The Reversal of Drug-Resistance in Tumors Using a Drug-Carrying Nanoparticular System


Abstract
:1. Introduction
2. Drug Resistance in Tumors
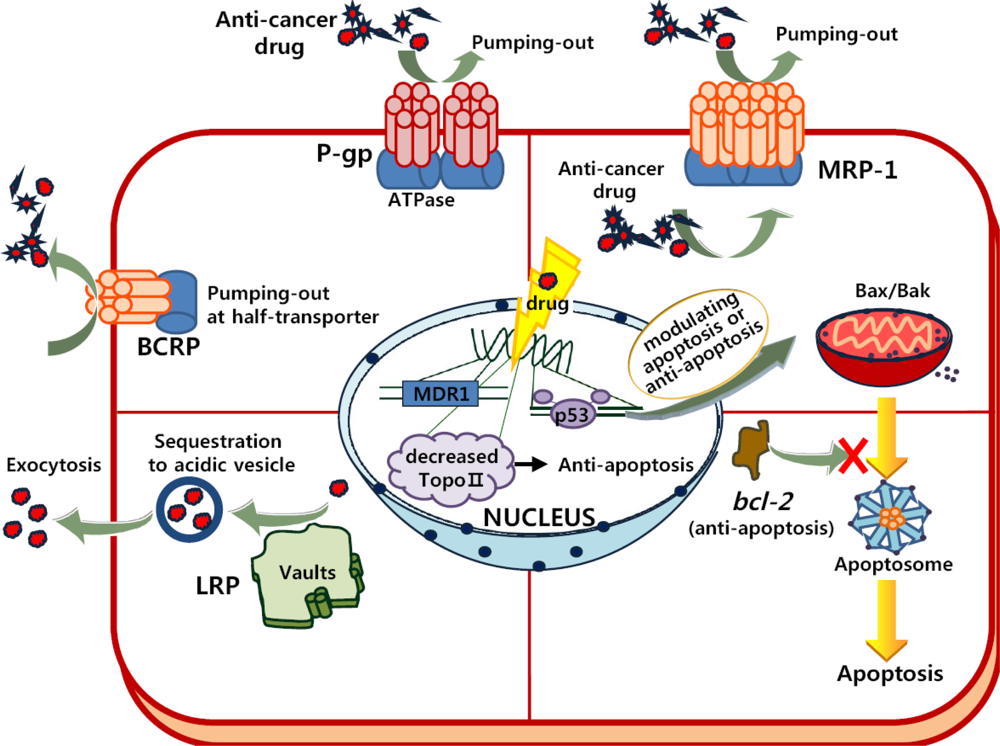
2.1. Multidrug Resistance (MDR) in Cells
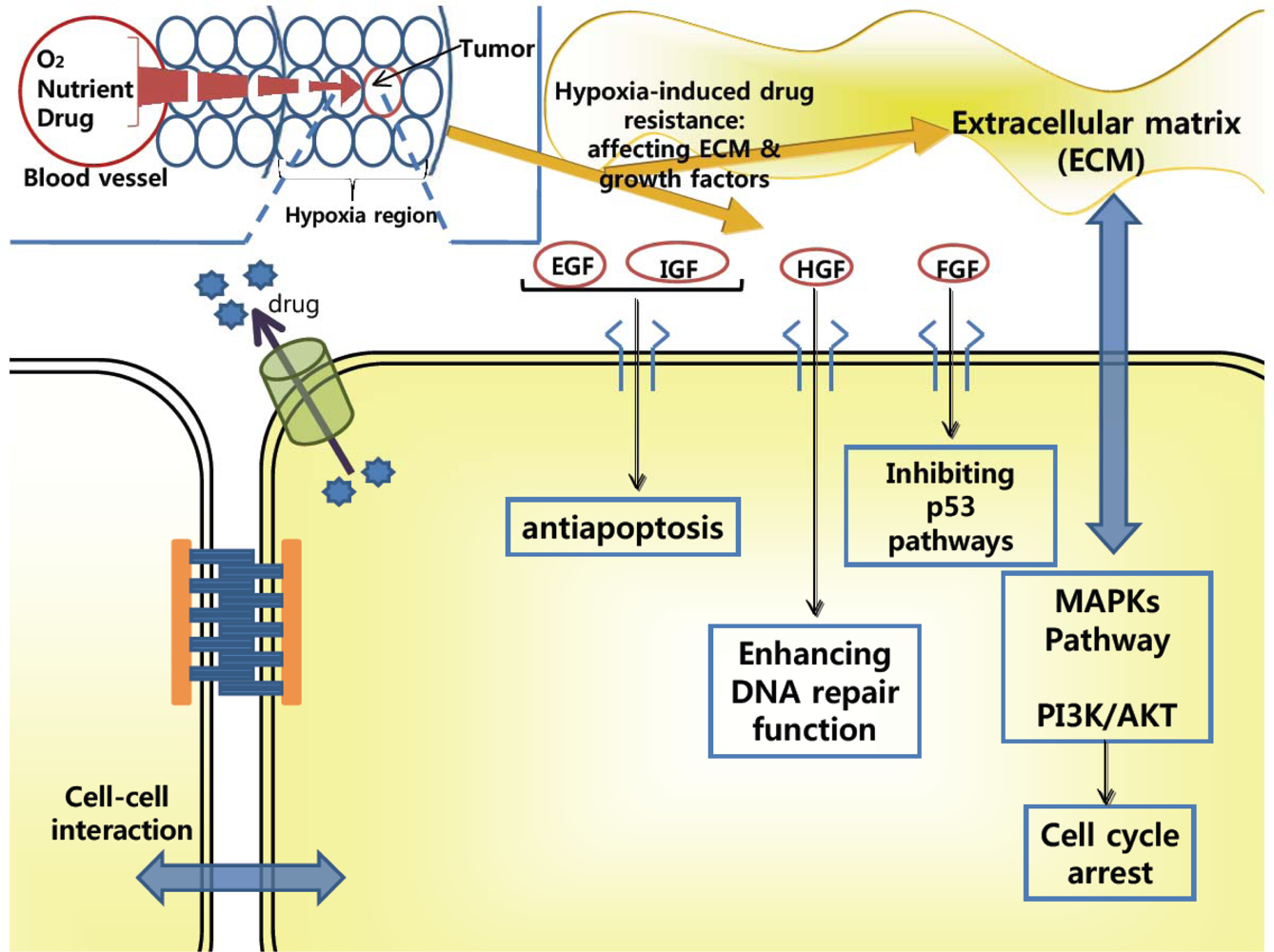
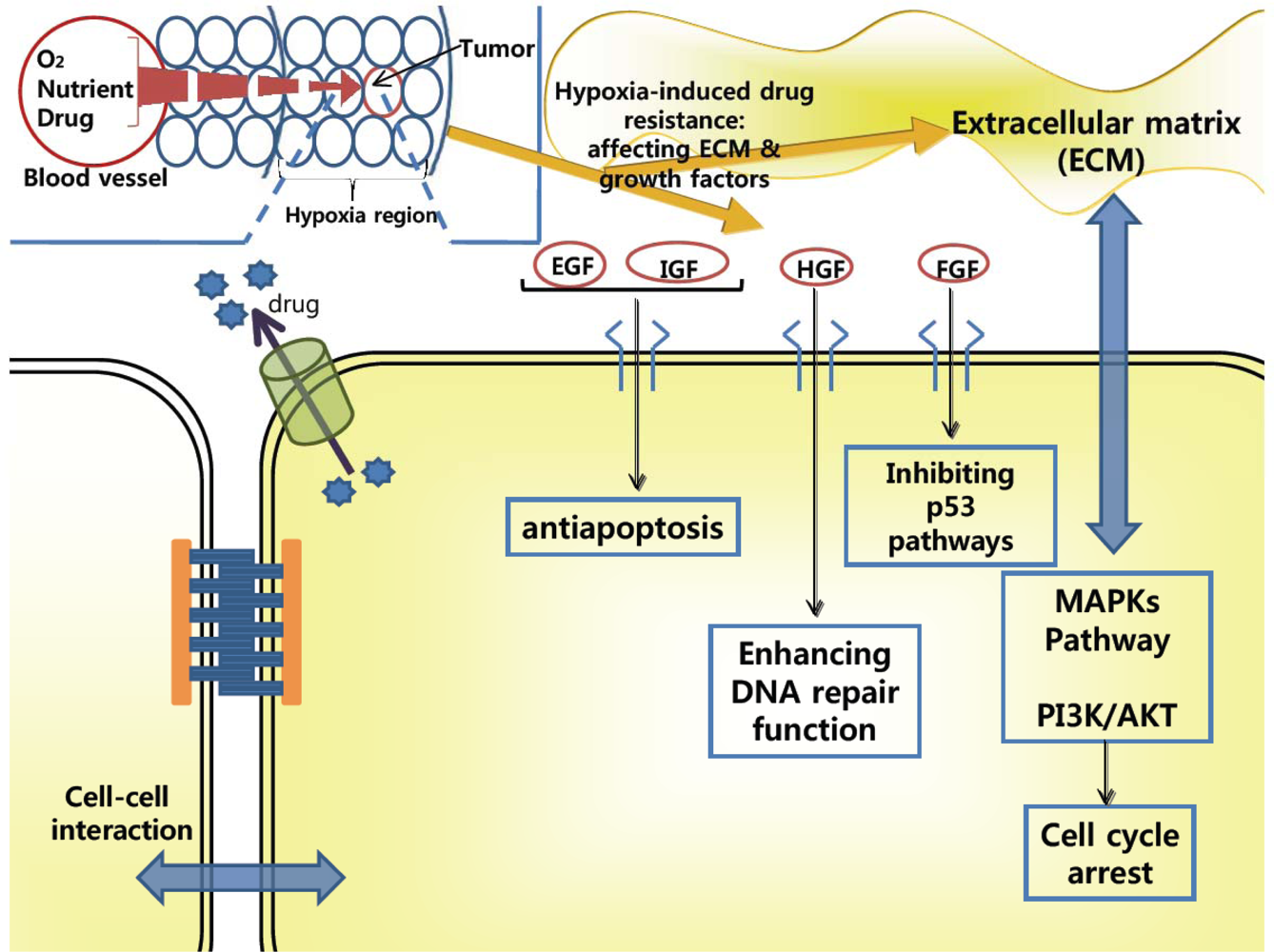
2.2. Drug-Resistance in Microenvironment of Tumors
3. Overcoming Drug-Resistance
3.1. Using P-gp Modulators
3.2. Using Nanoparticular Systems
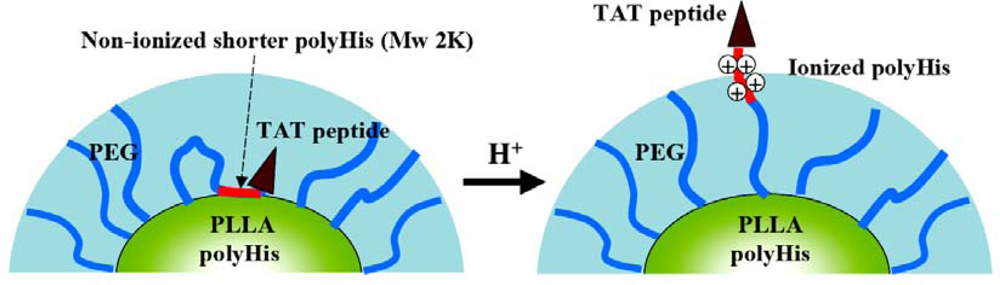
3.3. Smart Nanoparticular Systems
4. Conclusions
Acknowledgments
References and Note
- Duncan, R. Polymer conjugates as anticancer nanomedicines. Nat. Rev. Cancer 2006, 6, 688–701. [Google Scholar]
- Berger, W; Elbling, L; Micksche, M. Chemoresistance of human malignant melanoma: Cellular and molecular aspects. Onkologie 1998, 21, 105–110. [Google Scholar]
- Minko, T; Dharap, SS; Pakunlu, RI; Wang, Y. Molecular targeting of drug delivery systems to cancer. Curr. Drug Targets 2004, 5, 389–406. [Google Scholar]
- Crawford, J. Clinical uses of pegylated pharmaceuticals in oncology. Cancer Treat Rev 2002, 28, 7–11. [Google Scholar]
- Kim, R. Recent advances in understanding the cell death pathways activated by anticancer therapy. Cancer 2005, 103, 1551–1560. [Google Scholar]
- Wardwell, NR; Massion, PP. Novel strategies for the early detection and prevention of lung cancer. Semin. Oncol 2005, 32, 259–268. [Google Scholar]
- Gottesman, MM; Fojo, T; Bates, SE. Multidrug resistance in cancer: Role of ATP-dependent transports. Nat. Rev. Cancer 2000, 2, 48–58. [Google Scholar]
- Thomasand, H; Coley, HM. Overcoming multidrug resistance in cancer: An update on the clinical strategy of inhibiting p-glycoprotein. Cancer Control 2003, 10, 159–165. [Google Scholar]
- Choi, CH. ABC transporters as multidrug resistance mechanisms and the development of chemosensitizers for their reversal. Cancer Cell Int 2005, 5, 30–42. [Google Scholar]
- Ferrari, M. Cancer nanotechnology: Opportunities and challenges. Nat. Rev. Cancer 2005, 5, 161–171. [Google Scholar]
- Ferrari, M; Barker, A; Downing, G. A cancer nanotechnology strategy. NanoBioTechnology 2005, 1, 129–131. [Google Scholar]
- Gillies, ER; Frechet, JM. pH-Responsive copolymer assemblies for controlled release of doxorubicin. Bioconjug. Chem 2005, 16, 361–368. [Google Scholar]
- Heath, JR; Davis, ME. Nanotechnology and cancer. Annu. Rev. Med 2008, 59, 251–265. [Google Scholar]
- Ozben, T. Mechanisms and strategies to overcome multiple drug resistance in cancer. FEBS Lett 2006, 580, 2903–2909. [Google Scholar]
- Simon, S; Schindler, M. Cell biological mechanisms of multidrug resistance in tumors. Proc. Natl. Acad. Sci. USA 1994, 91, 3497–3504. [Google Scholar]
- Gottesman, MM. Mechanisms of cancer drug resistance. Ann. Rev. Med 2002, 53, 615–627. [Google Scholar]
- Leith, CP; Kopecky, KJ; Chen, IM; Eijdems, L; Slovak, ML; McConnell, TS; Head, DR; Weick, J; Grever, MR; Appelbaum, FR; Willman, CL. Frequency and clinical significance of the expression of the multidrug resistance proteins MDR1/P-glycoprotein, MRP1, and LRP in acute myeloid leukemia. A southwest oncology group study. Blood 1999, 94, 1086–1099. [Google Scholar]
- van der Zee, AG; Hollema, H; Suurmeijer, AJ; Krans, M; Sluiter, WJ; Willemse, PH; Aalders, JG; de Vries, EG. Value of P-glycoprotein, glutathione S-transferase pi, c-erbB-2, and p53 as prognostic factors in ovarian carcinomas. J. Clin. Oncol 1995, 13, 70–78. [Google Scholar]
- Dean, M; Rzhetsky, A; Allikmets, R. The human ATP-binding cassette (ABC) transporter superfamily. Genome Res 2001, 11, 1156–1166. [Google Scholar]
- Leonard, GD; Fojo, T; Bates, SE. The role of ABC transporters in clinical practice. Oncologist 2003, 8, 411–424. [Google Scholar]
- Fardel, O; Lecureur, V; Guillouzo, A. The P-glycoprotein multidrug transporter. Gen. Pharmac 1996, 27, 1283–1291. [Google Scholar]
- Borst, P; Evers, R; Kool, M; Wijnholds, J. A Family of drug transporters: The multidrug resistance-associated proteins. J. Natl. Cancer Inst 2000, 92, 1295–1302. [Google Scholar]
- Doyle, LA; Yang, W; Abruzzo, LV; Krogmann, T; Gao, Y; Rishi, AK; Ross, DD. A multidrug resistance transporter from human MCF-7 breast cancer cells. Proc. Natl. Acad. Sci. USA 1998, 95, 15665–15670. [Google Scholar]
- Gottesman, MM; Pastan, I. Biochemistry of multidrug resistance mediated by the multidrug transporter. Annu. Rev. Biochim 1993, 62, 385–361. [Google Scholar]
- Szakacs, G; Paterson, JK; Ludwig, JA; Booth-Genthe, C; Gottesman, MM. Targeting multidrug resistance in cancer. Nat. Rev. Drug Discov 2006, 5, 219–234. [Google Scholar]
- Scheffer, GL; Wijngaard, PLJ; Flens, MJ; Izquierdo, MA; Slovak, ML; Pinedo, HM; Meijer, CJLM; Clevers, HC; Scheper, RJ. The drug resistance-related protein LRP is the human major vault protein. Nat. Med 1995, 1, 578–582. [Google Scholar]
- Izquierdo, MA; Shoemaker, RH; Flens, MJ; Scheffer, GL; Wu, L; Prather, TR; Scheper, RJ. Overlapping phenotypes of multidrug resistance among panels of human cancer-cell lines. Int. J. Cancer 1996, 65, 230–237. [Google Scholar]
- Yang, HH; Ma, MH; Vescio, RA; Berenson, JR. Overcoming drug resistance in multiple myeloma: The emergence of therapeutic approaches to induce apoptosis. J. Clin. Oncol 2003, 21, 4239–4247. [Google Scholar]
- Hickman, JA. Apoptosis and chemotherapy resistance. Eur. J. Cancer 1996, 32A, 921–926. [Google Scholar]
- Bushand, JA; Li, G. Cancer chemoresistance: The relationship between p53 and multidrug transporters. Int. J. Cancer 2002, 98, 323–330. [Google Scholar]
- Kannan, K; Kaminski, N; Rechavi, G; Jakob-Hirsch, J; Amariglio, N; Givol, D. DNA microarray analysis of genes involved in p53 mediated apoptosis: Activation of Apaf-1. Oncogene 2001, 20, 3449–3455. [Google Scholar]
- Fisher, DE. Apoptosis in cancer therapy: Crossing the threshold. Cell 1994, 78, 539–542. [Google Scholar]
- Matsuo, K; Kohno, K; Takano, H; Sato, S; Kiue, A; Kuwano, M. Reduction of drug accumulation and DNA topoisomerase II activity in acquired teniposide-resistant human cancer kb cell lines. Cancer Res 1990, 50, 5819–5824. [Google Scholar]
- Minchinton, AI; Tannock, IF. Drug penetration in solid tumours. Nat. Rev. Cancer 2006, 6, 583–592. [Google Scholar]
- Frazier, MC; Simons, JW; Zhong, H; Mabjeesh, NJ. Hypoxia-directed cancer therapy. Expert Opin. Ther. Pat 2002, 12, 777–788. [Google Scholar]
- Zhou, J; Schmid, T; Schnitzer, S; Brune, B. Tumor hypoxia and cancer progression. Cancer Lett 2006, 237, 10–21. [Google Scholar]
- Chakravarti, A; Loeffler, JS; Dyson, NJ. Insulin-like growth factor receptor I mediates resistance to anti-epidermal growth factor receptor therapy in primary human glioblastoma cells through continued activation of phosphoinositide 3-kinase signaling. Cancer Res 2002, 62, 200–207. [Google Scholar]
- Konig, A; Menzel, T; Lynen, S; Wrazel, L; Rosen, A; Al-Katib, A; Raveche, E; Gabrilove, JL. Basic fibroblast growth factor (bFGF) upregulates the expression of bcl-2 in B cell chronic lymphocytic leukemia cell lines resulting in delaying apoptosis. Leukemia 1997, 11, 258–265. [Google Scholar]
- Wang, XW; Yeh, H; Schaeffer, L; Roy, R; Moncollin, V; Egly, JM; Wang, Z; Friedberg, EC; Evans, MK; Taffe, BG; Bohr, VA; Weeda, G; Hoeijmakers, JHJ; Forrester, K; Harris, CC. P53 modulation of TFIIH-associated nucleotide excision repair activity. Nat. Genet 1995, 10, 188–193. [Google Scholar]
- Hofmann, C; Obermeier, F; Artinger, M; Hausmann, M; Falk, W; Schoelmerich, J; Rogler, G; Grossmann, J. Cell-cell contacts prevent anoikis in primary human colonic epithelial cells. Gastroenterology 2007, 132, 587–600. [Google Scholar]
- Shain, KH; Dalton, WS. Cell adhesion is a key determinant in de novo multidrug resistance (mdr): New targets for the prevention of acquired MDR. Mol. Cancer Ther 2001, 1, 69–78. [Google Scholar]
- Mahadevan, D; List, AF. Targeting the multidrug resistance-1 transporter in AML: Molecular regulation and therapeutic strategies. Blood 2004, 104, 1940–1951. [Google Scholar]
- Fox, E; Bates, SE. Tariquidar (XR9576): A P-glycoprotein drug efflux pump inhibitor. Expert Rev. Anticancer Ther 2007, 7, 447–459. [Google Scholar]
- Golstein, PE; Boom, A; van Geffel, J; Jacobs, P; Masereel, B; Beauwens, R. P-glycoprotein inhibition by glibenclamide and related compounds. Pflugers Arch 1999, 437, 652–660. [Google Scholar]
- Advani, R; Lum, BL; Fisher, GA; Halsey, J; Chin, DL; Jacobs, CD; Sikic, BI. A phase I trial of liposomal doxorubicin, paclitaxel and valspodar (PSC-833), an inhibitor of multidrug resistance. Ann. Oncol 2005, 16, 1968–1973. [Google Scholar]
- Dantzig, AH; Shepard, RL; Cao, J; Law, KL; Ehlhardt, WJ; Baughman, TM; Bumol, TF; Starling, JJ. Reversal of P-glycoprotein-mediated multidrug resistance by a potent cyclopropyldibenzosuberane modulator, LY335979. Cancer Res 1996, 56, 4171–4179. [Google Scholar]
- Hyafil, F; Vergely, C; Du Vignaud, P; Grand-Perret, T. In vitro and in vivo reversal of multidrug resistance by GF120918, an acridone carboxamide derivative. Cancer Res 1993, 53, 4595–4602. [Google Scholar]
- Ferry, DR; Traunecker, H; Kerr, DJ. Clinical trials of P-glycoprtoein reversal in solid tumours. Eur. J. Cancer 1996, 31A, 1070–1081. [Google Scholar]
- Mahadevan, D; List, AF. Targeting the multidrug resistance-1 transporter in AML: Molecular regulation and therapeutic strategies. Blood 2004, 104, 1940–1951. [Google Scholar]
- Kabanov, AV; Alakhov, VY. Pluronic block copolymers in drug delivery: From micellar nanocontainers to biological response modifiers. Crit. Rev. Ther. Drug Carrier Syst 2002, 19, 1–72. [Google Scholar]
- Kabanov, AV; Batrakova, EV; Alakhov, VY. Pluronic block copolymers for overcoming drug resistance in cancer. Adv. Drug Deliv. Rev 2002, 54, 759–779. [Google Scholar]
- Batrakova, E; Lee, S; Li, S; Venne, A; Alakhov, V; Kabanov, A. Fundamental relationships between the composition of pluronic block copolymers and their hypersensitization effect in mdr cancer cells. Pharm. Res 1999, 16, 1373–1379. [Google Scholar]
- Li, X; Ruan, GR; Lu, WL; Hong, HY; Liang, GW; Zhang, YT; Liu, Y; Long, C; Ma, X; Yuan, L; Wang, JC; Zhang, X; Zhang, Q. A novel stealth liposomal topotecan with amlodipine: Apoptotic effect is associated with deletion of intracellular Ca2+ by amlodipine thus leading to an enhanced antitumor activity in leukemia. J. Control. Release 2006, 112, 186–198. [Google Scholar]
- Maeda, H. The enhanced permeability and retention (EPR) effect in tumor vasculature: The key role of tumor-selective macromolecular drug targeting. Adv. Enzyme Regul 2001, 41, 189–207. [Google Scholar]
- Bergstrand, N; Edwards, K. Effects of poly(ethylene oxide)-poly(propylene oxide)-poly(ethylene oxide) triblock copolymers on structure and stability of liposomal dioleoylphosphatidylethanolamine. J. Colloid Interface Sci 2004, 276, 400–407. [Google Scholar]
- Wang, JC; Liu, XY; Lu, WL; Chang, A; Zhang, Q; Goh, BC; Lee, HS. Pharmacokinetics of intravenously administered stealth liposomal doxorubicin modulated with verapamil in rats. Eur. J. Pharm. Biopharm 2006, 62, 44–51. [Google Scholar]
- Deng, WJ; Yang, XQ; Liang, YJ; Chen, LM; Yan, YY; Shuai, XT; Fu, LW. FG020326-loaded nanoparticle with PEG and PDLLA improved pharmacodynamics of reversing multidrug resistance in vitro and in vivo. Acta Pharmacol. Sin 2007, 28, 913–920. [Google Scholar]
- Pakunlu, RI; Wang, Y; Saad, M; Khandare, JJ; Starovoytov, V; Minko, T. In vitro and in vivo intracellular liposomal delivery of antisense oligonucleotides and anticancer drug. J. Control. Release 2006, 114, 153–162. [Google Scholar]
- Poupaert, JH; Couvreur, P. A computationally derived structural model of doxorubicin interacting with oligomeric polyalkylcyanoacrylate in nanoparticles. J. Control. Release 2003, 92, 19–26. [Google Scholar]
- Soma, CE; Dubernet, C; Bentolila, D; Benita, S; Couvreur, P. Reversion of multidrug resistance by co-encapsulation of doxorubicin and cyclosporin A in polyalkylcyanoacrylate nanoparticles. Biomaterials 2000, 21, 1–7. [Google Scholar]
- Lu, D; Wientjes, MG; Lu, Z; Au, JL. Tumor priming enhances delivery and efficacy of nanomedicines. J. Pharmacol. Exp. Ther 2007, 322, 80–88. [Google Scholar]
- van Vlerken, LE; Duan, Z; Seiden, MV; Amiji, MM. Modulation of intracellular ceramide using polymeric nanoparticles to overcome multidrug resistance in cancer. Cancer Res 2007, 67, 4843–4850. [Google Scholar]
- Rapoport, N. Combined cancer therapy by micellar-encapsulated drug and ultrasound. Int. J. Pharm 2004, 277, 155–162. [Google Scholar]
- Gao, Z; Fain, HD; Rapoport, N. Ultrasound-enhanced tumor targeting of polymeric micellar drug carriers. Mol. Pharm 2004, 1, 317–330. [Google Scholar]
- Yongzhong, W; Li, Y; Limei, H; Xianyi, S; Xiaoling, F. Difunctional pluronic copolymer micelles for paclitaxel delivery: Syngergistic effect of folate-mediated targeting and pluronic-mediated overcoming multidrug resistance in tumor cell lines. Int. J. Pharm 2007, 337, 63–73. [Google Scholar]
- Kim, D; Lee, ES; Oh, KT; Gao, ZG; Bae, YH. Doxorubicin-loaded polymeric micelle overcomes multidrug resistance of cancer by double-targeting folate receptor and early endosomal pH. Small 2008, 4, 2043–2050. [Google Scholar]
- Kobayashi, T; Ishida, T; Okada, Y; Ise, S; Harashima, H; Kiwada, H. Effect of transferrin receptor-targeted liposomal doxorubicin in p-glycoprotein-mediated drug resistant tumor cells. Int. J. Pharm 2007, 329, 94–102. [Google Scholar]
- Goren, D; Horowitz, AT; Tzemach, D; Tarshish, M; Zalipsky, S; Gabizon, A. Nuclear delivery of doxorubicin via folate-targeted liposomes with bypass of multidrug-resistance efflux pump. Clin. Cancer Res 2000, 6, 1949–1957. [Google Scholar]
- Lee, ES; Na, K; Bae, YH. Polymeric micelle for tumor pH and folate-mediated targeting. J. Control. Release 2003, 91, 103–113. [Google Scholar]
- Lee, ES; Oh, KT; Kim, D; Youn, YS; Bae, YH. Tumor pH-responsive flower-like micelles of poly(l-lactic acid)-b-poly(ethylene glycol)-b-poly(l-histidine). J. Control. Release 2007, 123, 19–26. [Google Scholar]
- Lee, ES; Na, K; Bae, YH. Doxorubicin loaded pH-sensitive polymeric micelles for reversal of resistant MCF-7 tumor. J. Control. Release 2005, 103, 405–418. [Google Scholar]
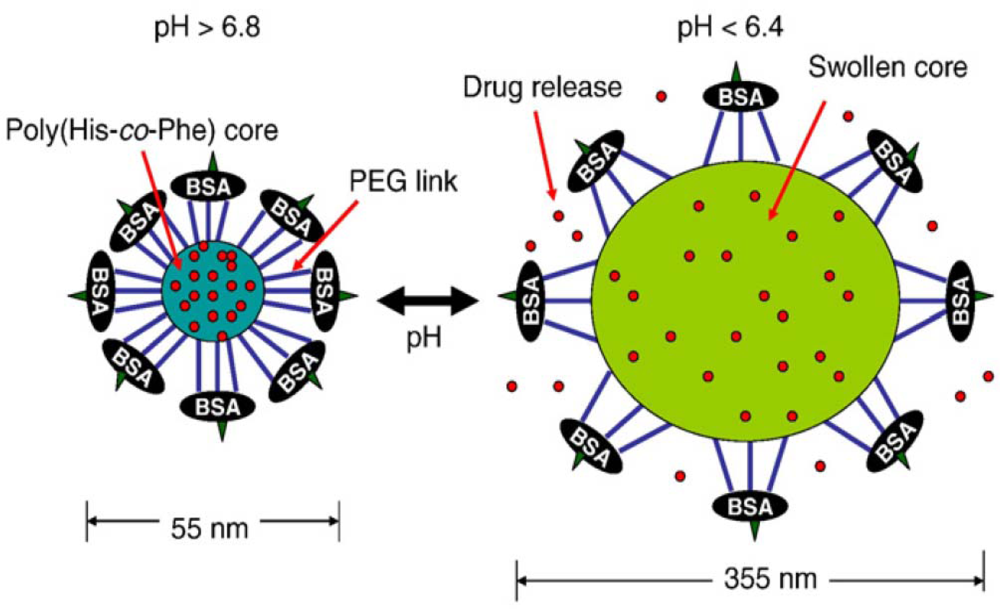
- Lee, ES; Na, K; Bae, YH. Super pH-sensitive multifunctional polymeric micelle. Nano Lett 2005, 5, 325–329. [Google Scholar]
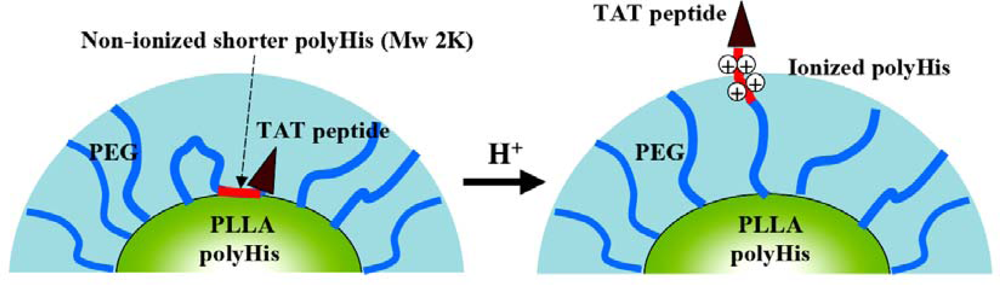
- Lee, ES; Gao, Z; Kim, D; Park, K; Kwon, IC; Bae, YH. Super pH-sensitive multifunctional polymeric micelle for tumor pH(e) specific TAT exposure and multidrug resistance. J. Control. Release 2008, 129, 228–236. [Google Scholar]
- Lewin, M; Carlesso, N; Tun, CH; Tang, XW; Cory, D; Scadden, DT; Weissleder, R. Tat peptide-derivatized magnetic nanoparticles allow in vivo tracking and recovery of progenitor cells. Nat. Biotechnol 2000, 18, 410–414. [Google Scholar]
- Lee, ES; Kim, D; Youn, YS; Oh, KT; Bae, YH. A virus-mimetic nanogel vehicle13. Angew. Chem. Int. Ed 2008, 47, 2418–2421. [Google Scholar]
- Lee, ES; Gao, Z; Bae, YH. Recent progress in tumor pH targeting nanotechnology. J. Control. Release 2008, 132, 164–170. [Google Scholar]
- Boussif, O; Lezoualc'h, F; Zanta, MA; Merqny, MD; Scherman, D; Demeneix, B; Behr, JP. A versatile vector for gene and oligonnucleotide transfer into cells in culture and in vivo: Polyethyleneimine. Proc. Natl. Acad. Sci. USA 1995, 92, 7297–7301. [Google Scholar]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Nanoparticular formulation | Drug-resistance target | Feature | Ref. |
|---|---|---|---|
| Pluronic® micelle with ultrasound treatment | Enhancing drug uptake by ultrasound treatment | Possible to treat wild and drug-resistant tumors | [63,64] |
| Paclitaxel loaded mixed micelle system of Pluronic® P105 and L101 | Inhibition of P-gp by Pluronic® | Combined mechanisms of FR-mediated endocytosis for tumor targeting | [65] |
| Liposomal formulation with doxorubicin/paclitaxel/valspodar | Inhibition of P-gp by valspodar | - | [44] |
| Liposomal topotecan with amlodipine | Inhibition of P-gp by amlodipine | - | [52] |
| Liposomal doxorubicin/verapamil | Inhibition of P-gp by verapamil | Verapamil affected pharmacokinetics of doxorubicin in vivo | [56] |
| Liposomal doxorubicin/Pluronic® F68 | Inhibition of P-gp by Pluronic® | - | [54] |
| Liposomal doxorubicin/antisense oligonucleotides | Targeted to bcl-2 mRNA and MDR1 mRNA | Overcoming bcl-2 and P-gp | [58] |
| Polyalkylcyanoacrylate nanoparticles with doxorubicin and cyclosporin A | Enhancing drug uptake by unknown mechanisms of polyalkylcyanoacrylate nanoparticles | Cyclosporin A can affect pharmacokinetics of doxorubicin | [59,60] |
| Daunorubicin loaded Fe3O4 nanoparticles | Enhancing drug uptake by Fe3O4 nanoparticles | Interaction between Fe3O4 and unknown biological active molecules on the membrane of leukemia cells, increased drug uptake | [74] |
| Poly(ethylene oxide)-modified poly(epsilon-caprolactone) (PEO-PCL) nanoparticle with ceramide and paclitaxel | Targeting to P-gp | Co-therapy (ceramide and paclitaxel) enhanced cytotoxicity for drug-resistant tumors | [62] |
| Transferrin receptor-targeting liposomal doxorubicin | Evading P-gp function by transferring receptor-mediated internalization pathway | - | [67] |
| Folate-conjugated liposomal doxorubicin | Evading P-gp function by FR-mediated internalization pathway | No significant tumor-growth inhibition effect in in vivo animal model | [68] |
| pH-sensitive poly(l-histidine)-based micelle system with folic acid | Enhancing cytoplasmic drug release due to proton-sponge effect of poly(l-histidine) | In vivo animal studies showed significant tumor regression effect in drug-resistant tumors | [69–73, 75] |
| PHSMpop-upTAT | Free DOX | |
|---|---|---|
| HL-60/MX2 a | 0.32 ± 0.07 μg/mL | 1.12 ± 0.08 μg/mL |
| HL-60 b | 0.10 ± 0.03 μg/mL | 0.42 ± 0.07 μg/mL |
| NCI-H69/AR c | 0.20 ± 0.06 μg/mL | 0.75 ± 0.08 μg/mL |
| A549 d | 0.75 ± 0.08 μg/mL | 6.60 ± 0.09 μg/mL |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Oh, K.T.; Baik, H.J.; Lee, A.H.; Oh, Y.T.; Youn, Y.S.; Lee, E.S. The Reversal of Drug-Resistance in Tumors Using a Drug-Carrying Nanoparticular System. Int. J. Mol. Sci. 2009, 10, 3776-3792. https://doi.org/10.3390/ijms10093776
Oh KT, Baik HJ, Lee AH, Oh YT, Youn YS, Lee ES. The Reversal of Drug-Resistance in Tumors Using a Drug-Carrying Nanoparticular System. International Journal of Molecular Sciences. 2009; 10(9):3776-3792. https://doi.org/10.3390/ijms10093776
Chicago/Turabian StyleOh, Kyung Taek, Hye Jung Baik, A Hyeong Lee, Young Taik Oh, Yu Seok Youn, and Eun Seong Lee. 2009. "The Reversal of Drug-Resistance in Tumors Using a Drug-Carrying Nanoparticular System" International Journal of Molecular Sciences 10, no. 9: 3776-3792. https://doi.org/10.3390/ijms10093776
APA StyleOh, K. T., Baik, H. J., Lee, A. H., Oh, Y. T., Youn, Y. S., & Lee, E. S. (2009). The Reversal of Drug-Resistance in Tumors Using a Drug-Carrying Nanoparticular System. International Journal of Molecular Sciences, 10(9), 3776-3792. https://doi.org/10.3390/ijms10093776