 | 252 | 199 | 914 | 146 | 64 | 2416 | 633 |
| T. rhothoeca | II | None | 638 | 224 | 154 | 267 | ||||
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bart, H.L., Jr.; Reneau, P.C.; Doosey, M.H.; Bell, C.D. Evolutionary Divergence of Duplicate Copies of the Growth Hormone Gene in Suckers (Actinopterygii: Catostomidae). Int. J. Mol. Sci. 2010, 11, 1090-1102. https://doi.org/10.3390/ijms11031090
Bart HL Jr., Reneau PC, Doosey MH, Bell CD. Evolutionary Divergence of Duplicate Copies of the Growth Hormone Gene in Suckers (Actinopterygii: Catostomidae). International Journal of Molecular Sciences. 2010; 11(3):1090-1102. https://doi.org/10.3390/ijms11031090
Chicago/Turabian StyleBart, Henry L., Jr., Paulette C. Reneau, Michael H. Doosey, and Charles D. Bell. 2010. "Evolutionary Divergence of Duplicate Copies of the Growth Hormone Gene in Suckers (Actinopterygii: Catostomidae)" International Journal of Molecular Sciences 11, no. 3: 1090-1102. https://doi.org/10.3390/ijms11031090
APA StyleBart, H. L., Jr., Reneau, P. C., Doosey, M. H., & Bell, C. D. (2010). Evolutionary Divergence of Duplicate Copies of the Growth Hormone Gene in Suckers (Actinopterygii: Catostomidae). International Journal of Molecular Sciences, 11(3), 1090-1102. https://doi.org/10.3390/ijms11031090