Biodegradable Polydepsipeptides

Abstract
:1. Introduction
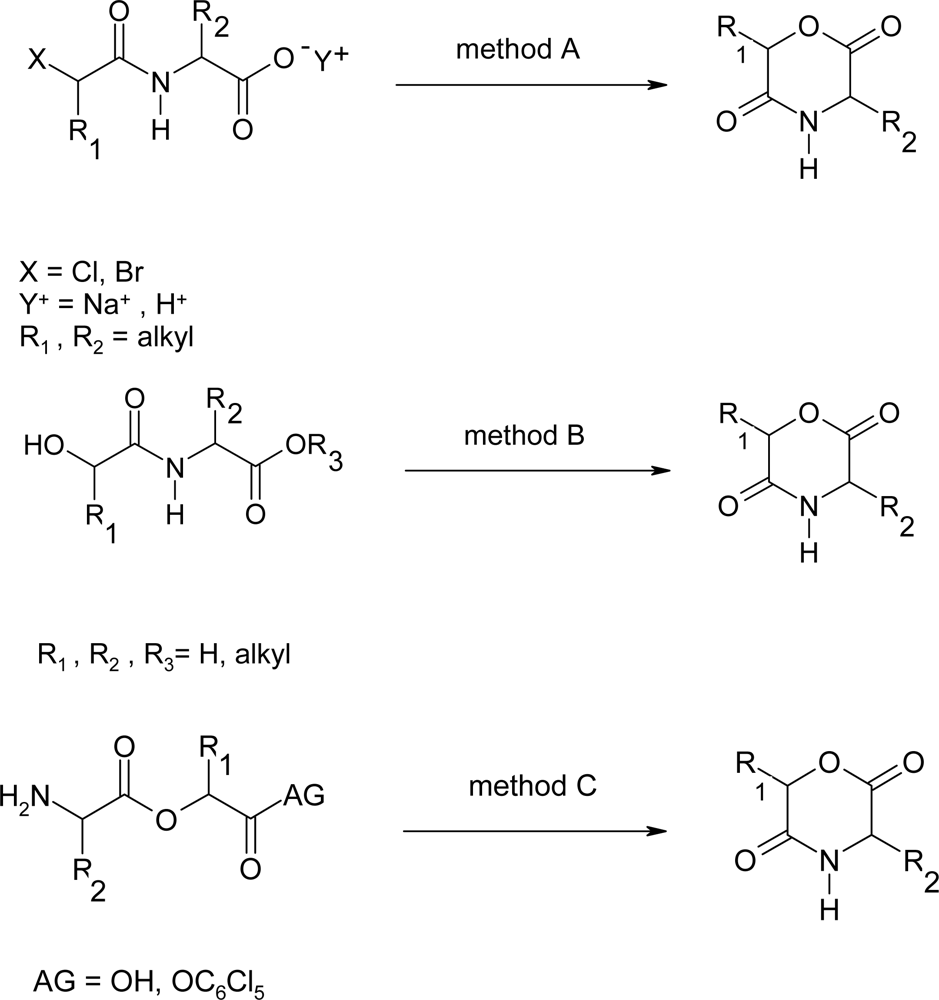
2. Synthesis of morpholine-2,5-dione derivatives
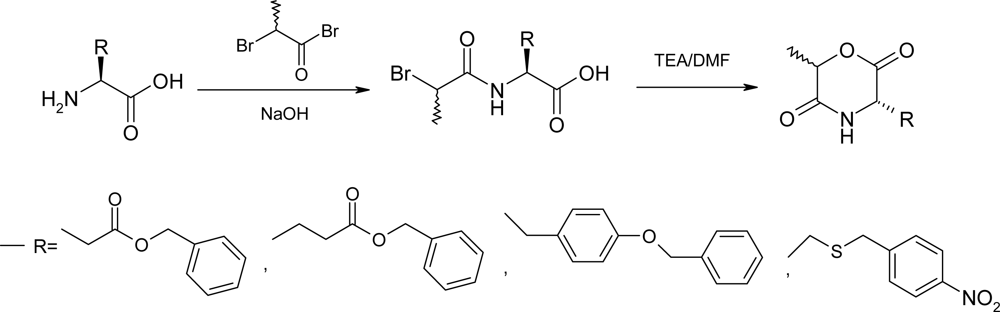
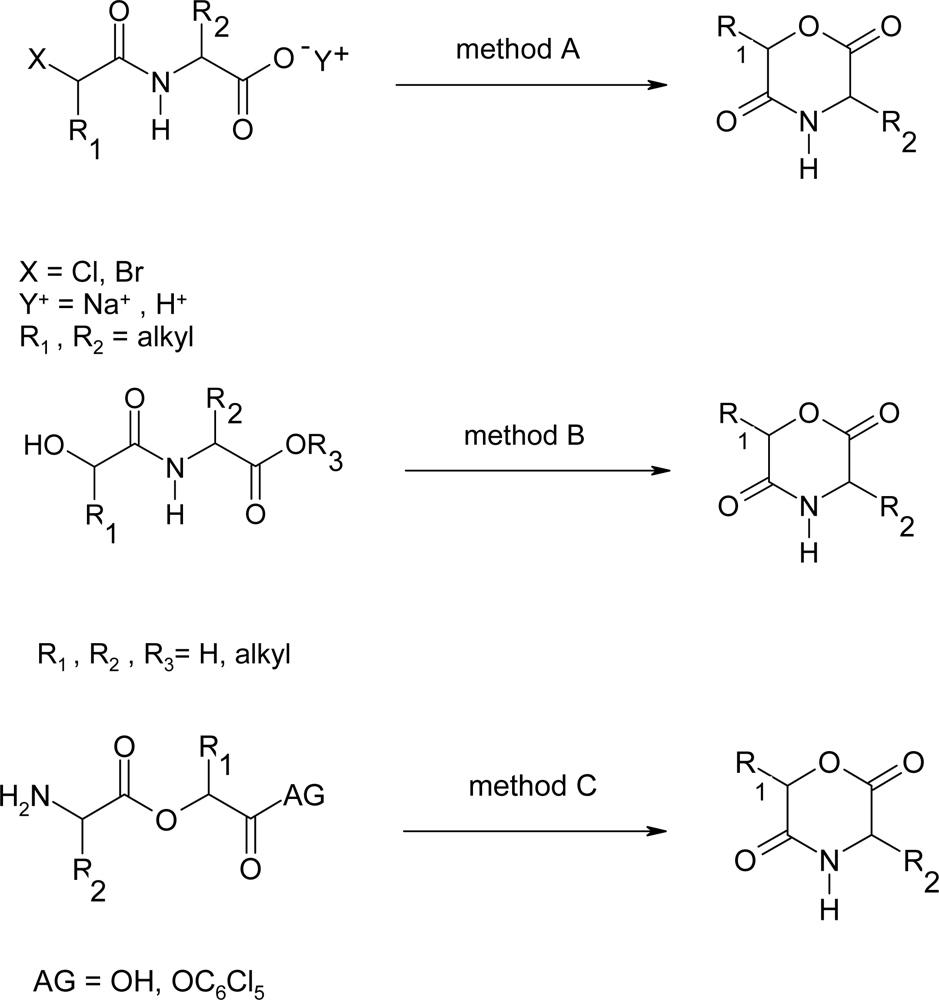
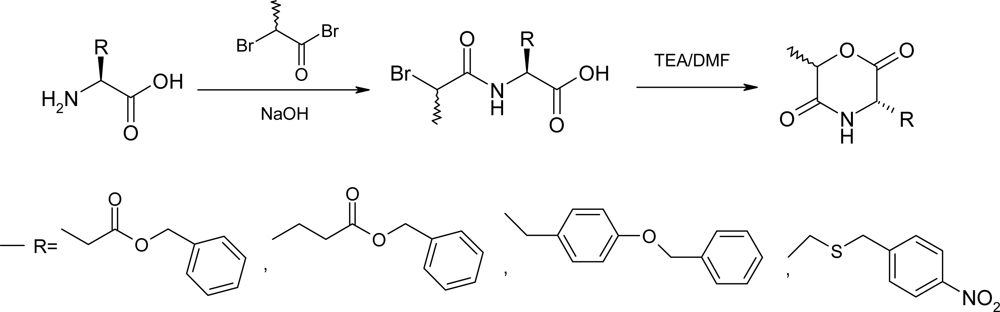
2.1. Method A
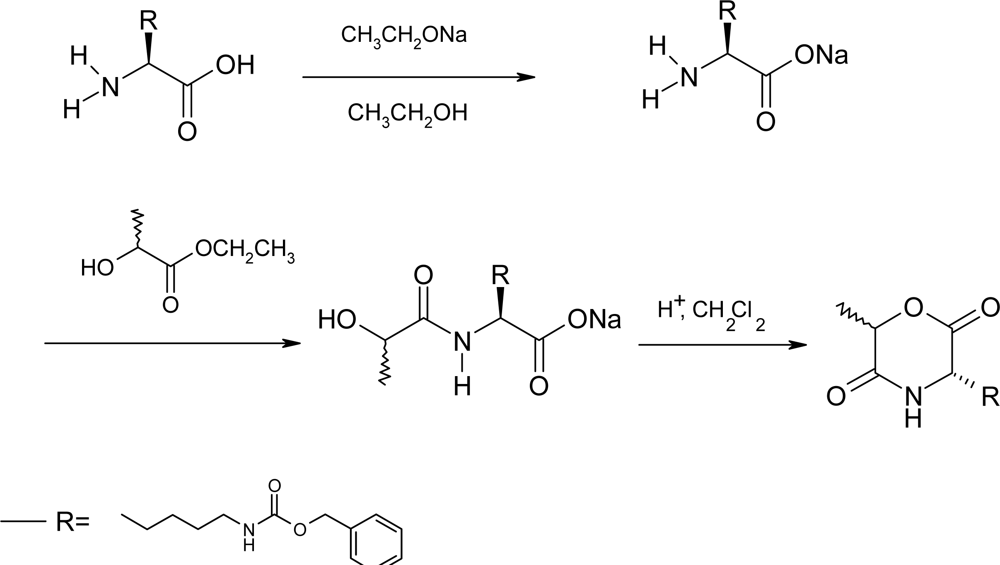
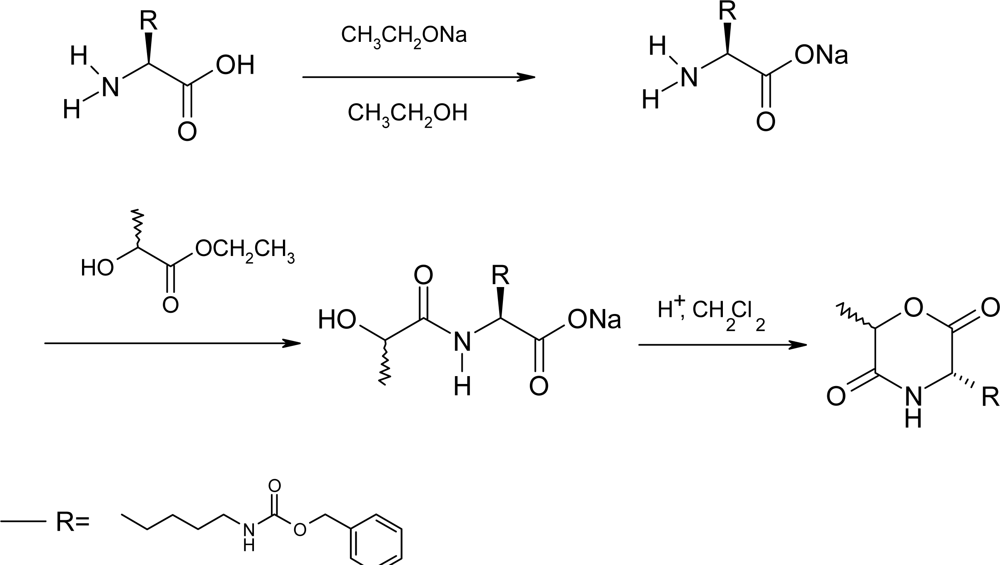
2.2. Method B
2.3. Method C
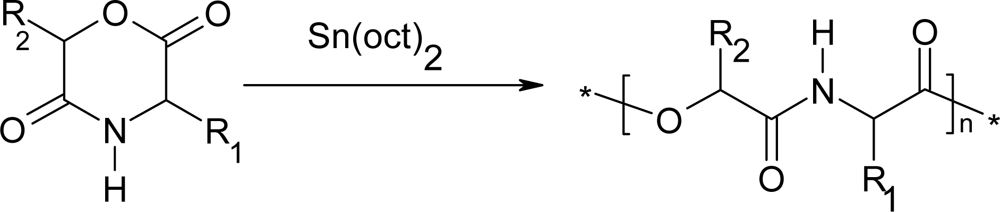
3. Ring-opening polymerization of morpholine-2,5-dione derivatives
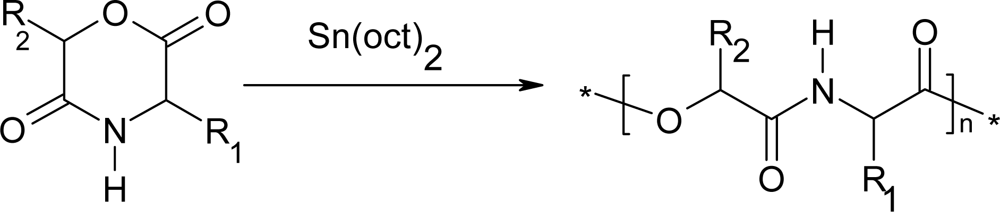
3.1. Ring-opening polymerization of morpholine-2,5-dione derivatives using organic Sn as catalyst
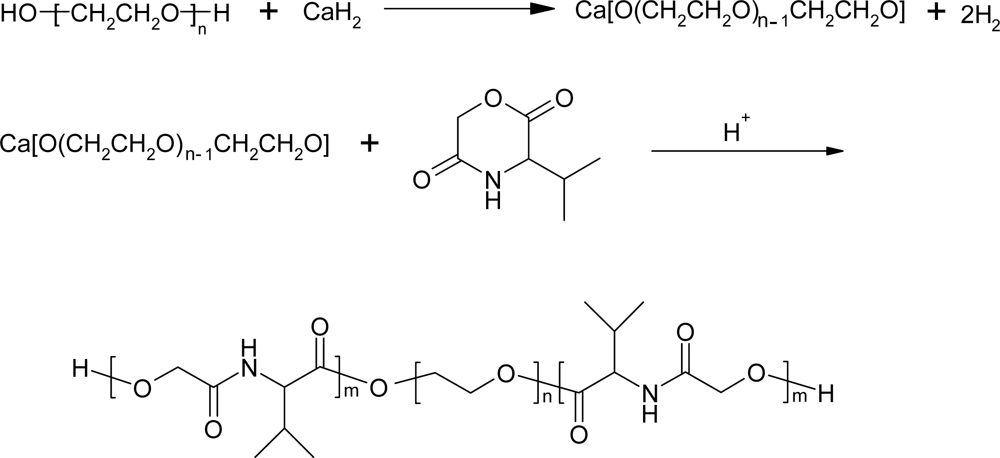
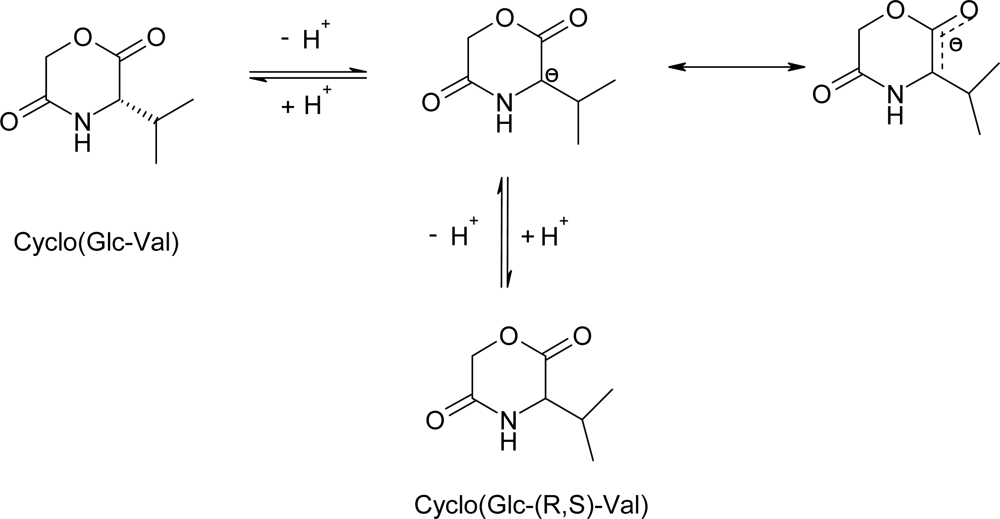
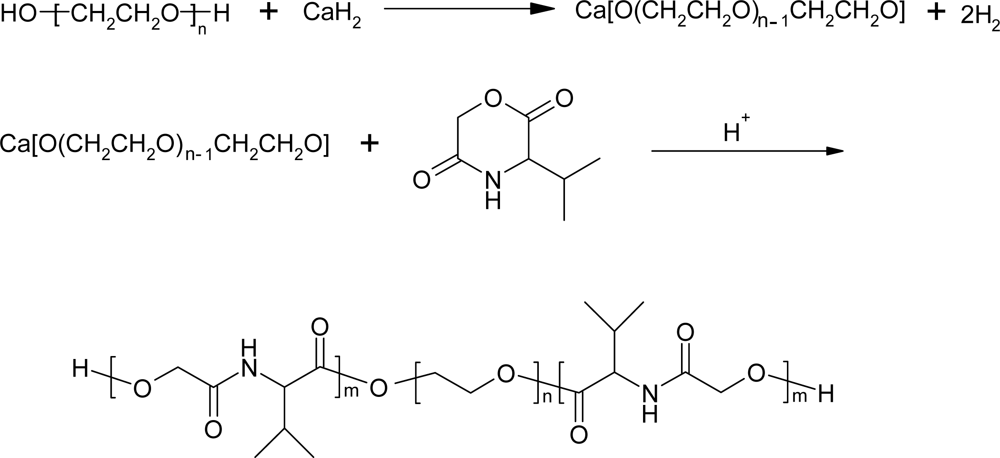
3.2. Ring-opening polymerization of morpholine-2,5-dione derivatives using CaH2 as catalyst
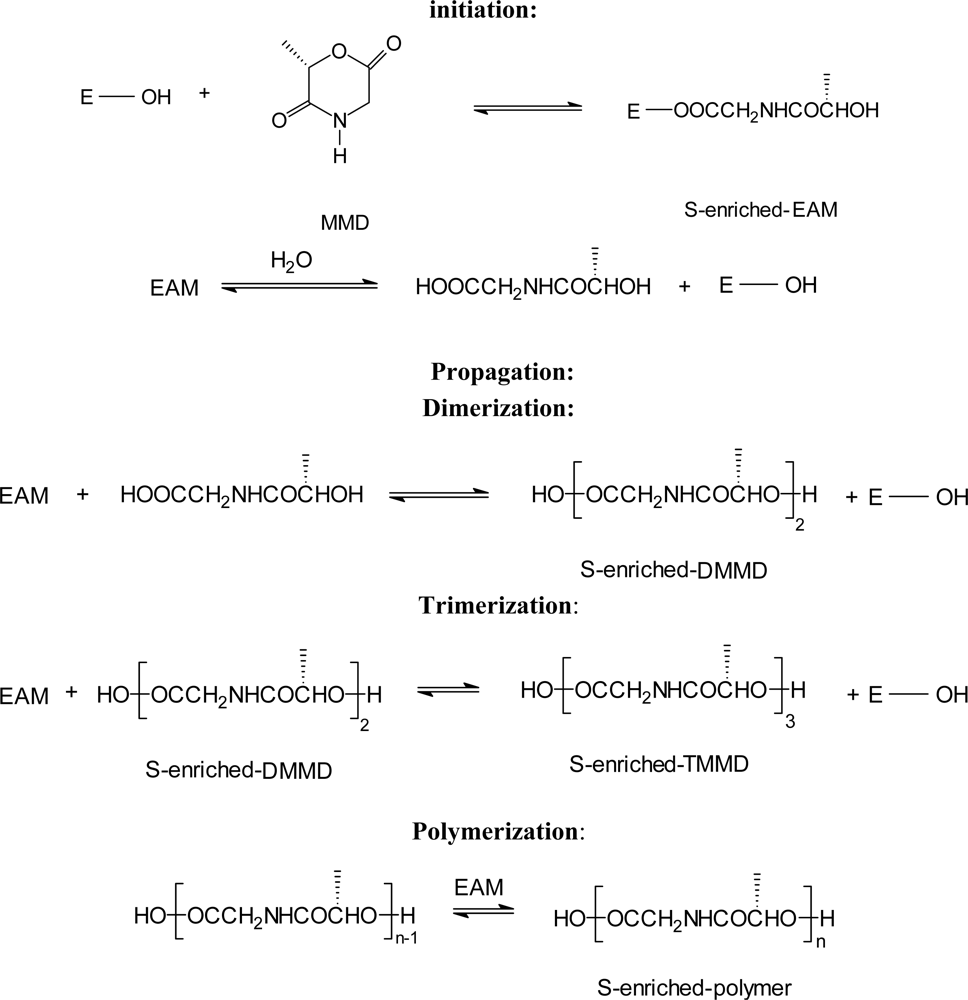
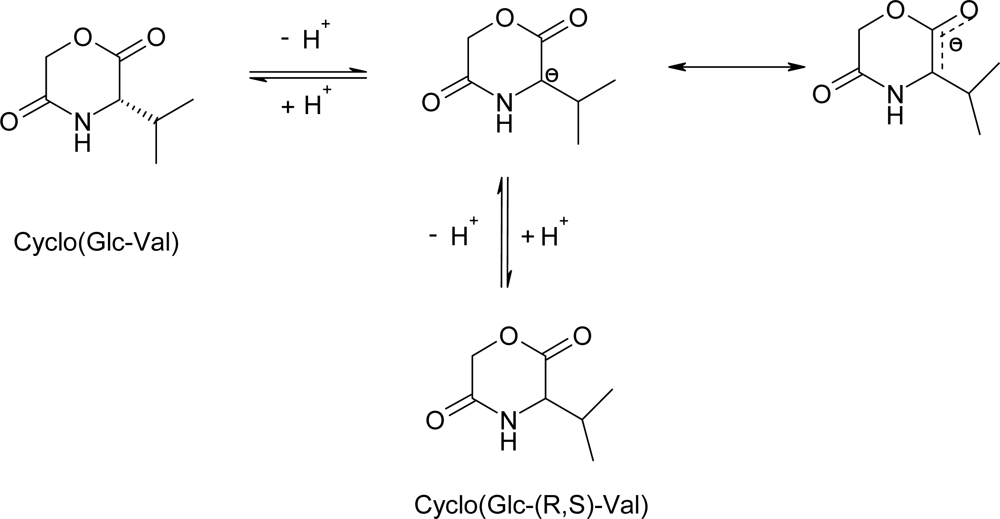
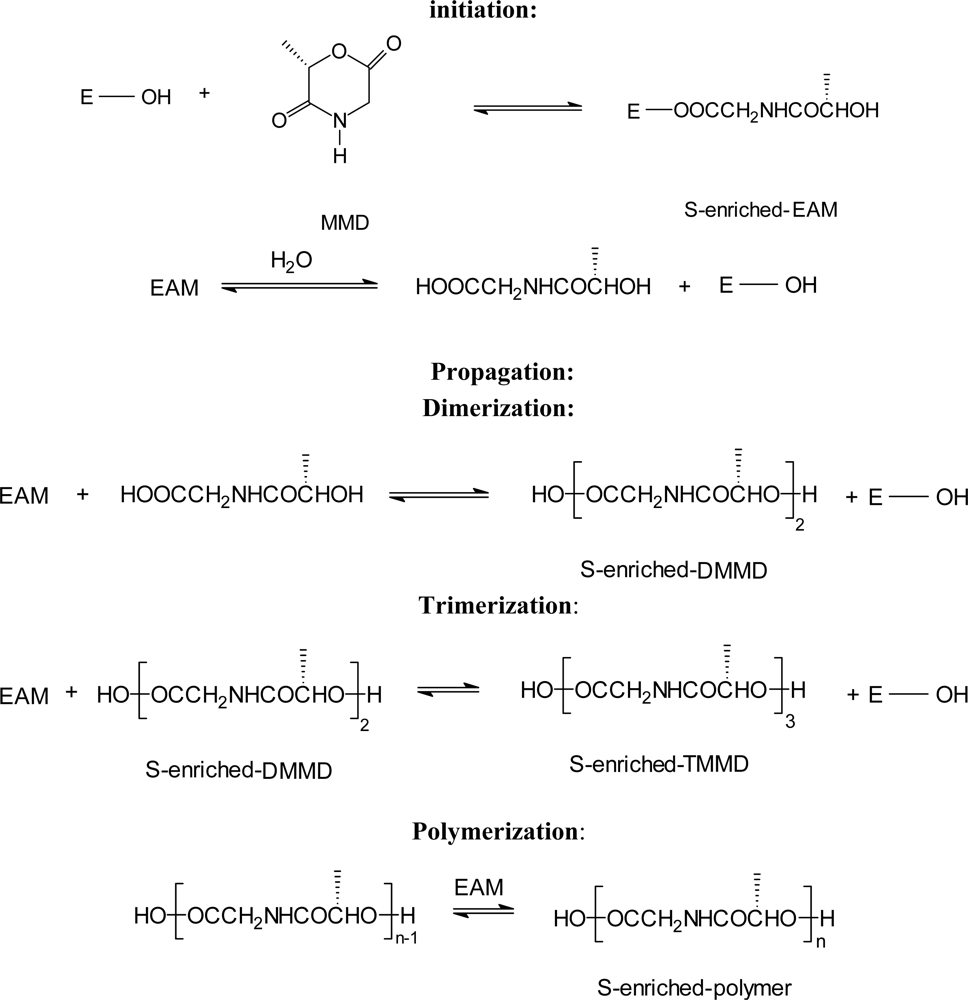
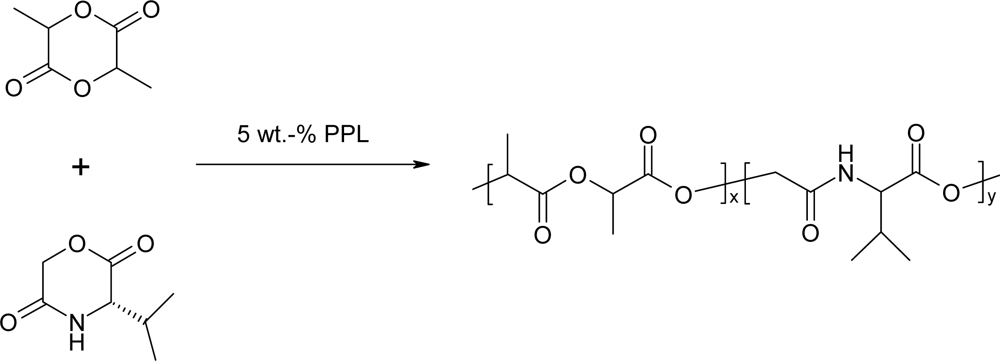
3.2. Ring-opening polymerization of morpholine-2,5-dione derivatives using enzymes as catalyst
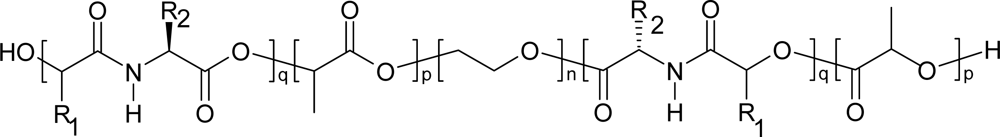
4. Biodegradation of polydepsipeptides and their copolymers
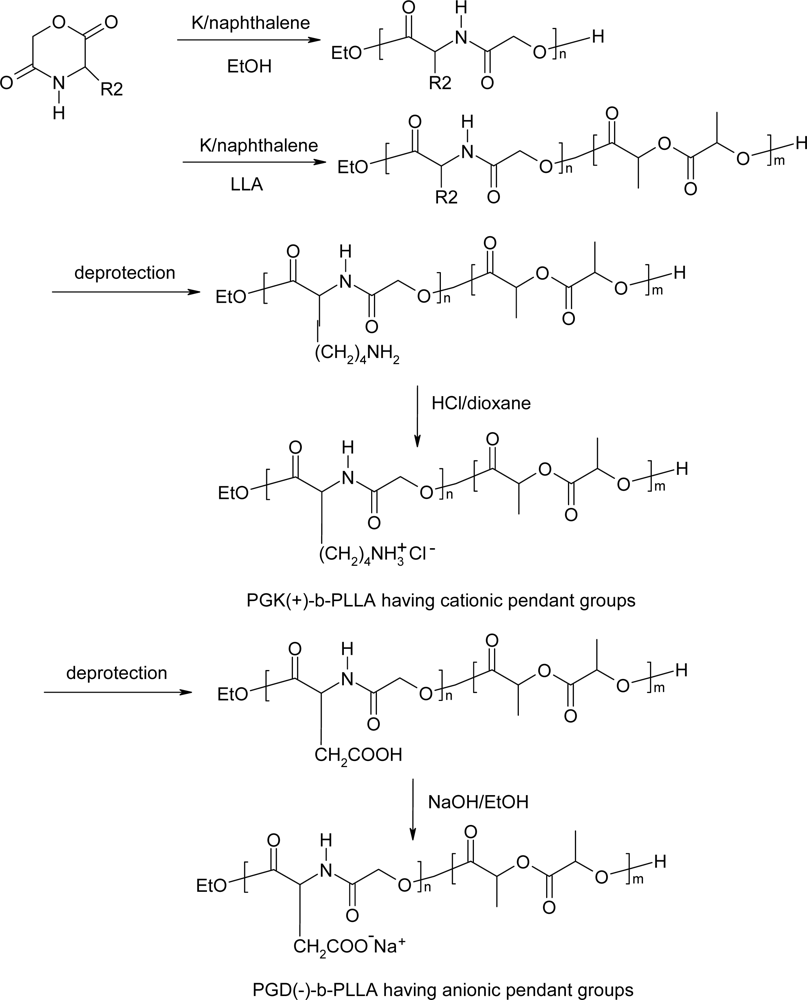
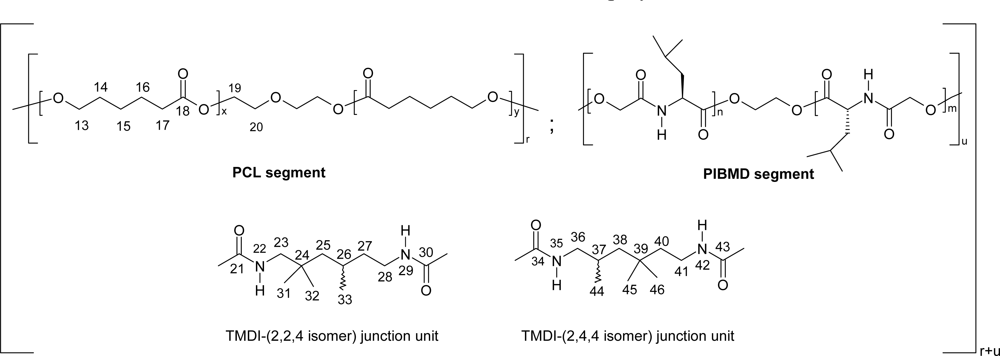
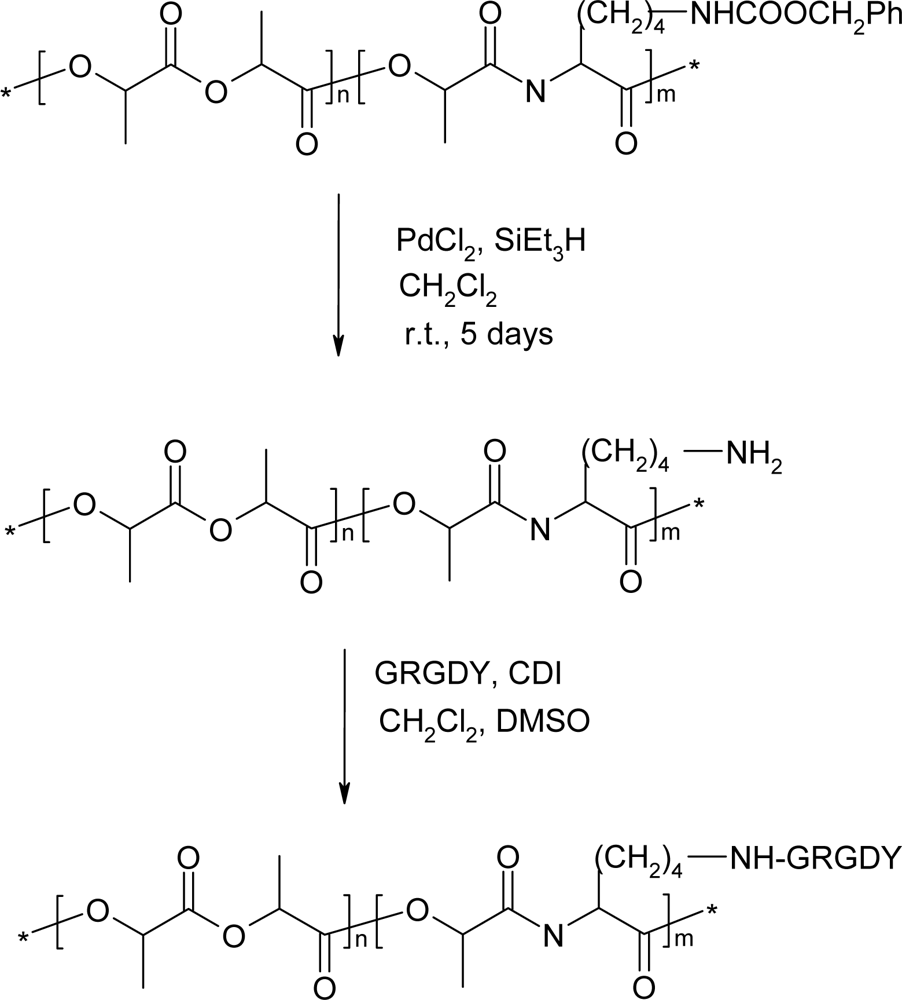
5. Polydepsipeptides and their copolymers used as biomaterials
5. Conclusions
Acknowledgments
References and Notes
- Lutz, JF; Andrieu, J; Uzgun, S; Rudolph, C; Agarwal, S. Biocompatible, thermoresponsive, and biodegradable: Simple preparation of “all-in-one” biorelevant polymers. Macromolecules 2007, 40, 8540–8543. [Google Scholar]
- Green, JJ; Zhou, BY; Mitalipova, MM; Beard, C; Langer, R; Jaenisch, R; Anderson, DG. Nanoparticles for gene transfer to human embryonic stem cell colonies. Nano Lett 2008, 8, 3126–3130. [Google Scholar]
- Kretlow, JD; Mikos, AG. From material to tissue: Biomaterial development, scaffold fabrication, and tissue engineering. AIChE J 2008, 54, 3048–3067. [Google Scholar]
- Wei, G; Ma, PX. Nanostructured biomaterials for regeneration. Adv. Funct. Mater 2008, 18, 3568–3582. [Google Scholar]
- Li, C; Wallace, S. Polymer-drug conjugates: Recent development in clinical oncology. Adv. Drug Deliv. Rev 2008, 60, 886–898. [Google Scholar]
- Xie, Z; Lu, T; Chen, X; Zheng, Y; Jing, X. Synthesis, self-assembly in water, and cytotoxicity of MPEG-block-PLLA/DX conjugates. J. Biomed. Mater. Res 2009, 88A, 238–245. [Google Scholar]
- Zhang, X; He, H; Yen, C; Ho, W; Lee, LJ. A biodegradable, immunoprotective, dual nanoporous capsule for cell-based therapies. Biomaterials 2008, 29, 4253–4259. [Google Scholar]
- Vert, M. Bioresorbable polymers for temporary therapeutic applications. Angew Makromol Chem 1989, (166/167), 155–168. [Google Scholar]
- Knüffermann, J. Synthesis and characterization of new bioresorbable polyesteramides, Dissertation, RWTH Aachen, 1999.
- Miller, RA; Brady, JM; Cutright, DE. Degradation rates of oral resorbable implants (polylactates and polyglycolates): Rate modification with changes in PLA/PGA copolymer ratios. J. Biomed. Mater. Res 1977, 11, 711–719. [Google Scholar]
- Tsuji, H; Ishida, T; Fukuda, N. Surface hydrophilicity and enzymatic hydrolyzability of biodegradable polyesters: 1. Effects of alkaline treatment. Polym. Int 2003, 52, 843–852. [Google Scholar]
- Tian, Y; Zhou, C; Zeng, Q; Yang, J; Han, F; Tian, J. Enhanced cell affinity of poly(L-lactide) film by immobilizing phosphonized chitosan. Appl. Surf. Sci 2008, 255, 446–448. [Google Scholar]
- Little, U; Buchanan, F; Harkin-Jones, E; McCaigue, M; Farrar, D; Dickson, G. Accelerated degradation behaviour of poly(ε-caprolactone) via melt blending with poly(aspartic acid-co-lactide) (PAL). Polym. Degrad. Stabil 2009, 94, 213–220. [Google Scholar]
- Gilding, DK; Reed, AM. Biodegradable polymers for use in surgery-polyglycolic/polylactic acid homo and copolymers. Polymer 1979, 20, 1459–1464. [Google Scholar]
- Borner, HG. Functional polymer-bioconjugates as molecular LEGO® bricks. Macromol. Chem. phys 2007, 208, 124–130. [Google Scholar]
- Lin, J; Zhu, J; Chen, T; Lin, S; Cai, C; Zhang, L; Zhuang, Y; Wang, X. Drug releasing behavior of hybrid micelles containing polypeptide triblock copolymer. Biomaterials 2009, 30, 108–117. [Google Scholar]
- Hamidi, M; Azadi, A; Rafiei, P. Hydrogel nanoparticles in drug delivery. Adv. Drug Deliv. Rev 2008, 60, 1638–1649. [Google Scholar]
- Deming, TJ. Synthetic polypeptides for biomedical applications. Prog. Polym. Sci 2007, 32, 858–875. [Google Scholar]
- Sokolsky-Papkov, M; Agashi, K; Olaye, A; Shakesheff, K; Domb, AJ. Polymer carriers for drug delivery in tissue engineering. Adv. Drug Deliv. Rev 2007, 59, 187–206. [Google Scholar]
- Ridge, B; Rydon, HN; Snell, CR. Polypeptides. Part XXI. Synthesis of some sequential macromolecular polypeptolides of L-leucine and L-2-hydroxy-4-methylpentanoic acid. J Chem Soc, Perkin Trans 1 1972, 2041–2046. [Google Scholar]
- Coin, I; Dolling, R; Krause, E; Bienert, M; Beyermann, M; Sferdean, CD; Carpino, LA. Depsipeptide methodology for solid-phase peptide synthesis: Circumventing side reactions and development of an automated technique via depsidipeptide units. J. Org. Chem 2006, 71, 6171–6177. [Google Scholar]
- Mathias, LJ; Fuller, WD; Nissen, D; Goodman, M. Polydepsipeptides. 6. Synthesis of sequential polymers containing varying ratios of L-alanine and L-lactic acid. Macromolecules 1978, 11, 534–540. [Google Scholar]
- Nobu, E; Hiroyuki, O; Keiichi, Y; Ryoichi, K. Synthesis of sequential polydepsipeptide microspheres as a controlled drug delivery system. Pept. Sci 2005, 2004, 621–624. [Google Scholar]
- Katakai, R; Kobayashi, K; Yamada, K; Oku, H; Emori, N. Synthesis of sequential polydepsipeptides utilizing a new approach for the synthesis of depsipeptides. Biopolymers 2004, 73, 641– 644. [Google Scholar]
- Katakai, R; Goodman, M. Polydepsipeptides. 9. Synthesis of sequential polymers containing some amino acids having polar side chains and (S)-lactic acid. Macromolecules 1982, 15, 25–30. [Google Scholar]
- Katakai, R. Synthesis of sequential polydepsipeptides involving depsipeptide formation by the 2-nitrophenylsulphenyl N-carboxy α-amino acid anhydride (Nps–NCA) method. J Chem Soc, Perkin Trans 1 1987, 2249–2251. [Google Scholar]
- Stawikowski, M; Cudic, P. Depsipeptide synthesis, in Peptide characterization and application protocols. In Series: Methods in Molecular Biology; Fields, GB, Ed.; Humana Press: Totowa, New Jersey USA, 2007; Volume 386, pp. 321–339. [Google Scholar]
- Yoshida, M; Asano, M; Kumakura, M; Katakai, R; Mashimo, T; Yuasa, H; Imai, K; Yamanaka, H. A new biodegradable polydepsipeptide microsphere for application in drug delivery systems. Colloid Polym. Sci 1990, 268, 726–730. [Google Scholar]
- Yoshida, M; Asano, M; Kumakura, M; Katakai, R; Mashimo, T; Yuasa, H; Yamanaka, H. Sequential polydepsipeptides containing tripeptide sequences and alpha-hydroxy acids as biodegradable carriers. Eur. Polym. J 1991, 27, 325–329. [Google Scholar]
- Chadwick, AF; Pascu, E. The resolution and rates of hydrolysis of d,l-α-bromopropionic acid and its glycine derivatives. J. Chem. Soc 1943, 65, 392–402. [Google Scholar]
- Wang, D; Feng, XD. Copolymerization of epsilon-caprolactone with (3S)-3-[(benzyloxycarbonyl)methyl]morpholine-2,5-dione and the C-13 NMR sequence analysis of the copolymer. Macromolecules 1998, 31, 3824–3831. [Google Scholar]
- Zhang, GD; Wang, D; Feng, XD. Preliminary study of hydrogen bonding in (3S)-3-[(benzyloxycarbonyl)methyl]morpholine-2,5-dione and its effect on polymerization. Macromolecules 1998, 31, 6390–6092. [Google Scholar]
- In′t Veld, PJA; Ye, WP; Klap, R; Dijkstra, PJ; Feijen, J. Copolymerization of ε-caprolactone and morpholine-2,5-dione derivatives. Makromol. Chem 1992, 193, 1927–1942. [Google Scholar]
- Vinsova, J. Morpholine-2,5-diones — Their preparation and exploitation. Chem. Listy 2001, 95, 22–27. [Google Scholar]
- Kricheldorf, HR; Hauser, K. Polylactones, 45, Homo- and copolymerizations of 3-methylmorpholine-2,5-dione initiated with a cyclic tin alkoxide. Macromol. Chem. Phys 2001, 202, 1219–1226. [Google Scholar]
- Hartwig, W; Schoellkopf, U. Asymmetrische Synthesen über heterocyclische Zwischenstufen, XVI. Enantioselektive Synthese von α-Alkyl-α-phenylglycinen durch Alkylieren von an C-6 chiral substituierten 3,6-Dihydro-3-phenyl-2H-1,4-oxazin-2-onen. Liebigs Ann Chem 1982, 1952–1970. [Google Scholar]
- Nakamura, CE; Cosimo, RD; Moran, JR. Process for the preparation of 3-, 6–substituted 2,5-morpholinediones, US Patent No. 5466801. 1995. [Google Scholar]
- Kardassis, G; Brungs, P; Nothhelfer, C; Steckhan, E. Electrogenerated chiral cationic glycine equivalents – Parts 2: Chiral 3-methoxy-2,5-morpholinediones from (S)-cz-hydroxy acids and dimethyl aminomalonate. Tetrahedron 1998, 54, 3479–3488. [Google Scholar]
- Nissen, D; Gilon, C; Goodmann, M. Polydepsipeptides. 4. Synthesis of the alternating polydepsipeptides poly(Ala-Lac) and poly(Val-Lac). Makromol. Chem. Suppl 1975, 1, 23–53. [Google Scholar]
- In’t Veld, PJA. Biodegradable polyesteramide. Dissertation University of Twente, Netherlands, 1992.
- Nicolaou, KC; Safina, BS; Winssinger, N. A new photolabile linker for the photoactivation of carboxyl groups. Syn Lett 2001, SI, 900–903. [Google Scholar]
- In′t Veld, PJA; Dijkstra, PJ; Feijen, J. Synthesis of biodegradable polyesteramides with pendant functional groups. Makromol. Chem 1992, 193, 2713–2730. [Google Scholar]
- Yao, J; Yang, Q; Fan, X; Wang, N; Guo, B. Synthesis and characterization of inexpensive 3-Nε-benzyloxycarbonyl-L-lysyl-morpholine-2,5-dione. Front. Chem. China 2007, 2, 183–187. [Google Scholar]
- Yao, J; Yang, Q; Fan, X; Wang, N; Guo, B. Synthesis and characterization of inexpensive 3-Nε-benzyloxycarbonyl-L-lysyl-morpholine-2,5-dione. J. Northwestern Polytech. Univ 2006, 24, 444–447. [Google Scholar]
- Helder, J; Kohn, FE; Sato, S; van den Berg, JW; Feijen, J. Synthesis of poly[oxyethylidenecarbonylimino(2-oxoethylene)] [poly(glycine-D,L-lactic acid)] by ring opening polymerization. Makromol. Chem., Rapid Commun 1985, 6, 9–14. [Google Scholar]
- Jörres, V. Biodegradable polyester amides-synthesis and polymerization of morpholine-2,5-diones. Dissertation, RWTH Aachen, 1997.
- John, G; Tsuda, S; Morita, M. Synthesis and modification of new biodegradable copolymers: Serine/glycolic acid based copolymers. J. Polym. Sci., Part A: Polym. Chem 1997, 35, 1901–1907. [Google Scholar]
- Ouchi, T; Nozaki, T; Okamoto, Y; Shiratani, M; Okya, Y. Synthesis and enzymatic hydrolysis of polydepsipeptides with functionalized pendant groups. Macromol. Chem. Phys 1996, 197, 1823–1833. [Google Scholar]
- Dijkstra, PJ; Feijen, J. Synthetic pathways to polydepsipeptides. Macromol. Sym 2000, 153, 67–76. [Google Scholar]
- Chisholm, MH; Galucci, J; Krempner, C; Wiggenhorn, C. Comments on the ring-opening polymerization of morpholine-2,5-dione derivatives by various metal catalysts and characterization of the products formed in the reactions involving R(2)SnX(2), where X = OPr(i) and NMe(2) and R = Bu(n), Ph and p-Me(2)NC(6)H(4). Dalton Trans 2006, 846–851. [Google Scholar]
- Shakaby, SW; Koelmel, DF. Copolymers of p-dioxanone and 2,5-morpholinediones and surgical devices formed therefrom having accelerated absorption characteristics. Eur Pat Appl 1983, 86613. [Google Scholar]
- Yonezawa, N; Toda, F; Hasegawa, M. Synthesis of polydepsipeptides: Ring-opening polymerization of 6-isopropylmorpholine-2,5-dione and 6-isopropyl-4-methylmorpholine-2,5-dione. Makromol. Chem., Rapid Commun 1985, 6, 607–611. [Google Scholar]
- Helder, J; Lee, SJ; Kim, SW; Feijen, J. Copolymers of D,L-lactic acid and glycine. Makromol. Chem., Rapid Commun 1986, 7, 193–198. [Google Scholar]
- Samyn, C; van Beylen, M. Polydespeptides: Ring-opening polymerization of 3-methyl-2,5-morpholinedione, 3,6-dimethyl-2,5-morpholinedione and copolymerization thereof with D,L-lactide. Makromol. Chem., Macromol. Sym 1988, 19, 225–234. [Google Scholar]
- Fung, FN; Glowaky, RC. Bioabsorbable polydepsipeptides, their preparation and use. Eur Pat Appl EP 1989, 322154. [Google Scholar]
- In′t Veld, PJA; Shen, ZR; Takens, GAJ; Dijkstra, PJ; Feijen, J. Glycine/Glycolic acid based copolymers. J. Polym. Sci., Part A: Polym. Chem 1994, 32, 1063–1069. [Google Scholar]
- Hrkach, JS; Ou, J; Lotan, N; Langer, R. Poly(L-lactic acid-co-amino acid) graft copolymers: A class of functional biodegradable biomaterials. In Hydrogels and Biodegradable Polymers for Bioapplications; Ottenbrite, RM, Huang, SJ, Park, K, Eds.; ACS Symposium SeriesVolume 627, American Chemical Society: Washington DC, USA, 1996; Ch. 8, pp. 93–102. [Google Scholar]
- Ouchi, T; Shiratani, M; Jinno, M; Hirao, M; Ohya, Y. Synthesis of poly[(glycolic acid)-alt-(L-aspartic acid)] and its biodegradation behavior in vitro. Makromol. Chem., Rapid Commun 1993, 14, 825–831. [Google Scholar]
- Barrera, DA; Zylstra, E; Lansbury, PT; Langer, R. Synthesis and RGD peptide modification of a new biodegradable copolymer: Poly(lactic acid-co-lysine). J. Am. Chem. Soc 1993, 115, 11010–11011. [Google Scholar]
- Ouchi, T; Nozaki, T; Ishikawa, A; Fujimoto, I; Ohya, Y. Synthesis and enzymatic hydrolysis of lactic acid-depsipeptide copolymers with functionalized pendant groups. J. Polym. Sci., Part A: Polym. Chem 1997, 35, 377–383. [Google Scholar]
- Ouchi, T; Hamada, A; Ohya, Y. Biodegradable microspheres having reactive groups prepared from L-lactic acid-depsipeptide copolymers. Macromol. Chem. Phys 1999, 200, 436–441. [Google Scholar]
- John, G; Morita, M. Biodegradable cross-linked microspheres from poly(ε-caprolactone-co-glycolic acid-co-L-serine) based polydepsipeptides. Macromol. Rapid Commun 1999, 20, 265–268. [Google Scholar]
- Feng, Y; Klee, D; Höcker, H. Biodegradable block copolymers with poly(ethylene oxide) and poly(glycolic acid-valine) blocks. J. Appl. Polym. Sci 2002, 86, 2916–2919. [Google Scholar]
- Ouchi, T; Toyohara, M; Arimura, H; Ohya, Y. Preparation of poly(L-lactide)-based microspheres having a cationic or anionic surface using biodegradable surfactants. Biomacromolecules 2002, 3, 885–888. [Google Scholar]
- Klibanov, AM. Enzyme Work in Organic Solvents. Chemtech 1986, 16, 354–359. [Google Scholar]
- Yoshizawa-Fujita, M; Saito, C; Takeoka, Y; Rikukawa, M. Lipase-catalyzed polymerization of l-lactide in ionic liquids. Polym. Advan. Technol 2008, 19, 1396–1400. [Google Scholar]
- Watterson, AC; Parmar, VS; Kumar, R; Sharma, SK; Shakil, NA; Tyagi, R; Sharma, AK; Samuelson, LA; Kumar, J; Nicolosi, R; Shea, T. Indo-U.S. collaborative studies on biocatalytic generation of novel molecular architectures. Pure Appl. Chem 2005, 77, 201–208. [Google Scholar]
- Lassalle, VL; Ferreira, ML. Lipase-catalyzed synthesis of polylactic acid: An overview of the experimental aspects. J. Chem. Technol. Biotechnol 2008, 83, 1493–1502. [Google Scholar]
- Numata, K; Srivastava, RK; Finne-Wistrand, A; Albertsson, AC; Doi, Y; Abe, H. Branched poly(lactide) synthesized by enzymatic polymerization: Effects of molecular branches and stereochemistry on enzymatic degradation and alkaline hydrolysis. Biomacromolecules 2007, 8, 3115–3125. [Google Scholar]
- Feng, Y; Knüfermann, J; Klee, D; Höcker, H. Enzyme-catalyzed ring-opening polymerization of 3(S)-isopropyl-morpholine-2,5-dione. Macromol. Rapid Commun 1999, 20, 88–90. [Google Scholar]
- Feng, Y; Knüfermann, J; Klee, D; Höcker, H. Lipase-catalyzed ring-opening polymerization of 3(S)-isopropyl-morpholine-2,5-dione. Macromol. Chem. Phys 1999, 200, 1506–1514. [Google Scholar]
- Cordova, A; Iversen, T; Hult, K; Martinelle, M. Lipase-catalysed formation of macrocycles by ring-opening polymerisation of ε-caprolactone. Polymer 1998, 39, 6519–6524. [Google Scholar]
- Feng, Y; Klee, D; Keul, H; Höcker, H. Lipase-catalyzed ring-opening polymerization of morpholine-2,5-dione derivatives: A novel route to the synthesis of poly(ester amide)s. Macromol. Chem. Phys 2000, 201, 2670–2675. [Google Scholar]
- Feng, Y; Klee, D; Höcker, H. Lipase catalyzed ring-opening polymerization of 6(S)-methyl-morpholine-2,5-dione. J. Polym. Sci., Part A: Polym. Chem 2005, 43, 3030–3039. [Google Scholar]
- Ritter, H. The use of enzymes in polymer synthesis. Trends Polym. Sci 1993, 1, 171–173. [Google Scholar]
- Namekawa, S; Suda, S; Uyama, H; Kobayashi, S. Lipase-catalyzed ring-opening polymerization of lactones to polyesters and its mechanistic aspects. Int. J. Biol. Macromol 1999, 25, 145–151. [Google Scholar]
- Svirkin, YY; Xu, J; Gross, RA; Kaplan, DL; Swift, G. Enzyme-catalyzed stereoelective ring-opening polymerization of α-methyl-β-propiolactone. Macromolecules 1996, 29, 4591–4597. [Google Scholar]
- Albertsson, AC; Srivastava, RK. Recent developments in enzyme-catalyzed ring-opening polymerization. Adv. Drug Deliv. Rev 2008, 60, 1077–1093. [Google Scholar]
- Feng, Y; Klee, D; Höcker, H. Lipase catalyzed copolymerization of 3(S)-isopropyl-morpholine-2,5-dione and D,L- lactide. Macromol. Biosci 2004, 4, 587–590. [Google Scholar]
- Schakenraad, JM; Nieuwenhuis, P; Molenaar, I; Helder, J; Dijkstra, PJ; Feijen, J. In vivo and in vitro degradation of glycine/DL-lactic acid copolymers. J. Biomed. Mater. Res 1989, 23, 1271–1288. [Google Scholar]
- Helder, J; Dijkstra, PJ; Feijen, J. In vitro degradation of glycin/DL-lactic acid copolymers. J. Biomed. Mater. Res 1990, 24, 1005–1020. [Google Scholar]
- Barrera, DA; Zylstre, E; Lansbury, PT; Langer, R. Copolymerization and degradation of poly(lactic acid-co-lysine). Macromolecules 1995, 28, 425–432. [Google Scholar]
- Feng, Y; Klee, D; Höcker, H. Synthesis of poly[(lactic acid)-alt- or co-((S)-aspartic acid)] from (3S, 6R,S)-3-[(benzyloxycarbonyl)methyl]-6-methylmorpholine-2,5-dione. Macromol. Chem. Phys 2002, 203, 819–824. [Google Scholar]
- Ouchi, T; Ohya, Y. Design of lactide copolymers as biomaterials. J. Polym. Sci., Part A: Polym. Chem 2004, 42, 453–462. [Google Scholar]
- Ouchi, T; Sasakawa, M; Arimura, H; Toyohara, M; Ohya, Y. Preparation of poly[DL-lactide-co-glycolide]-based microspheres containing protein by use of amphiphilic diblock copolymers of depsipeptide and lactide having ionic pendant groups as biodegradable surfactants by W/O/W emulsion method. Polymer 2004, 45, 1583–1589. [Google Scholar]
- Elisseeff, J; Anseth, K; Langer, R. Synthesis and characterization of photocrosslinked polymers based on poly(L-lactic acid-co-L-aspartic acid). Macromolecules 1997, 30, 2182–2184. [Google Scholar]
- John, G; Morita, M. Synthesis of polymer network scaffolds and microspheres based on poly(ε-caprolactone-co-glycolic acid-co-L-serine). Mater. Sci. Eng. C 2000, 13, 91–95. [Google Scholar]
- Zhao, Y; Hu, Y; Cheng, S; Chen, J. Degradation of morpholine-2,5-dione derivative copolymer in vitro. Polym. Bull 2008, 61, 35–41. [Google Scholar]
- Shirahama, H; Umemoto, K; Yasuda, H. Synthesis and enzymatic degradation of optically active depsipeptide copolymers. J. Biomater. Sci., Polym. Ed 1999, 10, 621–639. [Google Scholar]
- Ouchi, T; Nozaki, T; Ishikawa, A; Fujimoto, I; Ohya, Y. Synthesis and enzymatic hydrolysis of lactic acid-depsipeptide copolymers with functionalized pendant groups. J. Polym. Sci., Part A: Polym. Chem 1997, 35, 377–383. [Google Scholar]
- Ouchi, T; Seike, H; Nozaki, T; Ohya, Y. Synthesis and characteristics of polydepsipeptide with pendant thiol groups. J. Polym. Sci., Part A: Polym. Chem 1998, 36, 1283–1290. [Google Scholar]
- Ohya, Y; Toyohara, M; Sasakawa, M; Arimura, H; Ouchi, T. Thermosensitive biodegradable polydepsipeptide. Macromol. Biosci 2005, 5, 273–276. [Google Scholar]
- Feng, Y; Behl, M; Kelch, S; Lendlein, A. Biodegradable multiblock copolymers based on oligodepsipeptides having shape-memory properties. Macromol. Biosci 2009, 9, 45–54. [Google Scholar]
- Lendlein, A; Kelch, S. Shape-memory polymers as stimulisensitive implant materials. Clin. Hemorheol. Microcirc 2005, 32, 105–116. [Google Scholar]
- Tian, T; Chen, Q; Yu, C; Shen, J. Amino-terminated poly(ethylene glycol) as the initiator for the ring-opening polymerization of 3-methylmorpholine-2,5-dione. Eur. Polym. J 2003, 39, 1935–1938. [Google Scholar]
- Feng, Y; Klee, D; Höcker, H. Synthesis and characterization of new ABA triblock copolymers with poly[3(S)-isobutyl-morpholine-2,5-dione] and polyethylene oxide blocks. Macromol. Chem. Phys 1999, 200, 2276–2283. [Google Scholar]
- Feng, Y; Klee, D; Höcker, H. New biomaterial: Triblock copolymers of poly[3(S)-isobutyl-morpholine-2,5-dione]-poly(ethylene oxide). Materialwiss. Werkst 1999, 30, 862–868. [Google Scholar]
- Feng, Y; Klee, D; Keul, H; Höcker, H. Synthesis and characterization of new block copolymers with poly(ethylene oxide) and poly[3(S)-sec-butylmorpholine-2,5-dione] sequences. Macromol. Biosci 2001, 1, 30–39. [Google Scholar]
- Feng, Y; Klee, D; Keul, H; Höcker, H. Biodegradable copolymers based on poly(ethylene oxide), polylactide and polydepsipeptide sequences with functional groups. e-Polymers 2001, (3). [Google Scholar]
- Ouchi, T; Miyazaki, H; Arimura, H; Tasaka, F; Hamada, A; Ohya, Y. Synthesis of biodegradable amphiphilic AB-Type diblock copolymers of lactide and depsipeptide with pendant reactive groups. J. Polym. Sci., Part A: Polym. Chem 2002, 40, 1218–1225. [Google Scholar]
- Abayasinghe, NK; Perera, KPU; Thomas, C; Daly, A; Suresh, S; Burg, K; Harrison, GM; Smith, DW, Jr. Amido-modified polylactide for potential tissue engineering applications. J. Biomater. Sci., Polym Ed 2004, 15, 595–606. [Google Scholar]
- Zhao, YC; Liu, XB; Ge, JB; Gong, FR; Chen, GY; Hu, YJ; Cheng, SJ; Chen, J. A novel biodegradable copolymer for coronary artery stent: Preparation and its biocompatibility / in vivo. Chin. J. Biomed. Eng 2008, 27, 438–442. [Google Scholar]
- Ohya, Y; Nakai, T; Nagahama, K; Ouchi, T; Tanaka, S; Kato, K. The synthesis and biodegradability of poly(lactide-random-depsipeptide)-PEG-poly(lactide-random-depsipeptide) ABA-type triblock copolymers. J. Bioact. Compat. Polym 2006, 21, 557–577. [Google Scholar]
- Ohya, Y; Matsunami, H; Yamabe, E; Ouchi, T. Cell attachment and growth on films prepared from poly(depsipeptide-co-lactide) having various functional groups. J. Biomed. Mater. Res 2003, 65A, 79–88. [Google Scholar]
- Ohya, Y; Matsunami, H; Ouchi, T. Cell growth on the porous sponges prepared from poly(depsipeptide-co-lactide) having various functional groups. J. Biomater. Sci., Polym. Ed 2004, 15, 111–123. [Google Scholar]
- Yu, H; Guo, X; Qi, X; Liu, P; Shen, X; Duan, Y. Synthesis and characterization of arginine-glycine-aspartic peptides conjugated poly(lactic acid-co-L-lysine) diblock copolymer. J. Mater. Sci. Mater. Med 2008, 19, 1275–1281. [Google Scholar]
- Pierschbacher, MD; Ruoslahti, E. Cell attachment activity of fibronectin can be duplicated by small synthetic fragments of the molecule. Nature 1984, 309, 30–33. [Google Scholar]
- Kumar, CC. Integrin avb3 as a therapeutic target for blocking tumor-induced angiogenesis. Curr Drug Targets 2003, 4, 123–131. [Google Scholar]
- Weber, W; Haubner, R. Comment on “in vitro and in vivo evaluation of a technetium-99m-labeled cyclic RGD peptide as a specific marker of αVβ3 integrin for tumor imaging”. Bioconjugate Chem 2003, 14, 274. [Google Scholar]
- Yoshida, M; Asano, M; Kumakura, M; Katakai, R; Mashimo, T; Yuasa, H; Imai, K; Yamanaka, H. Sequential polydepsipeptides as biodegradable carriers for drug delivery systems. J. Biomed. Mater. Res 1990, 24, 1173–1184. [Google Scholar]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | Lipasea) | wt% | Temp. in °C | Time in h | Conv.b)in % | Mnb)103 | Mw/Mnb) |
|---|---|---|---|---|---|---|---|
| 1 | PPL | 1.5 | 100 | 72 | 3.2 | 6.1 | 1.10 |
| 2 | PPL | 5 | 100 | 72 | 26.2 | 10.4 | 1.05 |
| 3 | PPL | 10 | 100 | 72 | 53.6 | 11.0 | 1.08 |
| 4 | PS | 1.5 | 100 | 72 | 12.2 | 17.5 | 1.50 |
| 5 | PS | 4.7 | 100 | 168 | 73.8 | 12.5 | 3.33 |
| 6 | PC | 10 | 100 | 72 | 20.8 | 4.50 | 1.84 |
| 7 | CR | 10 | 100 | 72 | 46.3 | 3.50 | 1.99 |
| 8 | Novo-435 | 10 | 100 | 72 | 5.6 | ||
| 9 | blank | 0 | 100 | 168 | 0 |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Feng, Y.; Guo, J. Biodegradable Polydepsipeptides. Int. J. Mol. Sci. 2009, 10, 589-615. https://doi.org/10.3390/ijms10020589
Feng Y, Guo J. Biodegradable Polydepsipeptides. International Journal of Molecular Sciences. 2009; 10(2):589-615. https://doi.org/10.3390/ijms10020589
Chicago/Turabian StyleFeng, Yakai, and Jintang Guo. 2009. "Biodegradable Polydepsipeptides" International Journal of Molecular Sciences 10, no. 2: 589-615. https://doi.org/10.3390/ijms10020589
APA StyleFeng, Y., & Guo, J. (2009). Biodegradable Polydepsipeptides. International Journal of Molecular Sciences, 10(2), 589-615. https://doi.org/10.3390/ijms10020589