Non-Enzymatic Template-Directed Recombination of RNAs

Abstract
:
1. Introduction
2. Results and Discussion
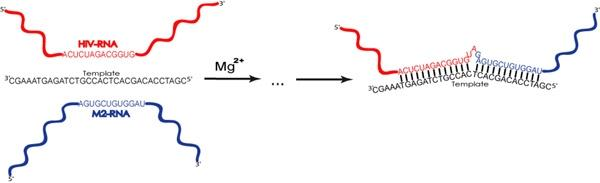
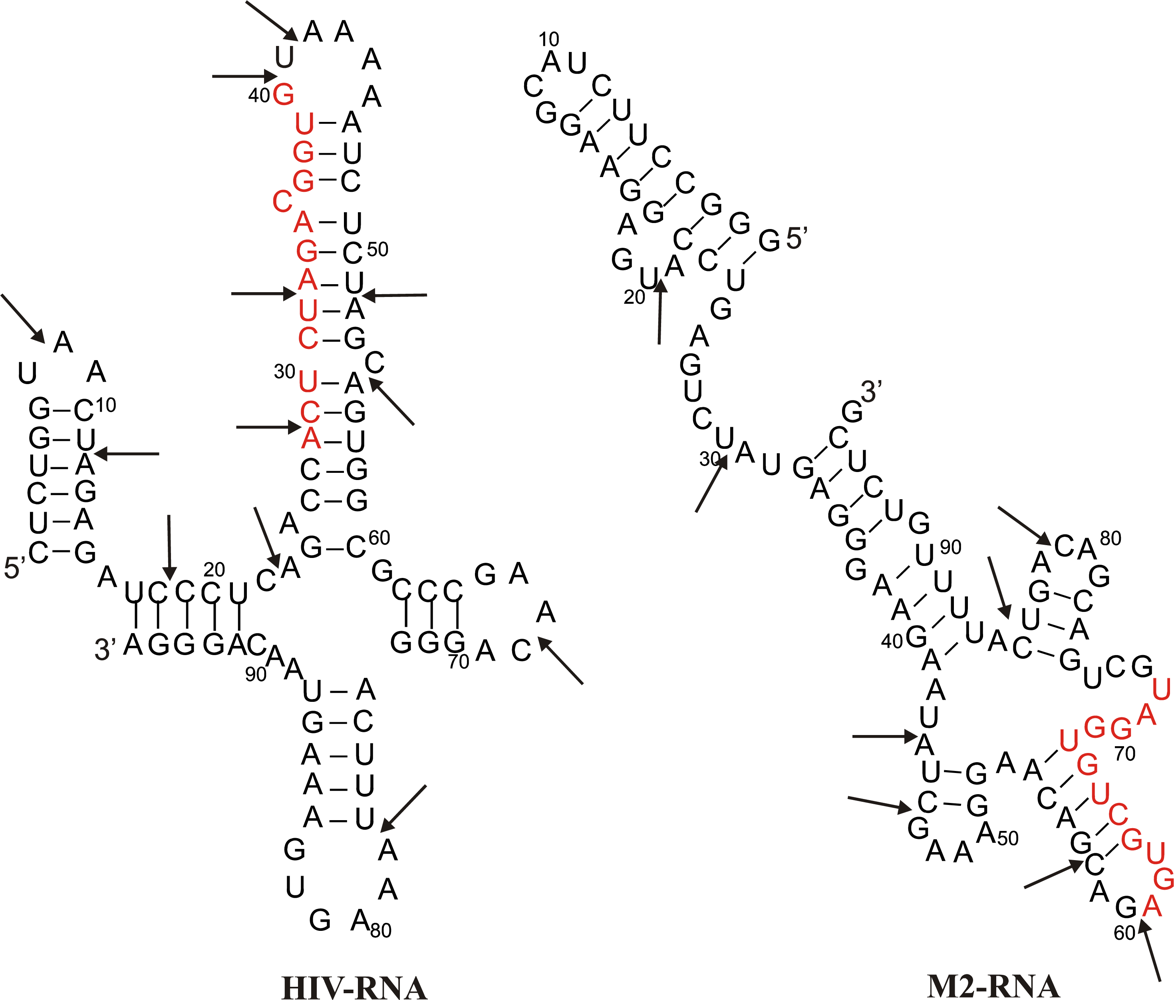
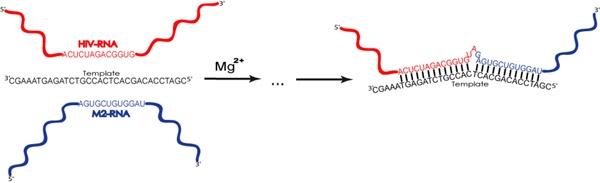
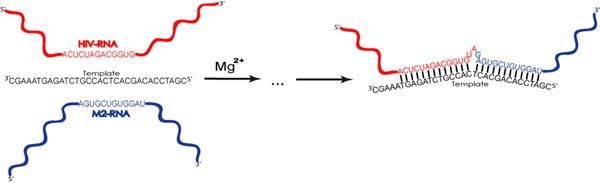
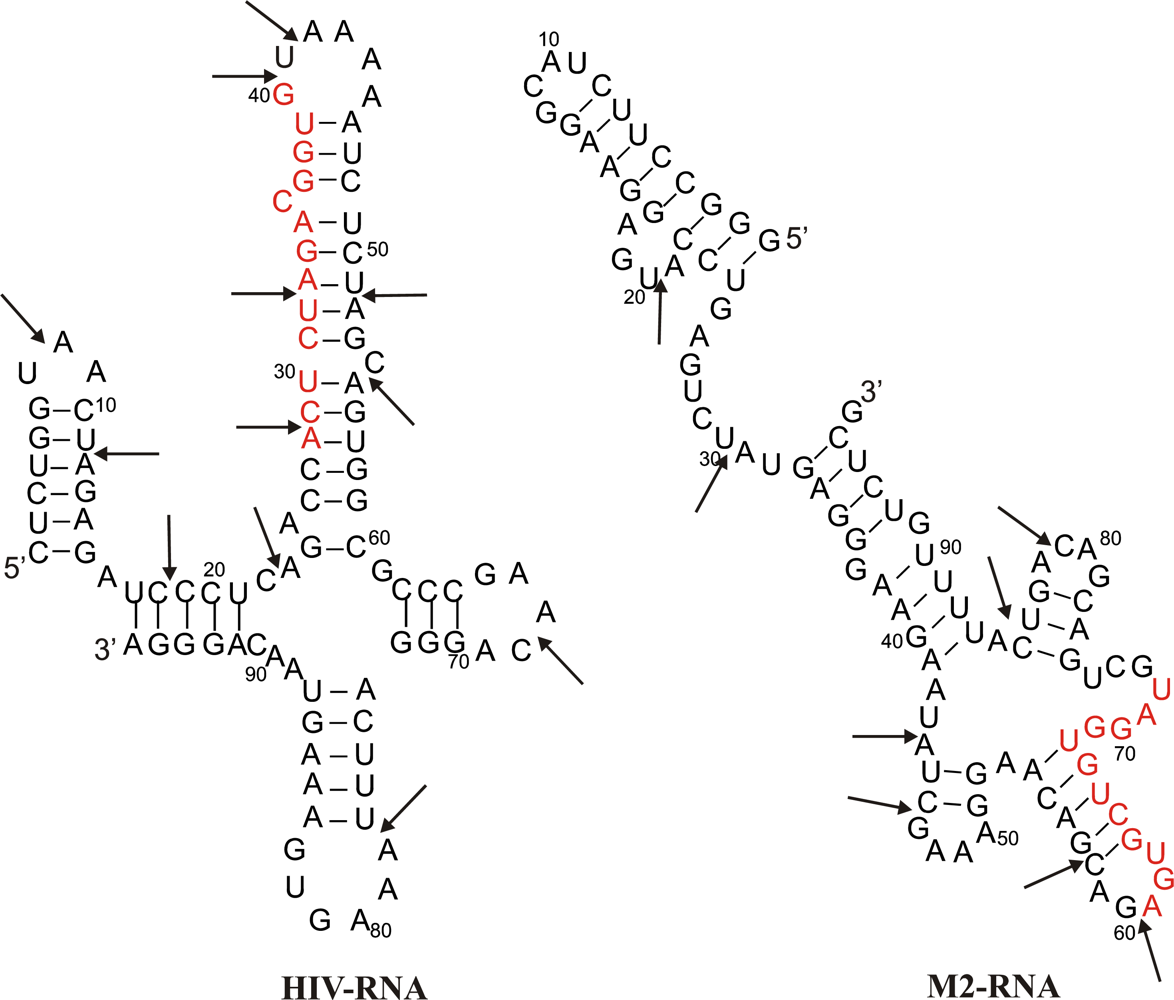
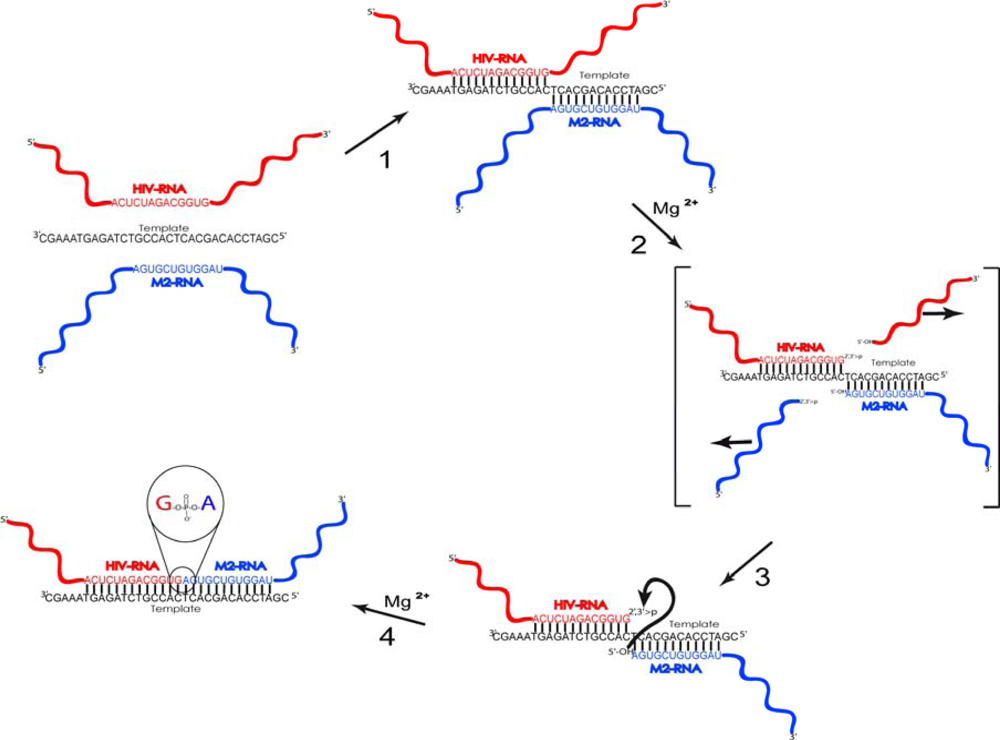
2.1. Description of model system and reaction scheme
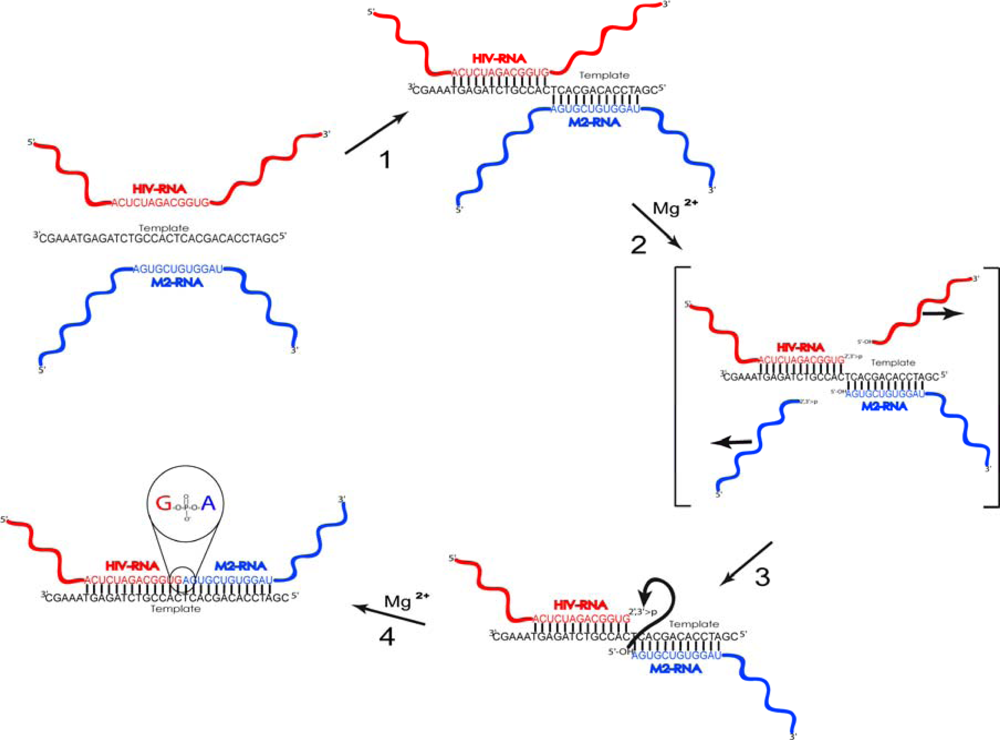
2.2. Reaction scheme and conditions
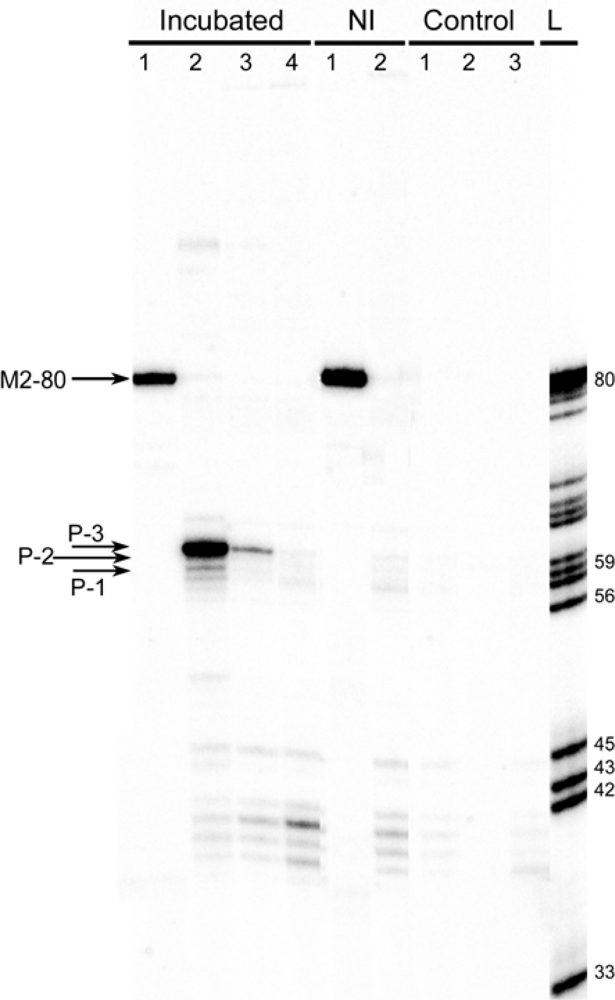
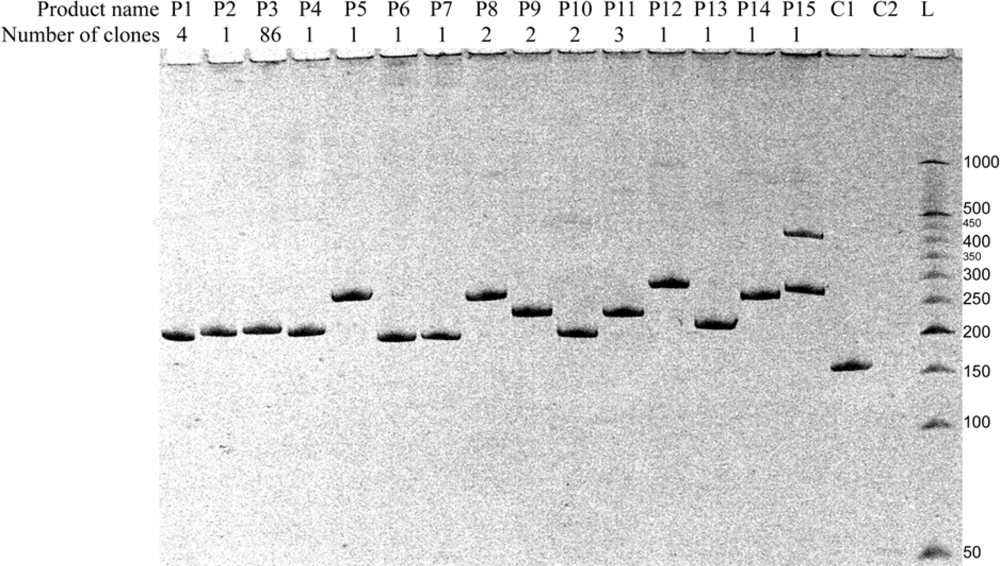
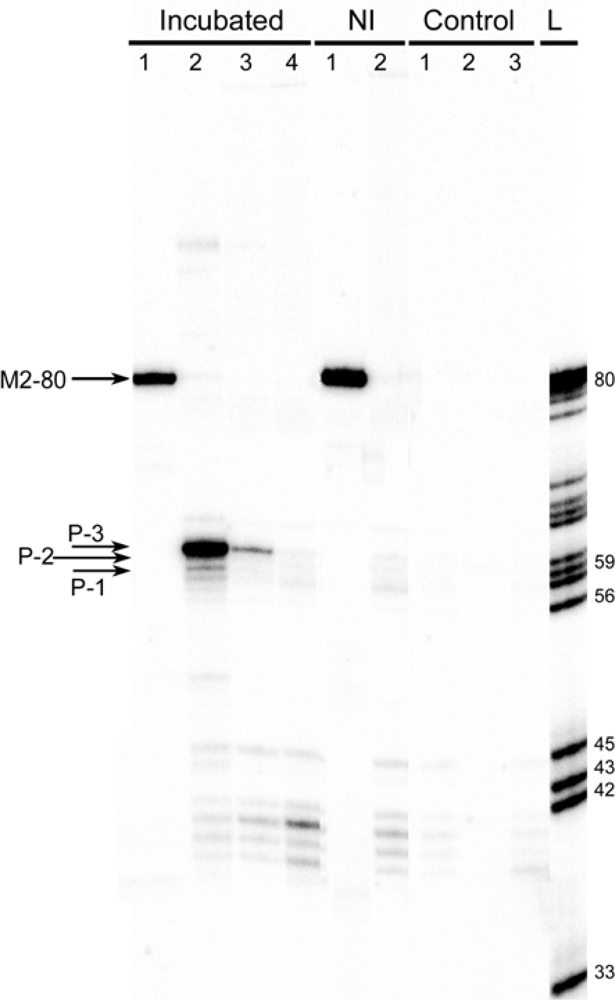
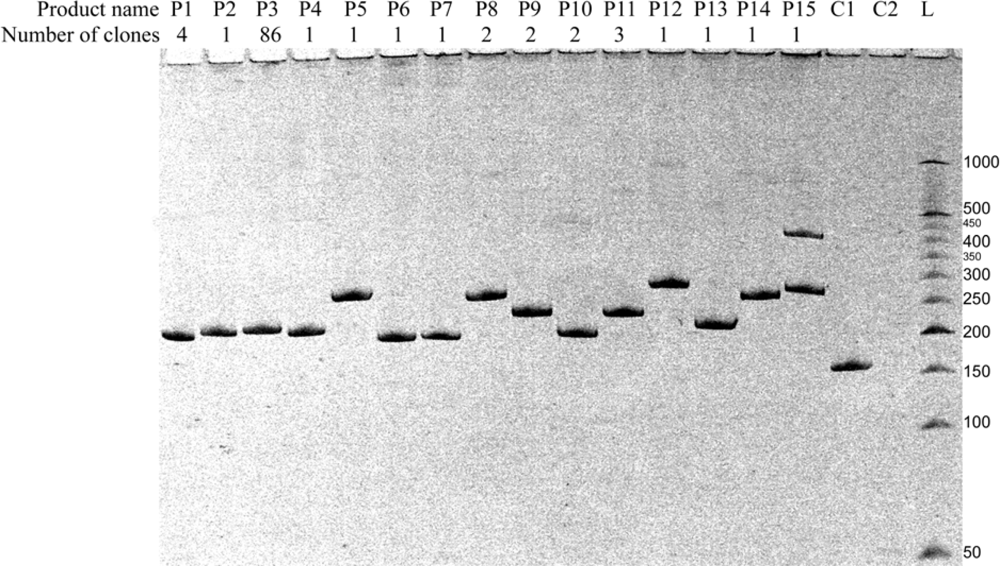
2.3. Identification of products of RNAs recombination
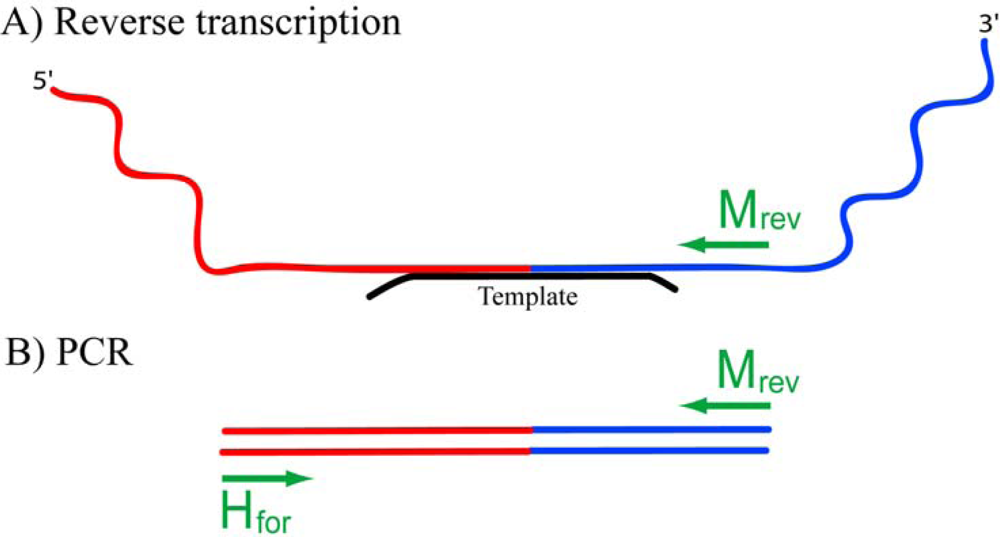
2.3.1. RT-PCR
2.3.2. Molecular cloning
2.3.3. Sequencing of TA-inserts
2.4. Discussion
2.5. Limitations of the detection method
3. Experimental Section
3.1. Enzymes and chemicals
3.2. Oligonucleotides
3.3. 5′-32P-oligonucleotide labeling
3.4. Plasmid linearization
3.5. Amplification of M2-DNA fragment
3.6. T7 transcription
3.7. Dephosphorylation of RNA
3.8. Non-enzymatic cleavage/ligation reaction
3.9. Reverse transcription and PCR of ligation products
3.10. TA cloning
3.11. Bacterial colony PCR
3.12. DNA sequencing
4. Conclusions
Acknowledgments
References and Notes
- Gilbert, W. The RNA World. Nature 1986, 319, 618. [Google Scholar]
- Ertem, G; Ferris, JP. Formation of RNA oligomers on montmorillonite: site of catalysis. Origins Life Evol. Biosphere 1998, 28, 485–499. [Google Scholar]
- Ertem, G; Ferris, JP. Sequence- and regio-selectivity in the montmorillonite-catalyzed synthesis of RNA. Origins Life Evol. Biosphere 2000, 30, 411–422. [Google Scholar]
- Orgel, LE. Some consequences of the RNA world hypothesis. Origins Life Evol. Biosphere 2003, 33, 211–218. [Google Scholar]
- Sreedhara, A; Cowan, JA. Structural and catalytic roles for divalent magnesium in nucleic acid biochemistry. Biometals 2002, 15, 211–223. [Google Scholar]
- Vlassov, VV; Zuber, G; Felden, B; Behr, JP; Giege, R. Cleavage of tRNA with imidazole and spermine imidazole constructs: a new approach for probing RNA structure. Nucleic Acids Res 1995, 23, 3161–3167. [Google Scholar]
- Kumar, P; Dhawan, G; Chandra, R; Gupta, KC. Polyamine-assisted rapid and clean cleavage of oligonucleotides from cis-diol bearing universal support. Nucleic Acids Res 2002, 30, e130. [Google Scholar]
- Emilsson, GM; Nakamura, S; Roth, A; Breaker, RR. Ribozyme speed limits. RNA 2003, 9, 907–918. [Google Scholar]
- Chetverina, HV; Demidenko, AA; Ugarov, VI; Chetverin, AB. Spontaneous rearrangements in RNA sequences. FEBS Lett 1999, 450, 89–94. [Google Scholar]
- Chetverin, AB; Chetverina, HV; Demidenko, AA; Ugarov, VI. Nonhomologous RNA recombination in a cell-free system: evidence for a transesterification mechanism guided by secondary structure. Cell 1997, 88, 503–513. [Google Scholar]
- Lutay, AV; Zenkova, MA; Vlassov, VV. Nonenzymatic recombination of RNA: possible mechanism for the formation of novel sequences. Chem. Biodivers 2007, 4, 762–767. [Google Scholar]
- Lutay, AV; Chernolovskaya, EL; Zenkova, MA; Vlassov, VV. The nonenzymatic template-directed ligation of oligonucleotides. Biogeosciences 2006, 3, 243–249. [Google Scholar]
- Kuznetsova, IL; Zenkova, MA; Gross, HJ; Vlassov, VV. Enhanced RNA cleavage within bulge-loops by an artificial ribonuclease. Nucleic Acids Res 2005, 33, 1201–1212. [Google Scholar]
- Mironova, NL; Pyshnyi, DV; Ivanova, EM; Zenkova, MA; Gross, HJ; Vlassov, VV. Covalently attached oligodeoxyribonucleotides induce RNase activity of a short peptide and modulate its base specificity. Nucleic Acids Res 2004, 32, 1928–1936. [Google Scholar]
- Mironova, NL; Pyshnyi, DV; Shtadler, DV; Fedorova, AA; Vlassov, VV; Zenkova, MA. RNase T1 mimicking artificial ribonuclease. Nucleic Acids Res 2007, 35, 2356–2367. [Google Scholar]
- Kovalev, NA; Medvedeva, DA; Zenkova, MA; Vlassov, VV. Cleavage of RNA by an amphiphilic compound lacking traditional catalytic groups. Bioorg. Chem 2008, 36, 33–45. [Google Scholar]
- O’Brien, EJ; MacEwan, AW. Molecular and crystal structure of the polynucleotide complex: Polyinosinic acid plus polydeoxycytidylic acid. J. Mol. Biol 1970, 48, 243–261. [Google Scholar]
- Lima, WF; Monia, BP; Ecker, DJ; Freier, SM. Implication of RNA structure on antisense oligonucleotide hybridization kinetics. Biochemistry 1992, 31, 12055–12061. [Google Scholar]
- Petyuk, VA; Zenkova, MA; Giege, R; Vlassov, VV. Hybridization of antisense oligonucleotides with the 3′part of tRNA(Phe). FEBS Lett 1999, 444, 217–221. [Google Scholar]
- Patiuk, VA; Giege, R; Vlasov, VV; Zenkova, MA. Mechanism of oligonucleotide hybridization with the 3′-terminal region of yeast tRNA(Phe). Mol. Biol. (Mosk) 2000, 34, 879–886. [Google Scholar]
- Husken, D; Goodall, G; Blommers, MJ; Jahnke, W; Hall, J; Haner, R; Moser, HE. Creating RNA bulges: cleavage of RNA in RNA/DNA duplexes by metal ion catalysis. Biochemistry 1996, 35, 16591–16600. [Google Scholar]
- Orgel, LE. Prebiotic chemistry and the origin of the RNA world. Crit. Rev. Biochem. Mol. Biol 2004, 39, 99–123. [Google Scholar]
- Pino, S; Ciciriello, F; Costanzo, G; Di Mauro, E. Nonenzymatic RNA ligation in water. J. Biol. Chem 2008, 283, 36494–36503. [Google Scholar]
- Soukup, GA; Breaker, RR. Relationship between internucleotide linkage geometry and the stability of RNA. RNA 1999, 5, 1308–1325. [Google Scholar]
- Ciesiolka, J; Michalowski, D; Wrzesinski, J; Krajewski, J; Krzyzosiak, WJ. Patterns of cleavages induced by lead ions in defined RNA secondary structure motifs. J. Mol. Biol 1998, 275, 211–220. [Google Scholar]
- Breaker, RR; Emilsson, GM; Lazarev, D; Nakamura, S; Puskarz, IJ; Roth, A; Sudarsan, N. A common speed limit for RNA-cleaving ribozymes and deoxyribozymes. RNA 2003, 9, 949–957. [Google Scholar]
- Usher, DA; McHale, AH. Hydrolytic stability of helical RNA: a selective advantage for the natural 3′,5′-bond. Proc. Natl. Acad. Sci. USA 1976, 73, 1149–1153. [Google Scholar]
- Hermann, T; Patel, DJ. RNA bulges as architectural and recognition motifs. Structure 2000, 8, R47–54. [Google Scholar]
- Carter-O’Connell, I; Booth, D; Eason, B; Grover, N. Thermodynamic examination of trinucleotide bulged RNA in the context of HIV-1 TAR RNA. RNA 2008, 14, 2550–2556. [Google Scholar]
- Turner, DH. Bulges in nucleic acids. Curr. Opin. Struct. Biol 1992, 2, 334–337. [Google Scholar]
- Gralla, J; Crothers, DM. Free energy of imperfect nucleic acid helices. 3. Small internal loops resulting from mismatches. J. Mol. Biol 1973, 78, 301–319. [Google Scholar]
- Lorsch, JR; Bartel, DP; Szostak, JW. Reverse transcriptase reads through a 2′–5′ linkage and a 2′-thiophosphate in a template. Nucleic Acids Res 1995, 23, 2811–2814. [Google Scholar]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Amplicon | Sequences of amplified regions of recombinant RNA molecules2) | Number of detected clones3) | Comments | ||
|---|---|---|---|---|---|
| Name1) | Length | Bact. colony PCR | Sequencing | ||
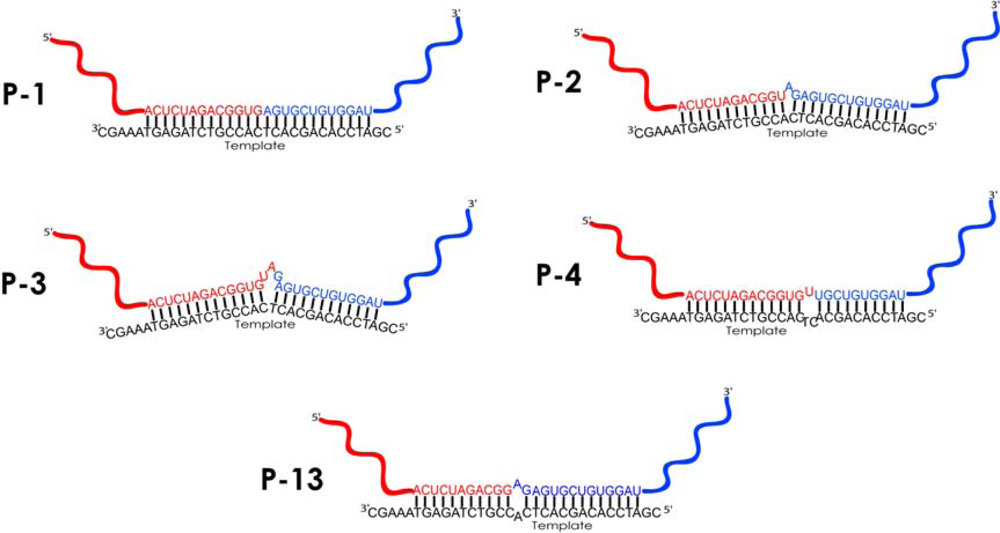
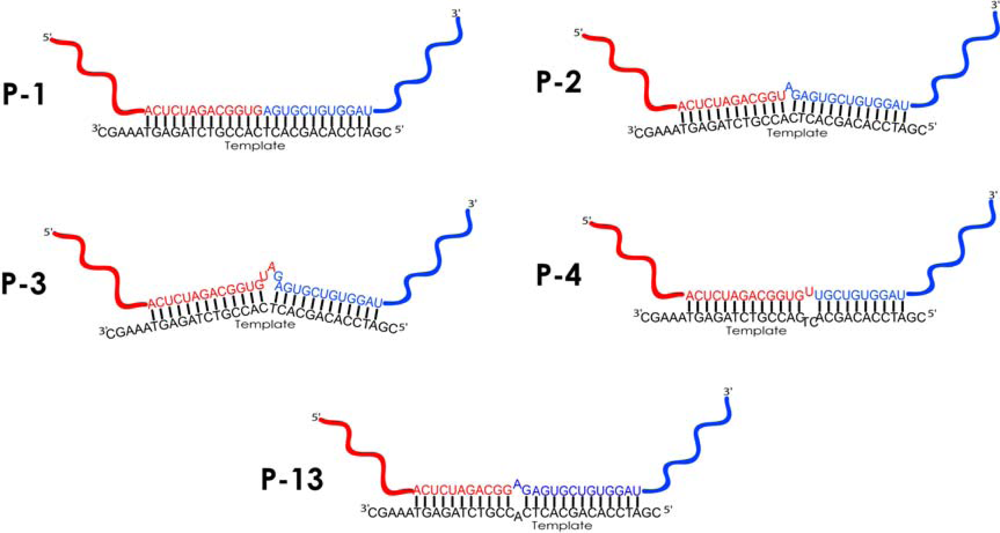
| P1 | 55 | [HFOR] CUAGACGGUGAGUGCUGUG [MREV]4) | 4 | 4 | Ligation in butt-to-butt manner* |
| P2 | 56 | [HFOR] CUAGACGGUAGAGUGCUGUG [MREV] | 1 | 1 | 1 nt RNA bulge loop* |
| P3 | 58 | [HFOR] CUAGACGGUGUAGAGUGCUGUG [MREV] | 86 | 8 | 3 nts RNA bulge loop* |
| P4 | 54 | [HFOR] CUAGACGGUGUUGCUGUG [MREV] | 1 | 1 | Asymmetric 3 nts internal loop * |
| P5 | 108 | [HFOR] CUAGACGGUGUAAAAAUCUCUAGC AGUGGCGCCAUGAGGGAAGAAUAUCGAAAGGAA CAGCAGAGUGCUGUG [MREV] | 1 | 1 | Template-independent recombination |
| P6 | 38 | [HFOR]AU[MREV] | 1 | 1 | |
| P7 | 39 | [HFOR] CUA[MREV] | 1 | 1 | |
| P8 | 107 | [HFOR] CUAGACGGUGUAAAAAUCUCUAGC AGUGGCGCAUGAGGGAAGAAUAUCGAAAGGAAC AGCAGAGUGCUGUG [MREV] | 2 | 2 | |
| P9 | 73 | [HFOR] CUAGACGGUGUAUCGAAAGGAACA GCAGAGUGCUGUG [MREV] | 2 | 2 | |
| P10 | 42 | [HFOR] CUAGAU[MREV] | 2 | 2 | |
| P11 | 76 | [HFOR] [HFOR] CUAGACGGUGUAGAGUGCU GUG [MREV] | 3 | 3 | 3 nts RNA bulge loop, double forward primer |
| P12 | 119 | [HFOR] UAGACGGUGUAAAAAUCUCUAGCA GUGGCGCCCGAACAGGGACAUGAGGGAAGAAUA UCGAAAGGAACAGCAGAGUGCUGUG [MREV] | 1 | 1 | Template-independent recombination |
| P13 | 55 | [HFOR] CUAGACGGAGAGUGCUGUG [MREV] | 1 | 1 | Symmetric 2 nts internal loop* |
| P14 | 105 | [HFOR] CUAGACGGUGUAAAAAUCUCUAGC AGUGGCGCGAGGGAAGAAUAUCGAAAGGAACAG CAGAGUGCUGUG [MREV] | 1 | 1 | Template-independent recombination |
| P15 | 109 | [HFOR] UAGACGGUGUAAAAAUCUCUAGCA GUGGCGCCCGAACAGGGACUUUAUCGGGAGGAA CAGCAGAGUGCUGUG [MREV] | 1 | 1 | Replacement of AA by GG (template-independent recombination) |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Nechaev, S.Y.; Lutay, A.V.; Vlassov, V.V.; Zenkova, M.A. Non-Enzymatic Template-Directed Recombination of RNAs. Int. J. Mol. Sci. 2009, 10, 1788-1807. https://doi.org/10.3390/ijms10041788
Nechaev SY, Lutay AV, Vlassov VV, Zenkova MA. Non-Enzymatic Template-Directed Recombination of RNAs. International Journal of Molecular Sciences. 2009; 10(4):1788-1807. https://doi.org/10.3390/ijms10041788
Chicago/Turabian StyleNechaev, Sergey Y., Alexei V. Lutay, Valentin V. Vlassov, and Marina A. Zenkova. 2009. "Non-Enzymatic Template-Directed Recombination of RNAs" International Journal of Molecular Sciences 10, no. 4: 1788-1807. https://doi.org/10.3390/ijms10041788
APA StyleNechaev, S. Y., Lutay, A. V., Vlassov, V. V., & Zenkova, M. A. (2009). Non-Enzymatic Template-Directed Recombination of RNAs. International Journal of Molecular Sciences, 10(4), 1788-1807. https://doi.org/10.3390/ijms10041788