Lysophosphatidic Acid Level and the Incidence of Silent Brain Infarction in Patients with Nonvalvular Atrial Fibrillation

Abstract
:1. Introduction
2. Results and Discussion
2.1. Results
2.1.1. Baseline Characteristics of the Study Groups
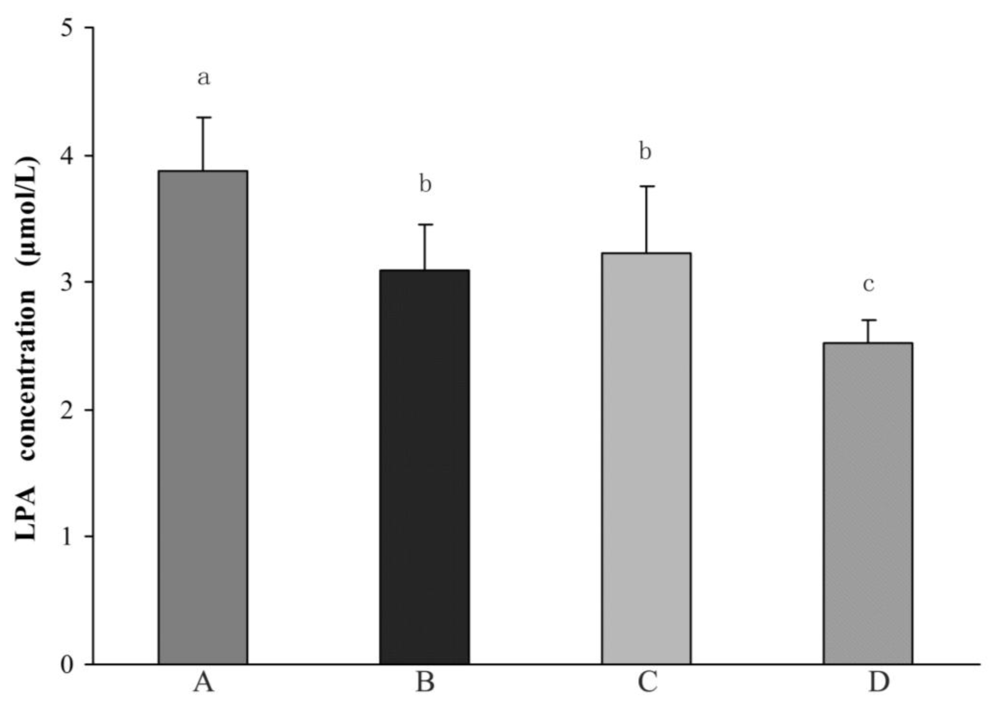
2.1.2. Comparison of Plasma LPA Levels between Different Groups
2.1.3. Incidence of NVAF with SBI
2.1.4. Comparison in the Positive Rate of Platelet Activation
2.2. Discussion
3. Materials and Methods
3.1. Study Subjects
3.2. Sample Collection and LPA Analysis
3.3. Estimation of Platelet Activation Degree
3.4. Statistical Analysis
4. Conclusions
Acknowledgements
References
- Kopecky, SL; Gersh, BJ; McGoon, MD; Whisnant, JP; Holmes, DR, Jr; Ilstrup, DM; Frye, RL. The natural history of lone atrial fibrillation: a population-based study over three decades. N. Engl. J. Med 1987, 7, 669–674. [Google Scholar]
- Ma, CS; Du, X; Jiang, CX. Atrial fibrillation in China: a brief review. Chin. Med. J. (Engl.) 2009, 122, 2803–2806. [Google Scholar]
- Hart, RG; Pearce, LA. Current status of stroke risk stratification in patients with atrial fibrillation. Stroke 2009, 40, 2607–2610. [Google Scholar]
- Ahmad, O; Ahmad, KE; Dear, KB; Harvey, I; Hughes, A; Lueck, CJ. Atrial fibrillation and anticoagulation in a stroke unit population. Intern. Med. J 2009, 39, 752–756. [Google Scholar]
- Kamath, S; Blann, AD; Lip, GY. Platelets and atrial fibrillation. Eur. Heart J 2001, 22, 2233–2242. [Google Scholar]
- Pamuklar, Z; Federico, L; Liu, S; Umezu-Goto, M; Dong, A; Panchatcharam, M; Fulerson, Z; Berdyshev, E; Natarajan, V; Fang, X; van Meeteren, LA; Moolenaar, WH; Mills, GB; Morris, AJ; Smyth, SS. Autotaxin/Lysopholipase D and Lysophosphatidic acid regulate murine hemostasis and thrombosis. J. Biol. Chem 2009, 284, 7385–7394. [Google Scholar]
- Smyth, SS; Cheng, HY; Miriyala, S; Panchatcharam, M; Morris, AJ. Roles of lysophosphatidic acid in cardiovascular physiology and disease. Biochim. Biophys. Acta 2008, 1781, 563–570. [Google Scholar]
- Eichholtz, T; Jalink, K; Fahrenfort, I; Moolenaar, WH. The bioactive phospholipid lysophosphatidic acid is released from activated platelets. Biochem. J 1993, 291, 677–680. [Google Scholar]
- Rother, E; Brandl, R; Baker, DL; Goyal, P; Gebhard, H; Tigyi, G; Siess, W. Subtype-selective antagonists of lysophosphatidic acid receptors inhibit platelet activation triggered by the lipid core of atherosclerotic plaques. Circulation 2003, 108, 741–747. [Google Scholar]
- Siess, W; Zangl, KJ; Essler, M; Bauer, M; Brandl, R; Corrinth, C; Bittman, R; Tigyi, G; Aepfelbacher, M. Lysophosphatidic acid mediates the rapid activation of platelets and endothelial cells by mildly oxidized low density lipoprotein and accumulates in human atherosclerotic lesions. Proc. Natl. Acad. Sci. USA 1999, 96, 6931–6936. [Google Scholar]
- Aoki, J; Taira, A; Takanezawa, Y; Kishi, Y; Hama, K; Kishimoto, T; Mizuno, K; Saku, K; Taguchi, R; Arai, H. Serum lysophosphatidic acid is produced through diverse phospholipase pathways. J. Biol. Chem 2002, 277, 48737–48744. [Google Scholar]
- Boucharaba, A; Serre, CM; Gres, S; Saulnier-Blache, JS; Bordet, JC; Guglielmi, J; Clezardin, P; Peyruchaud, O. Platelet-derived lysophosphatidic acid supports the progression of osteolytic bone metastases in breast cancer. J. Clin. Invest 2004, 114, 1714–1725. [Google Scholar]
- Vermeer, SE; Longstreth, WT, Jr; Koudstaal, PJ. Silent brain infarcts: a systematic review. Lancet Neurol 2007, 6, 611–619. [Google Scholar]
- Matsui, T; Arai, H; Yuzuriha, T; Yao, H; Miura, M; Hashimoto, S; Higuchi, S; Matsushita, S; Morikawa, M; Kato, A; Sasaki, H. Elevated plasma homocysteine levels and risk of silent brain infarction in elderly people. Stroke 2001, 32, 1116–1119. [Google Scholar]
- Uehara, T; Tabuchi, M; Mori, E. Risk factors for silent cerebral infarsts in subcortical white matter and basal ganglia. Stroke 1999, 30, 378–382. [Google Scholar]
- Lee, SC; Park, SJ; Ki, HK; Gwon, HC; Chung, CS; Byun, HS; Shin, KJ; Shin, MH; Lee, WR. Prevalence and risk factors of silent cerebral infarction in apparently normal adults. Hypertension 2000, 36, 73–77. [Google Scholar]
- Adachi, T; Kobayashi, S; Yamaguchi, S; Okada, K. MRI findings of small subcortical ‘lacunar-like’. infarction resulting from large vessel disease. J. Neurol 2000, 247, 280–285. [Google Scholar]
- Narumiya, T; Sakamaki, T; Sato, Y; Kanmatsuse, K. Relationship between left atrial appendage function and left atrial thrombus in patients with nonvalvular chronic atrial fibrillation and atrial flutter. Circ. J 2003, 67, 68–72. [Google Scholar]
- Kobayasi, S; Okada, K; Koide, H; Bokua, H; Yamaguti, S. Subcortical silent brain infarction as a risk factor for clinical stroke. Stroke 1997, 28, 1932–1939. [Google Scholar]
- Coull, BM; Malinow, MR; Beamer, N; Sexton, G; Nordt, F; de Garmo, P. Elevated plasma homocyst(e)ine concentration as a possible independent risk factor for stroke. Stroke 1990, 21, 572–576. [Google Scholar]
- Giles, WH; Croft, JB; Greenlund, KJ; Ford, ES; Kittner, SJ. Total homocysteine concentration and the likelihood of nonfatal stroke: Results from the Third National Health and Nutrition Examination Survey, 1988–1994. Stroke 1998, 29, 2473–2477. [Google Scholar]
- Boushey, CJ; Beresford, SA; Omenn, GS; Motulsky, AG. A quantitative assessment of plasma homocysteine as a risk factor for vascular disease. Probable benefits of increasing folic acid intakes. JAMA 1995, 274, 1049–1057. [Google Scholar]
- Moller, J; Nielsen, GM; Tvedegaard, KC; Andersen, NT; Jorgensen, PE. A meta analysis of cerebrovascular disease and hyperhomocystinemia. Scand. J. Clin. Lab. Invest 2000, 60, 491–499. [Google Scholar]
- Hankey, GJ; Eikelboom, JW. Homocysteine and stroke. Curr. Opin. Neurol 2001, 14, 95–102. [Google Scholar]
- Homocysteine Studies Collaboration. Homocysteine and risk of ischemic heart disease and stroke: a meta-analysis. JAMA 2002, 288, 2015–2022.
- Wald, DS; Law, M; Morris, JK. Homocysteine and cardiovascular disease: evidence on causality from a meta-analysis. BMJ 2002, 325, 1202–1206. [Google Scholar]
- Anan, F; Takahashi, N; Shimomura, T; Imagawa, M; Yufu, K; Nawata, T; Nakagawa, M; Yonemochi, H; Eshima, N; Saikawa, T; Yoshimatsu, H. Hyperhomocysteinemia is a significant risk factor for silent cerebral infarction in patients with chronic renal failure undergoing hemodialysis. Metabolism 2006, 55, 656–661. [Google Scholar]
- Kamath, S; Blann, AD; Chin, BS; Lanza, F; Aleil, B; Cazenave, JP; Lip, GY. A study of platelet activation in atrial fibrillation and the effects of antithrombotic therapy. Eur. Heart J 2002, 23, 1788–1795. [Google Scholar]
- Shinkawa, A; Ueda, K; Kiyohara, Y; Kato, I; Sueishi, K; Tsuneyoshi, M; Fujishima, M. Silent cerebral infarction in a community-based autopsy series in Japan. The Hisayama Study. Stroke 1995, 26, 380–385. [Google Scholar]
- Price, TR; Manolio, TA; Kronmal, RA; Kittner, SJ; Yue, NC; Robbins, J; Anton-Culver, H; Oleary, DH. Silent brain infarction on magnetic resonance imaging and neurological abnormalities in community dwelling older adults: the Cardiovascular Health Study. CHS collaborative research group. Stroke 1997, 28, 1158–1164. [Google Scholar]
- Gartner, W; Zierhut, B; Mineva, I; Sodeck, G; Leutmezer, F; Domanovits, H; Prayer, D; Wolf, F; Base, W; Weissel, M; Wagner, L. Brain natriuretic peptide correlates with the extent of atrial fibrillation-associated silent brain lesions. Clin. Biochem 2008, 41, 1434–1439. [Google Scholar]
- Minamino, T; Kitakaze, M; Sato, H; Asanuma, H; Funaya, H; Koretsune, Y; Hori, M. Plasma levels of nitrite/nitrate and platelet cGMP levels are decreased in patients with atrial fibrillation. Arterioscler. Thromb. Vasc. Biol 1997, 17, 3191–3195. [Google Scholar]
- Minamino, T; Kitakaze, M; Sanada, S; Asanuama, H; Kurotobi, T; Koretsune, Y; Fukunami, M; Kuzuya, T; Hoki, N; Hori, M. Increased expression of P-selectin on platelets is a risk factor for silent cerebral infarction in patients with atrial fibrillation: role of nitric oxide. Circulation 1998, 98, 1721–1727. [Google Scholar]
- Noris, M; Morigi, M; Donadelli, R; Aiello, S; Foppolo, M; Todeschini, M; Orisio, S; Remuzzi, G; Remuzzi, A. Nitric oxide synthesis by cultured endothelial cell is modulated by flow condition. Circ. Res 1995, 76, 536–543. [Google Scholar]
- Topper, J; Cai, J; Falb, D; Gimborne, M. Identification of vascular endothelial genes differentially responsive to fluid mechanical stimuli: cyclooxygenase-2, manganese superoxide dismutase, and endothelial cell nitric oxide synthase are selectively up-regulated by steady laminar shear stress. Proc. Natl. Acad. Sci. USA 1996, 93, 10417–10422. [Google Scholar]
- Uehara, T; Tabuchi, M; Mori, E. Risk factors for silent cerebral infarsts in subcortical white matter and basal ganglia. Stroke 1999, 30, 378–382. [Google Scholar]
- Lindgren, A; Roijer, A; Rudling, O; Norrving, B; Larsson, EM; Eskilsson, J; Wallin, L; Olsson, B; Johansson, BB. Cerebral lesions on magnetic resonance imaging, heart disease, vascular risk factors in subjects without stroke: a population-based study. Stroke 1994, 25, 929–934. [Google Scholar]
- Ylikoski, A; Erkinjuntti, T; Raininko, R; Sarna, S; Sulkava, R; Tilvis, R. White matter hyperintensities on MRI in the neurologically nondiseased elderly. Analysis of cohorts of consecutive subjects aged 55 to 85 years living at home. Stroke 1995, 26, 1171–1177. [Google Scholar]
- Longstreth, WT, Jr; Manolio, TA; Arnold, A; Burke, GL; Bryan, N; Jungreis, CA; Enright, PL; OLeary, D; Fried, L. Clinical correlates of white matter findings on cranial magnetic resonance imaging of 3301 elderly people: the cardiovascular health study. Stroke 1996, 27, 1274–1282. [Google Scholar]
- Hoshi, T; Kitagawa, K; Yamagami, H; Furukado, S; Hougaku, H; Hori, M. Relations of serum high-sensitivity C-reactive protein and interleukin-6 levels with silent brain infarction. Stroke 2005, 36, 768–772. [Google Scholar]
- Baker, DL; Desiderio, DM; Miller, DD; Tolley, B; Tigyi, GJ. Direct quantitative analysis of lysophosphatidic acid molecular species by stable isotope dilution electrospray ionization liquid chromatography-mass spectrometry. Anal. Biochem 2001, 292, 287–295. [Google Scholar]
- Li, ZG; Yu, ZC; Wang, DZ; Ju, WP; Zhan, X; Wu, QZ; Wu, XJ; Cong, HM; Man, HH. Influence of acetylsalicylate on plasma lysophosphatidic acid level in patients with ischemic cerebral vascular diseases. Neurol. Res 2008, 30, 366–369. [Google Scholar]


{kind=link}
| Variable | NVAF + SBI (+) (n = 74) | NVAF + SBI (−) (n = 161) | SBI + NVAF (−) (n = 116) | Control (n = 120) | p |
|---|---|---|---|---|---|
| Age, years (mean + SD) | 64.6 ± 6.5 | 65.6 ± 7.9 | 62.5 ± 7.6 | 66.9 ± 5.8 | ns |
| Male | 39 (52.7%) | 83 (51.6%) | 65 (56.0%) | 64 (53.3%) | ns |
| Hypertension | 38 (51.4%) | 78 (48.4%) | 59 (50.9%) | 51 (42.5%) | ns |
| Diabetes mellitus | 17 (23.0%) | 36 (22.4%) | 25 (21.6%) | 22 (18.3%) | ns |
| Dyslipidemia | 20 (27.0%) | 40 (24.8%) | 28 (24.1%) | 26 (21.7%) | ns |
| Smoking | 13 (17.6%) | 27 (16.7%) | 20 (17.2%) | 18 (15.0%) | ns |
| Platelet count (×109) | 197.1 ± 38.7 | 180.3 ± 33.6 | 203.2 ± 40.5 | 190.4 ± 38.8 | ns |
| Variables | Cut off Values |
|---|---|
| Respiratory, PaO2/FiO2 (mmHg) | ≤300 |
| Coagulation, platelets ×103/μmol | ≤150 |
| Liver, bilirubin (mg/dL)a | ≥2.0 |
| Cardiovascular, hypotension | Mean arterial pressure <70 mmHg |
| Renal creatinine (mg/dL)b or | >2.0 or |
| Urine output (mL/d) | <500 mL |
| Glomerular filtration rate (GFR) | ≤70 mL/min |
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Li, Z.-G.; Yu, Z.-C.; Yu, Y.-P.; Ju, W.-P.; Wang, D.-Z.; Zhan, X.; Wu, X.-J.; Zhou, L. Lysophosphatidic Acid Level and the Incidence of Silent Brain Infarction in Patients with Nonvalvular Atrial Fibrillation. Int. J. Mol. Sci. 2010, 11, 3988-3998. https://doi.org/10.3390/ijms11103988
Li Z-G, Yu Z-C, Yu Y-P, Ju W-P, Wang D-Z, Zhan X, Wu X-J, Zhou L. Lysophosphatidic Acid Level and the Incidence of Silent Brain Infarction in Patients with Nonvalvular Atrial Fibrillation. International Journal of Molecular Sciences. 2010; 11(10):3988-3998. https://doi.org/10.3390/ijms11103988
Chicago/Turabian StyleLi, Zhen-Guang, Zhan-Cai Yu, Yong-Peng Yu, Wei-Ping Ju, Dao-Zhen Wang, Xia Zhan, Xi-Juan Wu, and Li Zhou. 2010. "Lysophosphatidic Acid Level and the Incidence of Silent Brain Infarction in Patients with Nonvalvular Atrial Fibrillation" International Journal of Molecular Sciences 11, no. 10: 3988-3998. https://doi.org/10.3390/ijms11103988
APA StyleLi, Z.-G., Yu, Z.-C., Yu, Y.-P., Ju, W.-P., Wang, D.-Z., Zhan, X., Wu, X.-J., & Zhou, L. (2010). Lysophosphatidic Acid Level and the Incidence of Silent Brain Infarction in Patients with Nonvalvular Atrial Fibrillation. International Journal of Molecular Sciences, 11(10), 3988-3998. https://doi.org/10.3390/ijms11103988