Theoretical Investigation of the NO3 Radical Addition to Double Bonds of Limonene

Abstract
:
1. Introduction
2. Theoretical Methods
3. Results and Discussion
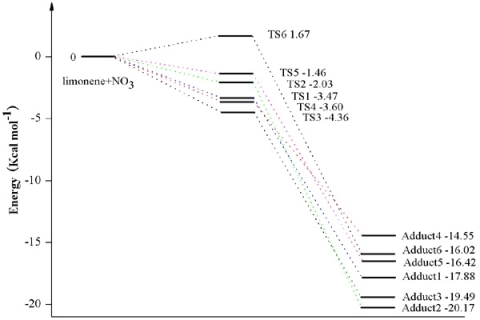
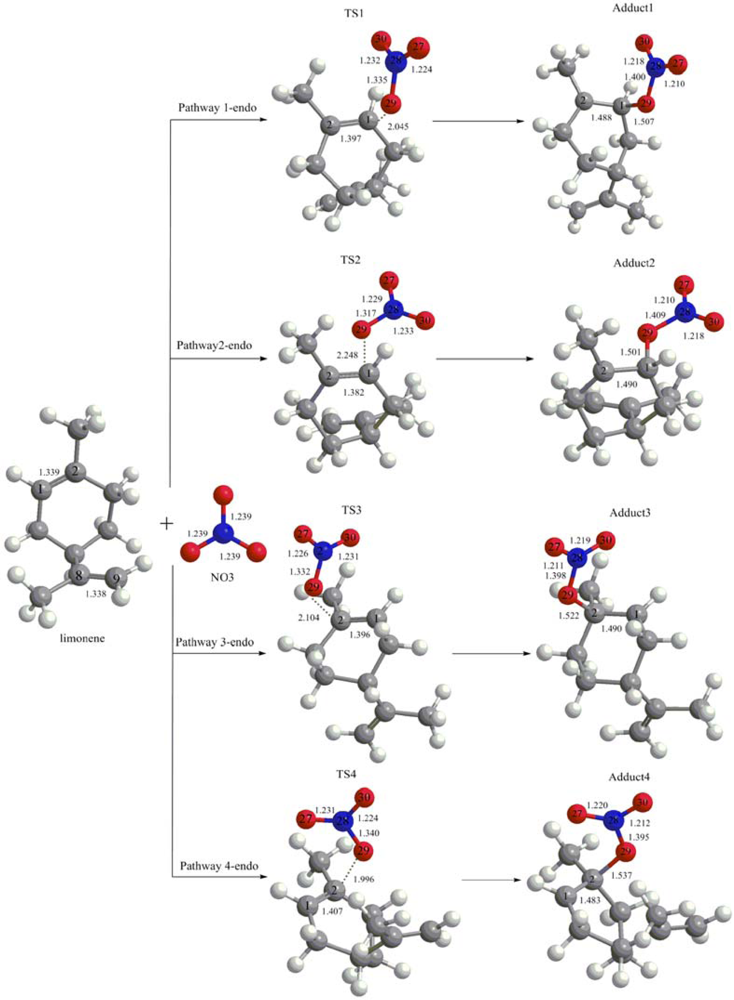
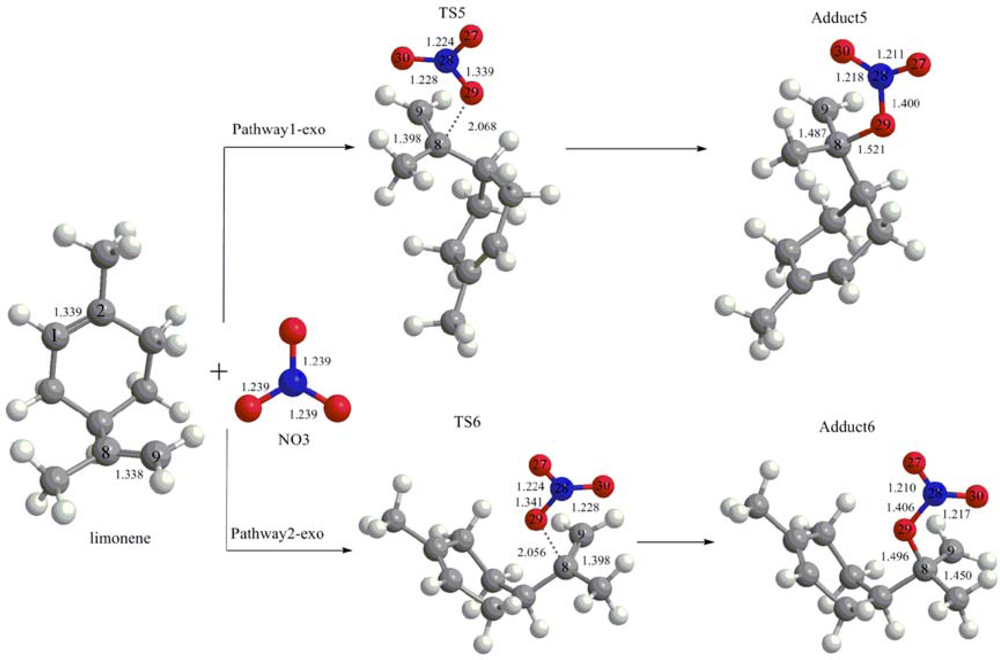
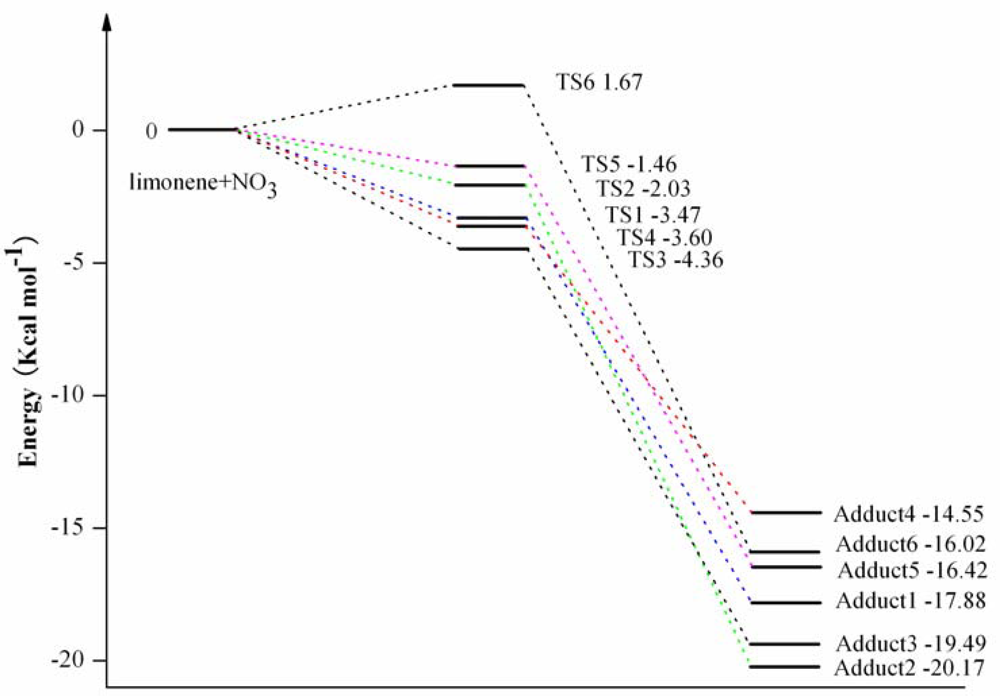
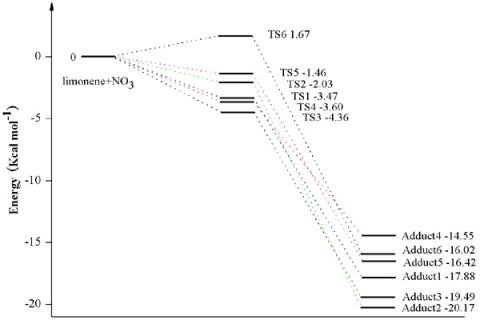
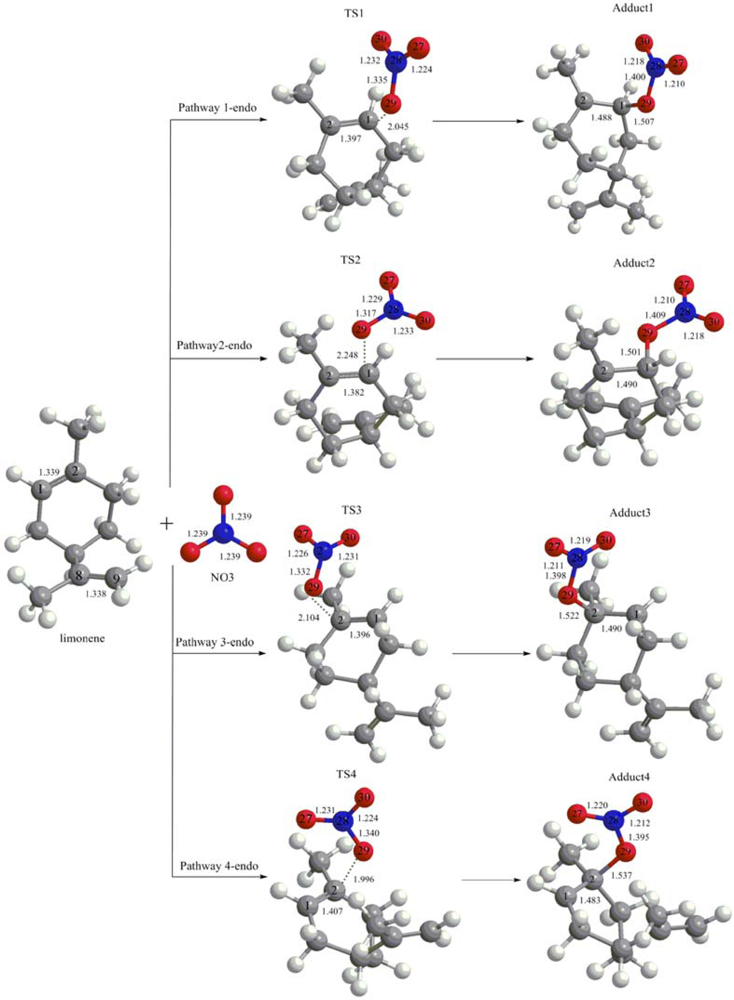
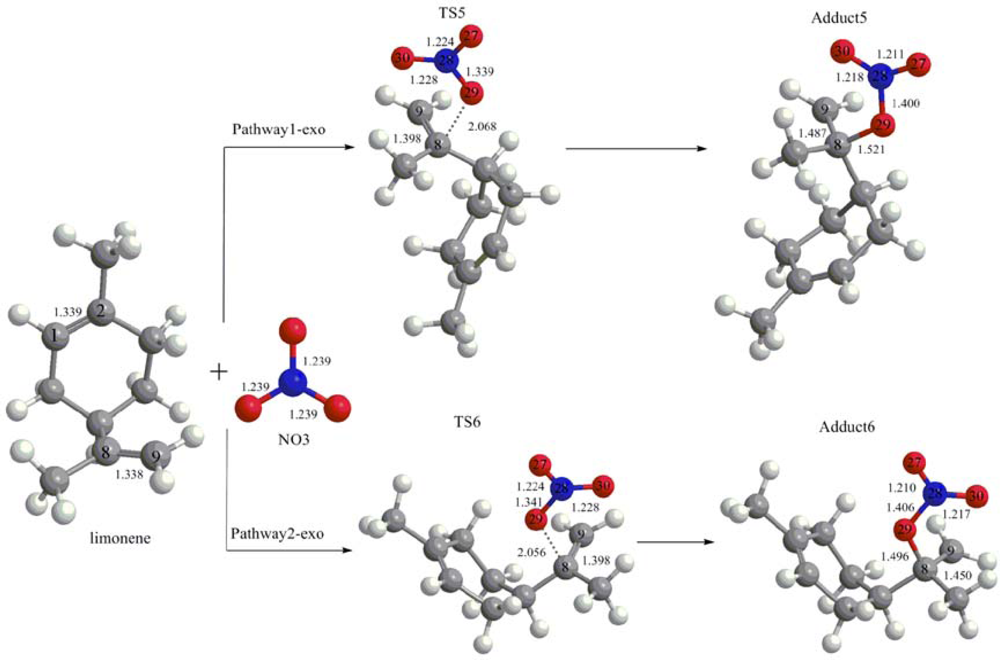
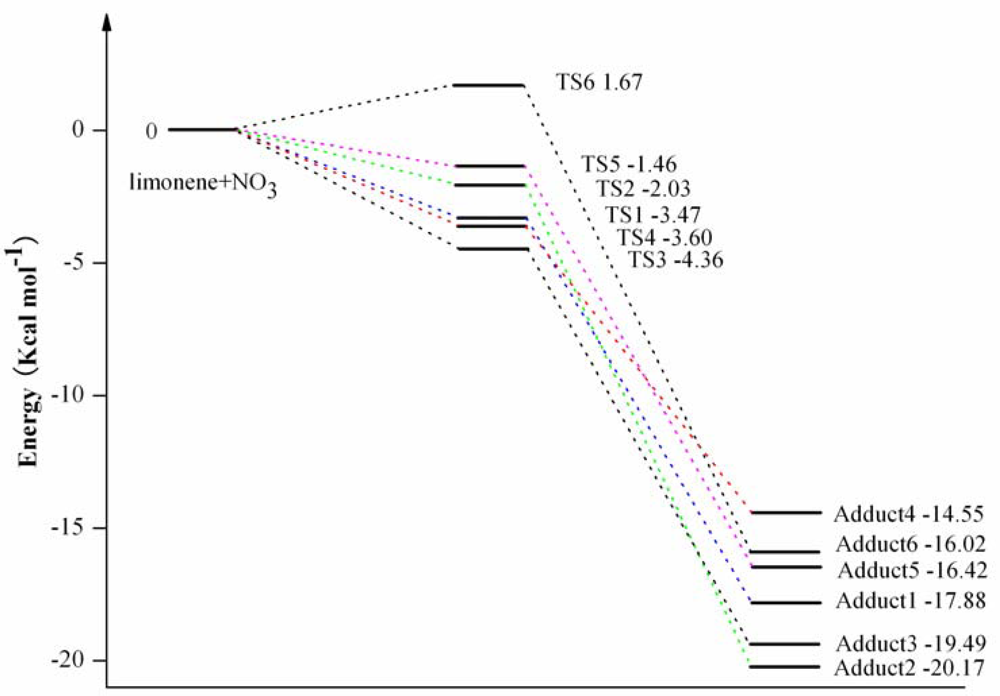
3.1. Reaction Mechanism
3.2. Thermochemical Analysis
4. Conclusions
Acknowledgments
References
- Gill, KJ; Hites, RA. Rate constants for the gas-phase reactions of the hydroxyl radical with isoprene, α-and β-pinene, and limonene as a function of temperature. J. Phys. Chem. A 2002, 106, 2538–2544. [Google Scholar]
- Guenther, A; Hewitt, CN; Erickson, D; Fall, R; Geron, C; Graedel, T; Harley, P; Klinger, L; Lerdau, M; McKay, WA; Pierce, T; Scholes, B; Steinbrecher, R; Tallamraju, R; Taylor, J; Zimmerman, PJ. A global model of natural volatile organic compound emissions. J. Geophys. Res 1995, 100, 8873–8892. [Google Scholar]
- Grosjean, D; Williams, EL; Grosjean, E; Andino, JM; Seinfeld, JH. Atmospheric oxidation of biogenic hydrocarbons, reactions of ozone with β-pinene, d-limonene and transcaryophylene. Environ. Sci. Technol 1993, 27, 2754–2758. [Google Scholar]
- Kleindienst, TE; Harris, GH; Pitts, JN, Jr. Rates and temperature dependence of the reaction of OH with isoprene, its oxidation products, and selected terpenes. Environ. Sci. Technol 1982, 16, 844–846. [Google Scholar]
- Shu, Y; Atkinson, R. Rate constants for the gas-phase reactions of O3 with a series of terpenes and OH radical formation from the O3 reactions with sesquiterpenes at 296 ± 2 K. Int. J. Chem. Kinet 1994, 26, 1193–1205. [Google Scholar]
- Atkinson, R. Gas-phase tropospheric chemistry of organic compounds. J. Phys. Chem. Ref. Data, Monogr 1994, 2, 1–216. [Google Scholar]
- Wayne, RP. The nitrate radical: Physics, chemistry and the atmosphere. Atmos. Environ 1991, 25A, 1–206. [Google Scholar]
- Atkinson, R; Plum, CN; Carter, WPL; Winer, AM; Pitts, JN, Jr. Kinetics of the gas-phase reactions of NO3 radicals with a series of alkanes at 296 ± 2K. J. Phys. Chem 1984, 88, 2361–2364. [Google Scholar]
- Cantrell, CA; Davidson, JA; Busarow, KL; Calvert, JG. The CH3CHO-NO3 reaction and possible night-time PAN generation. J. Geophys. Res 1986, 91, 5347–5353. [Google Scholar]
- Bandow, H; Okuda, M; Akimoto, H. Mechanism of the gas-phase reactions of propylene and nitroxy radicals. J. Phys. Chem 1980, 84, 3604–3608. [Google Scholar]
- Guenther, A; Geron, C; Pierce, T; Lamb, B; Harley, P; Fall, R. Natural emissions of non-methane volatile organic compounds, carbon monoxide, and oxides of nitrogen from North America. Atmos. Environ 2000, 34, 2205–2230. [Google Scholar]
- He, C; Murray, F; Lyons, T. Monoterpene and isoprene emissions from 15 eucalyptus species in Australia. Atmos. Environ 2000, 34, 645–655. [Google Scholar]
- Spittler, M; Barnes, I; Bejan, I; Brockmann, KJ; Benter, Th; Wirtz, K. Reactions of NO3 radicals with limonene and α-pinene: Product and SOA formation. Atmos. Environ 2006, 40, S116–S127. [Google Scholar]
- Frisch, MJ; Trucks, GW; Schlegel, HB; Scuseria, GE; Robb, MA; Cheeseman, JR; Montgomery, JA, Jr; Vreven, T; Kudin, KN; Burant, JC; Millam, JM; Iyengar, SS; Tomasi, J; Barone, V; Mennucci, B; Cossi, M; Scalmani, G; Rega, N; Petersson, GA; Nakatsuji, H; Hada, M; Ehara, M; Toyota, K; Fukuda, R; Hasegawa, J; Ishida, M; Nakajima, T; Honda, Y; Kitao, O; Nakai, H; Klene, M; Li, X; Knox, JE; Hratchian, HP; Cross, JB; Bakken, V; Adamo, C; Jaramillo, J; Gomperts, R; Stratmann, RE; Yazyev, O; Austin, AJ; Cammi, R; Pomelli, C; Ochterski, JW; Ayala, PY; Morokuma, K; Voth, GA; Salvador, P; Dannenberg, JJ; Zakrzewski, VG; Dapprich, S; Daniels, AD; Strain, MC; Farkas, O; Malick, DK; Rabuck, AD; Raghavachari, K; Foresman, JB; Ortiz, JV; Cui, Q; Baboul, AG; Clifford, S; Cioslowski, J; Stefanov, BB; Liu, G; Liashenko, A; Piskorz, P; Komaromi, I; Martin, RL; Fox, DJ; Keith, T; Al-Laham, MA; Peng, CY; Nanayakkara, A; Challacombe, M; Gill, PMW; Johnson, B; Chen, W; Wong, MW; Gonzalez, C; Pople, JA. Gaussian 03; Revision E01; Gaussian, Inc: Wallingford, CT, USA, 2004. [Google Scholar]
- Pérez-Casany, MP; Nebot-Gil, I; Sánchez-Marín, J. Ab initio study on the mechanism of tropospheric reactions of the nitrate radical with alkenes: Propene. J. Phys. Chem. A 2000, 104, 6277–6286. [Google Scholar]
- Suh, I; Lei, W; Zhang, R. Experimental and theoretical studies of isoprene reaction with NO3. J. Phys. Chem. A 2001, 105, 6471–6478. [Google Scholar]
- Lei, W; Zhang, R; McGivern, WS; Derecskei-Kovacs, A; North, SW. Theoretical study of isomeric branching in the isoprene-OH reaction: Implications to final product yields in isoprene oxidation. Chem. Phys. Lett 2000, 326, 109–114. [Google Scholar]
- Vereecken, L; Peeters, J. Theoretical study of the formation of acetone in the OH-initiated atmospheric oxidation of α-pinene. J. Phys. Chem. A 2000, 104, 11140–11146. [Google Scholar]
- Gutbrod, R; Kraka, E; Schindler, RN; Cremer, D. Kinetic and theoretical investigation of the gas-phase ozonolysis of isoprene: Carbonyl oxides as an important source for OH radicals in the atmosphere. J. Am. Chem. Soc 1997, 119, 7330–7342. [Google Scholar]
- Olzmann, M; Kraka, E; Cremer, D; Gutbrod, R; Andersson, S. Energetics, kinetics, and product distributions of the reactions of ozone with ethene and 2,3-dimethyl-2-butene. J. Phys. Chem. A 1997, 101, 9421–9429. [Google Scholar]
- Aplincourt, P; Ruiz-López, MF. Theoretical study of formic acid anhydride formation from carbonyl oxide in the atmosphere. J. Phys. Chem. A 2000, 104, 380–388. [Google Scholar]
- Lei, W; Derecskei-Kovacs, A; Zhang, R. Ab initio study of OH addition reaction to isoprene. J. Chem. Phys 2000, 113, 5354–5360. [Google Scholar]
- Zhang, D; Zhang, R. Mechanism of OH formation from ozonolysis of isoprene: A quantum-chemical study. J. Am. Chem. Soc 2002, 124, 2692–2703. [Google Scholar]
- Jiang, L; Wang, W; Xu, Y. Ab initio investigation of O3 addition to double bonds of limonene submitted for publication.
- Martínez, E; Cabañas, B; Aranda, A; Martín, P; Salgado, S. Absolute rate coefficients for the gas-phase reactions of NO3 radical with a series of monoterpenes at T=298 to 433 K. J. Atmos. Chem 1999, 33, 265–282. [Google Scholar]
- Shu, Y; Kwok, ESC; Tuazon, EC; Atkinson, R; Arey, J. Products of the gas-phase reactions of linalool with OH radicals, NO3 radicals, and O3. Environ. Sci. Technol 1997, 31, 896–904. [Google Scholar]
- Lee, TJ; Taylor, PR. A diagnostic for determining the quality of single-reference electron correlation methods. Int. J. Quantum Chem. Symp 1989, 23, 199–207. [Google Scholar]
- Rienstra-Kiracofe, JC; Allen, WD; Schaefer, HF, III. The C2H5 + O2 reaction mechanism: High-level ab initio characterizations. J. Phys. Chem. A 2000, 104, 9823–9840. [Google Scholar]
- Pérez-Casany, MP; Nebot-Gil, I; Sánchez-Marín, J. DFT theoretical study on the reaction mechanism of the nitrate radical with alkenes: 2-Butene, isobutene, 2-methyl-2-butene, and 2,3-dimethyl-2-butene. J. Phys. Chem. A 2000, 104, 10721–10730. [Google Scholar]
- Alvarez-Idaboy, JR; Mora-Díez, N; Vivier-Bunge, A. A quantum chemical and classical transition state theory explanation of negative activation energies in OH addition to substituted ethenes. J. Am.Chem. Soc 2000, 122, 3715–3720. [Google Scholar]
- Mozurkewich, M; Lamb, JJ; Benson, SW. Negative activation energies and curved Arrhenius plots. 2. Hydroxyl + carbon monoxide. J. Phys. Chem 1984, 88, 6435–6441. [Google Scholar]
- Ramírez-Ramírez, VM; Nebot-Gil, I. Theoretical study of the OH addition to the endocyclic and exocyclic double bonds of the d-limonene. Chem. Phys. Lett 2005, 409, 23–28. [Google Scholar]
- Díaz-Acosta, I; Alvarez-Idaboy, JR; Vivier-Bunge, A. Mechanism of the OH-propene-O2 reaction: An ab initio study. Int. J. Chem. Kinet 1999, 31, 29–36. [Google Scholar]
- García-Cruz, I; Ruiz-Santoyo, ME; Alvarez-Idaboy, JR; Vivier-Bunge, A. Ab-initio study of initial atmospheric oxidation reactions of C3 and C4 alkanes. J. Comput. Chem 1999, 20, 845–856. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Species | CCSD(T)/6–31G(d) | UB3LYP/6–31G(d,p) | UB3LYP/6–31G(d,p) |
|---|---|---|---|
| T1 | <S2> | <S2>A | |
| limonene | 0.0100 | 0.000 | 0.000 |
| NO3 | 0.0228 | 0.755 | 0.750 |
| TS1 | 0.0211 | 0.761 | 0.750 |
| Adduct 1 | 0.0148 | 0.754 | 0.750 |
| TS2 | 0.0289 | 0.760 | 0.750 |
| Adduct 2 | 0.0149 | 0.754 | 0.750 |
| TS3 | 0.0230 | 0.764 | 0.750 |
| Adduct 3 | 0.0149 | 0.754 | 0.750 |
| TS4 | 0.0202 | 0.764 | 0.750 |
| Adduct 4 | 0.0156 | 0.754 | 0.750 |
| TS5 | 0.0220 | 0.768 | 0.750 |
| Adduct 5 | 0.0146 | 0.754 | 0.750 |
| TS6 | 0.0215 | 0.768 | 0.750 |
| Adduct 6 | 0.0145 | 0.754 | 0.750 |
| Method | RE1-endo | RE2-endo | RE3-endo | RE4-endo | RE1-exo | RE2-exo |
|---|---|---|---|---|---|---|
| PMP2/6–31G(d) | −9.76 | −12.19 | −11.41 | −5.68 | −9.15 | −8.72 |
| PMP2/6–311++G(d,p) | −7.06 | −9.46 | −8.99 | −3.97 | −6.45 | −6.18 |
| B3LYP/6–31G(d,p) | −15.26 | −16.90 | −14.00 | −8.73 | −11.26 | −10.08 |
| B3LYP/6–311 + G(3df,2pd) | −12.04 | −13.15 | −10.43 | −4.91 | −7.25 | −6.02 |
| CCSD(T)/6–31G(d) | −20.58 | −22.90 | −21.90 | −16.26 | −19.12 | −18.55 |
| CCSD(T)/6–31G(d) + CF | −17.88 | −20.17 | −19.49 | −14.55 | −16.42 | −16.02 |
| Method | ΔE1-endo | ΔE2-endo | ΔE3-endo | ΔE4-endo | ΔE1-exo | ΔE2-exo |
|---|---|---|---|---|---|---|
| PMP2/6–31G(d) | 7.20 | 13.68 | 7.68 | 7.15 | 8.62 | 11.06 |
| PMP2/6–311++G(d,p) | 10.06 | 16.06 | 10.55 | 9.41 | 11.94 | 14.50 |
| B3LYP/6–31G(d,p) | −8.17 | −8.01 | −7.72 | −4.72 | −4.34 | −0.92 |
| B3LYP/6–311 + G(3df,2pd) | −6.47 | −5.35 | −5.46 | −2.02 | −1.30 | 2.27 |
| CCSD(T)/6–31G(d) | −6.34 | −4.41 | −7.23 | −5.86 | −4.78 | −1.77 |
| CCSD(T)/6–31G(d) + CF | −3.47 | −2.03 | −4.36 | −3.60 | −1.46 | 1.67 |
| Method | 1-endo | 2-endo | 3-endo | 4-endo | 1-exo | 1-exo |
|---|---|---|---|---|---|---|
| ΔH | −15.59 | −17.42 | −14.70 | −9.32 | −11.73 | −10.55 |
| ΔG | −3.64 | −4.69 | −0.97 | 3.61 | 1.80 | 2.83 |
| ΔS | −40.08 | −42.70 | −46.05 | −43.37 | −45.38 | −44.88 |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Jiang, L.; Wang, W.; Xu, Y.-S. Theoretical Investigation of the NO3 Radical Addition to Double Bonds of Limonene. Int. J. Mol. Sci. 2009, 10, 3743-3754. https://doi.org/10.3390/ijms10093743
Jiang L, Wang W, Xu Y-S. Theoretical Investigation of the NO3 Radical Addition to Double Bonds of Limonene. International Journal of Molecular Sciences. 2009; 10(9):3743-3754. https://doi.org/10.3390/ijms10093743
Chicago/Turabian StyleJiang, Lei, Wei Wang, and Yi-Sheng Xu. 2009. "Theoretical Investigation of the NO3 Radical Addition to Double Bonds of Limonene" International Journal of Molecular Sciences 10, no. 9: 3743-3754. https://doi.org/10.3390/ijms10093743
APA StyleJiang, L., Wang, W., & Xu, Y.-S. (2009). Theoretical Investigation of the NO3 Radical Addition to Double Bonds of Limonene. International Journal of Molecular Sciences, 10(9), 3743-3754. https://doi.org/10.3390/ijms10093743