Contributions of the C-Terminal Helix to the Structural Stability of a Hyperthermophilic Fe-Superoxide Dismutase (TcSOD)


Abstract
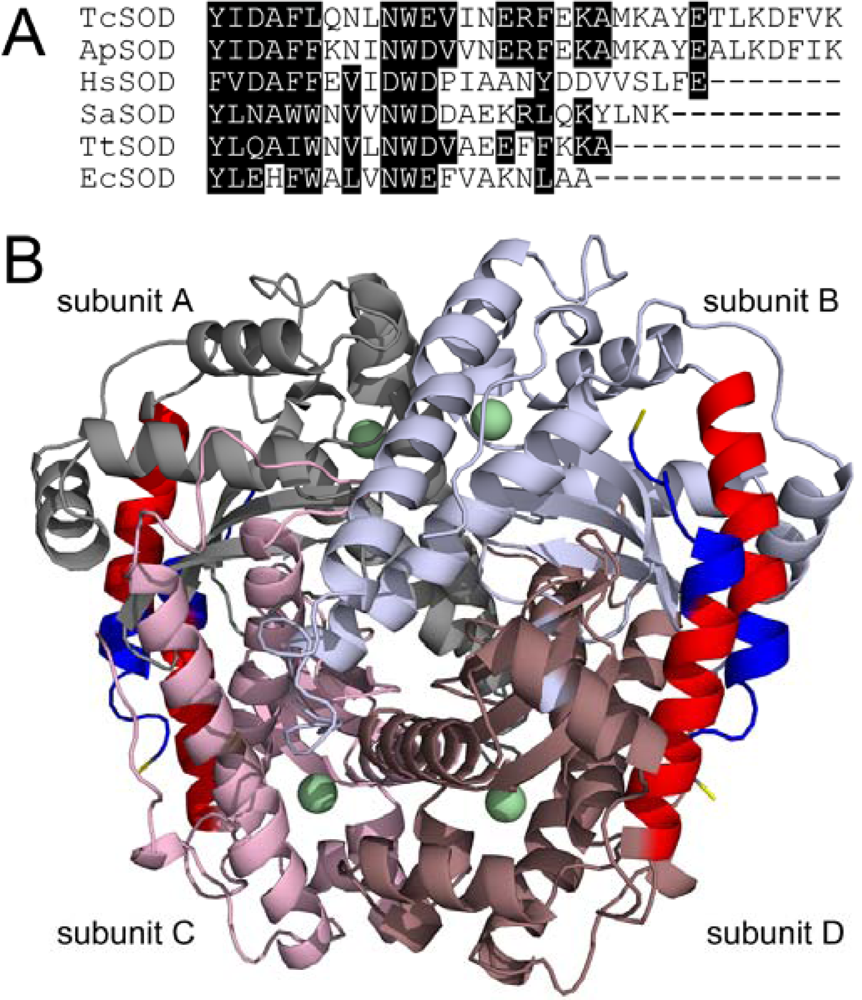
:
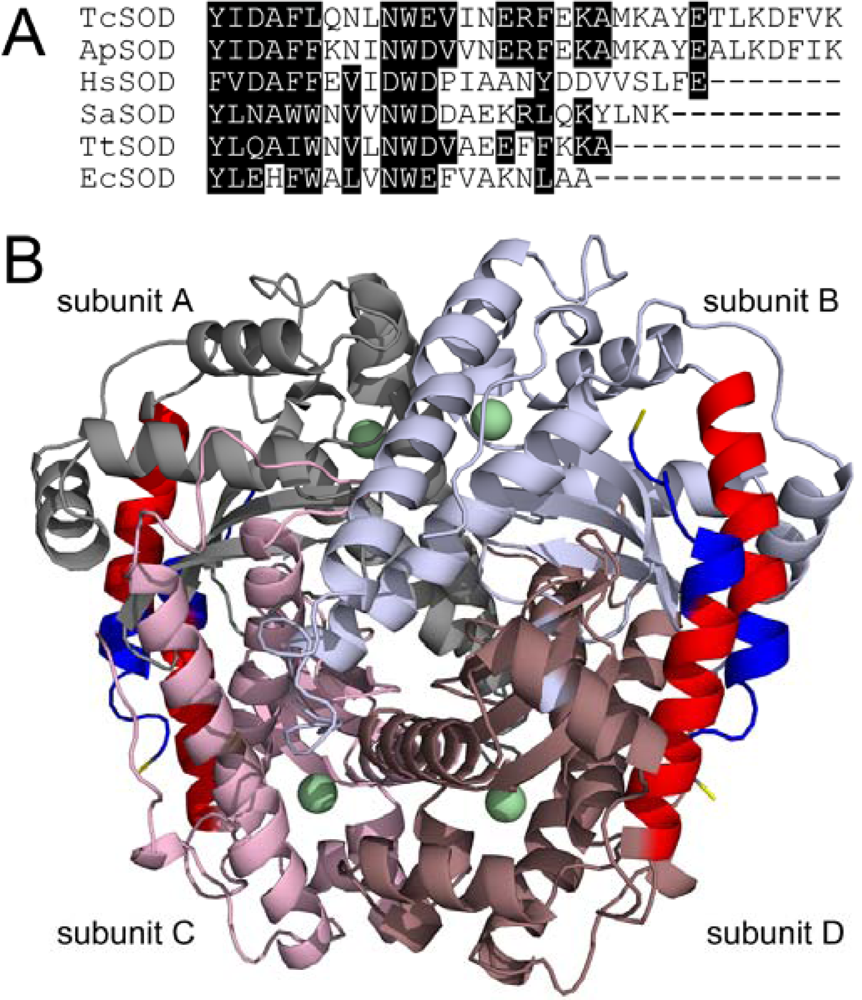
1. Introduction
2. Results and Discussion
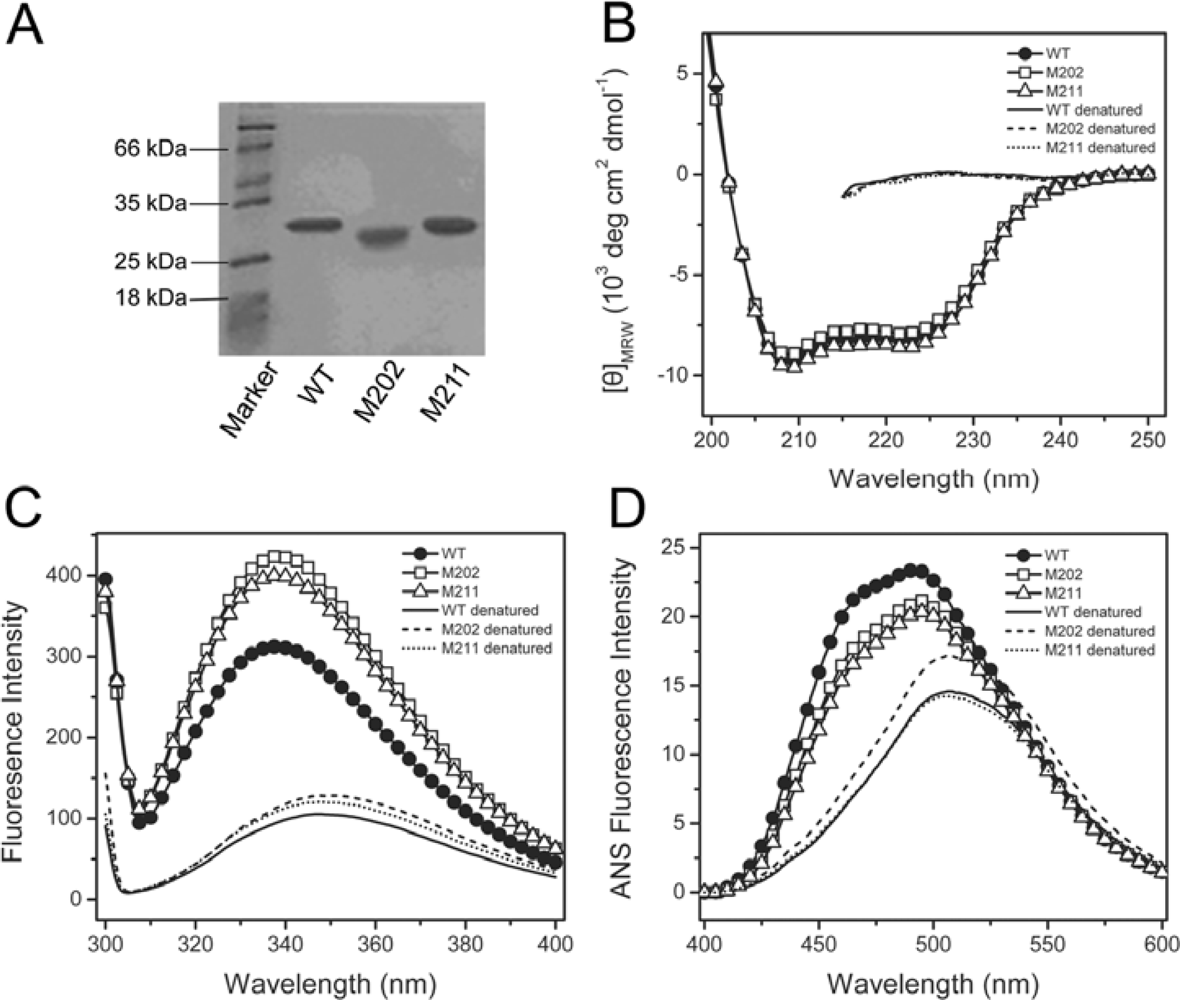
2.1. Characterization of the WT and Mutated TcSODs
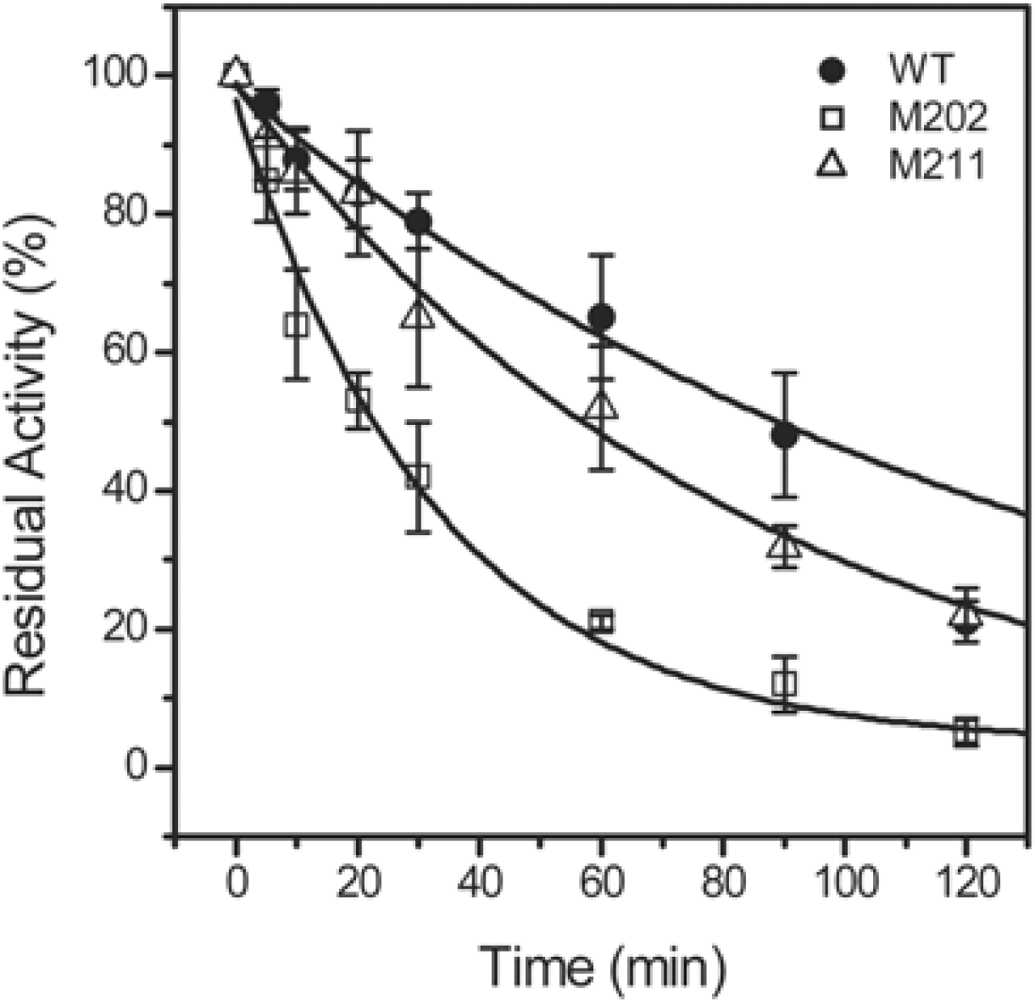
2.2. Thermal Stability of the WT and Mutated TcSODs
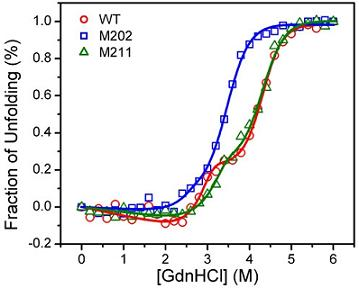
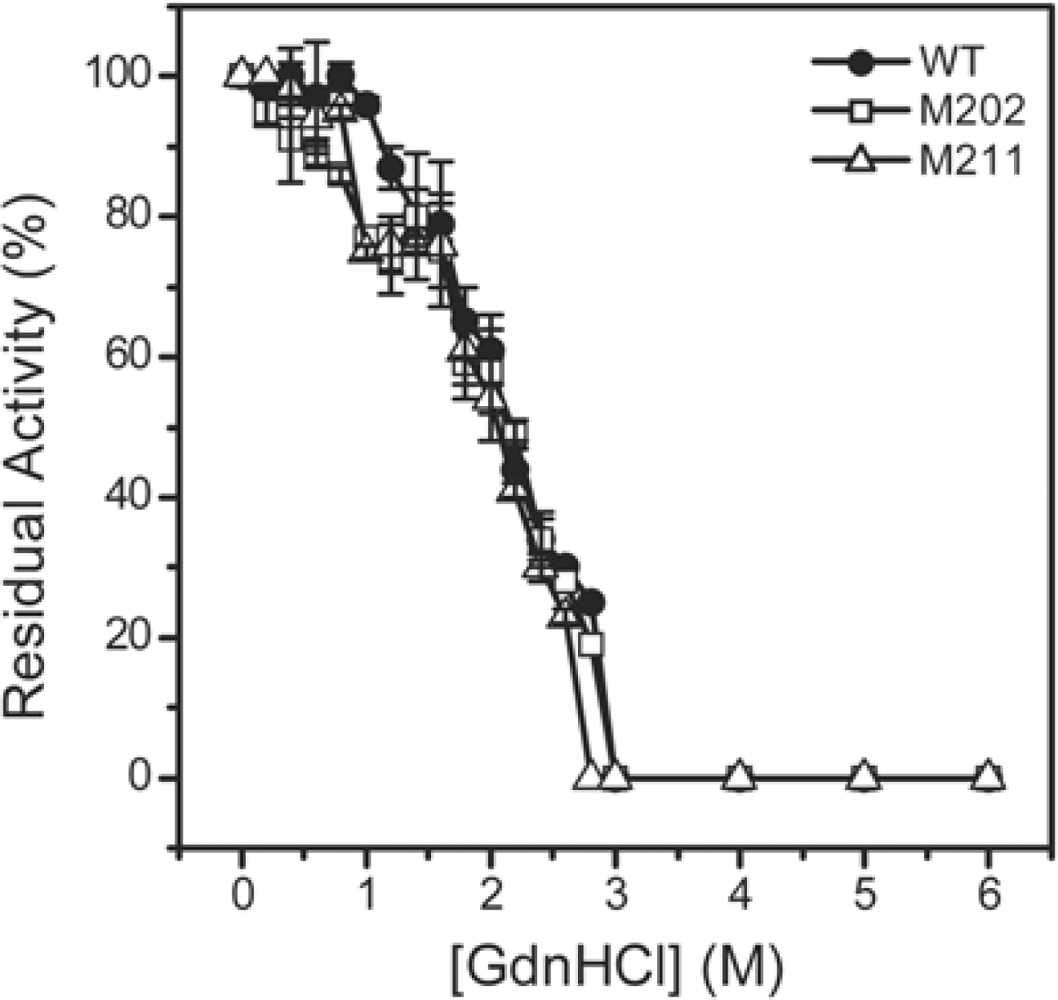
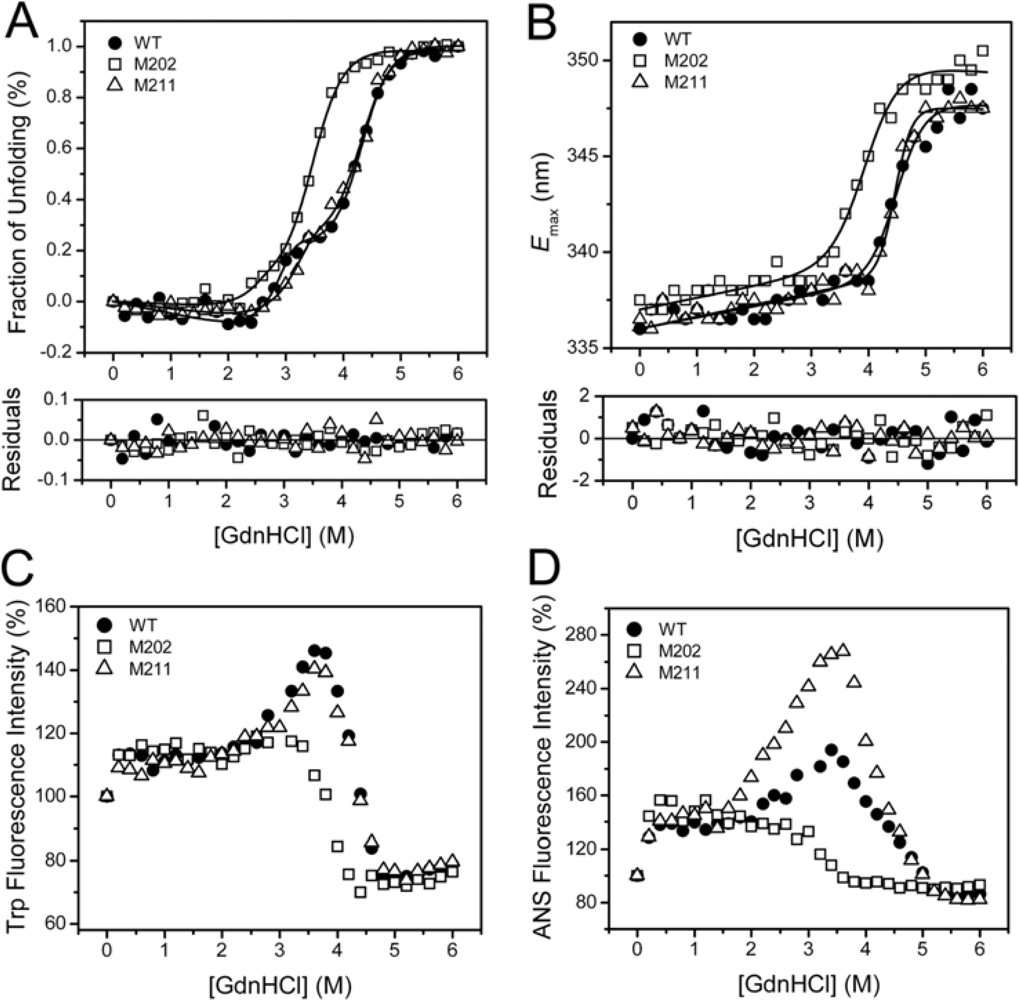
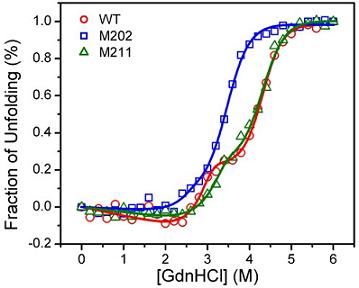
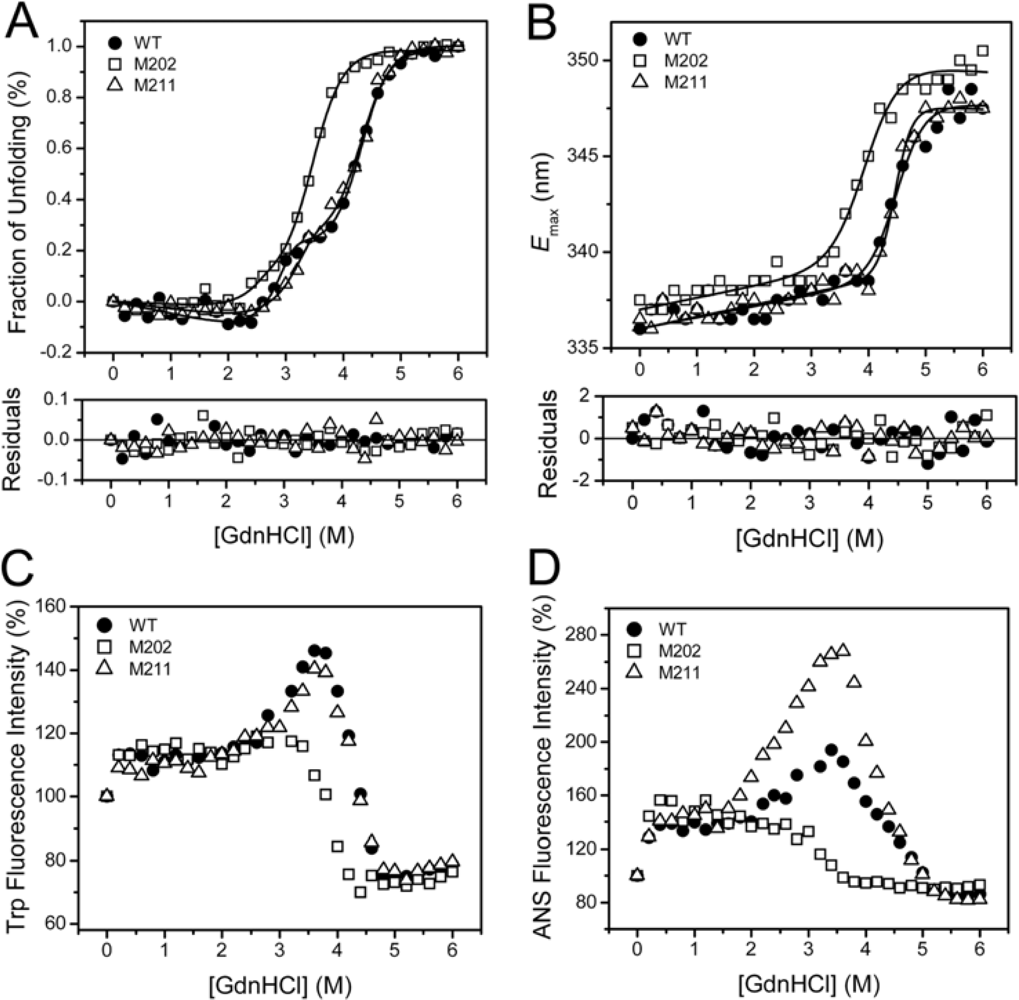
2.3. GdnHCl-induced Inactivation and Equilibrium Unfolding GdnHCl-induced Inactivation and Equilibrium Unfolding
2.4. Discussion
3. Experimental Section
3.1. Materials
3.2. Site-directed Mutagenesis
3.3. Protein Expression, Purification and Characterization
3.4. Size-exclusion Chromatography
3.5. Spectroscopic Experiments
3.6. Data Analysis
3.7. Thermal- and GdnHCl- Induced Inactivation
4. Conclusions
Acknowledgments
References
- Vieille, C; Zeikus, GJ. Hyperthermophilic enzymes: Sources, uses, and molecular mechanisms for thermostability. Microbiol. Mol. Biol. Rev 2001, 65, 1–43. [Google Scholar]
- Scandurra, R; Consalvi, V; Chiaraluce, R; Politi, L; Engel, PC. Protein thermostability in extremophiles. Biochimie 1998, 80, 933–941. [Google Scholar]
- Vetriani, C; Maeder, DL; Tolliday, N; Yip, KS; Stillman, TJ; Britton, KL; Rice, DW; Klump, HH; Robb, FT. Protein thermostability above 100 °C: A key role for ionic interactions. Proc Natl Acad Sci USA 1998, 95, 12300–12305. [Google Scholar]
- Unsworth, LD; van der Oost, J; Koutsopoulos, S. Hyperthermophilic enzymes-stability, activity and implementation strategies for high temperature applications. FEBS J 2007, 274, 4044–4056. [Google Scholar]
- Kumar, S; Nussinov, R. How do thermophilic proteins deal with heat? Cell. Mol. Life Sci 2001, 58, 1216–1233. [Google Scholar]
- Grabarse, W; Vaupel, M; Vorholt, JA; Shima, S; Thauer, RK; Wittershagen, A; Bourenkov, G; Bartunik, HD; Ermler, U. The crystal structure of methenyltetrahydromethanopterin cyclohydrolase from the hyperthermophilic archaeon Methanopyrus kandleri. Structure 1999, 7, 1257–1268. [Google Scholar]
- Ursby, T; Adinolfi, BS; Al-Karadaghi, S; de Vendittis, E; Bocchini, V. Iron superoxide dismutase from the archaeon Sulfolobus solfataricus: Analysis of structure and thermostability. J. Mol. Biol 1999, 286, 189–205. [Google Scholar]
- Thoma, R; Hennig, M; Sterner, R; Kirschner, K. Structure and function of mutationally generated monomers of dimeric phosphoribosylanthranilate isomerase from Thermotoga maritima. Structure 2000, 8, 265–276. [Google Scholar]
- Tanaka, Y; Tsumoto, K; Yasutake, Y; Umetsu, M; Yao, M; Fukada, H; Tanaka, I; Kumagai, I. How oligomerization contributes to the thermostability of an archaeon protein. Protein l-isoaspartyl-O-methyltransferase from Sulfolobus tokodaii. J. Biol. Chem 2004, 279, 32957–32967. [Google Scholar]
- Wang, S; Liu, W-F; He, Y-Z; Zhang, A; Huang, L; Dong, Z-Y; Yan, Y-B. Multistate folding of a hyperthermostable Fe-superoxide dismutase (TcSOD) in guanidinium hydrochloride: The importance of the quaternary structure. Biochim. Biophys. Acta: Prot. Proteom 2008, 1784, 445–454. [Google Scholar]
- Brookes, PS; Yoon, YS; Robotham, JL; Anders, MW; Sheu, SS. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol.: Cell Physiol 2004, 287, C817–C833. [Google Scholar]
- Fridovich, I. Superoxide radical and superoxide dismutases. Annu. Rev. Biochem 1995, 64, 97–112. [Google Scholar]
- Miller, A-F. Superoxide dismutases: Active sites that save, but a protein that kills. Curr. Opin. Chem. Biol 2004, 8, 162–168. [Google Scholar]
- Lim, J-H; Yu, YG; Choi, I-G; Ryu, J-R; Ahn, B-Y; Kim, S-H; Han, Y-S. Cloning and expression of superoxide dismutase from Aquifex pyrophilus, a hyperthermophilic bacterium. FEBS Lett 1997, 406, 142–146. [Google Scholar]
- Knapp, S; Kardinahl, S; Hellgren, N; Tibbelin, G; Schäfer, G; Ladenstein, R. Refined crystal structure of a superoxide dismutase from the hyperthermophilic archaeon Sulfolobus acidocaldarius at 2.2 Å resolution. J. Mol. Biol 1999, 285, 689–702. [Google Scholar]
- Whittaker, MM; Whittaker, JW. Recombinant superoxide dismutase from a hyperthermophilic archaeon, Pyrobaculum aerophilium. J. Biol. Inorg. Chem 2000, 5, 402–408. [Google Scholar]
- He, Y-Z; Fan, K-Q; Jia, C-J; Wang, Z-J; Pan, W-B; Huang, L; Yang, K-Q; Dong, Z-Y. Characterization of a hyperthermostable Fe-superoxide dismutase from hot spring. Appl. Microbiol. Biotechnol 2007, 75, 367–376. [Google Scholar]
- Lim, J-H; Yu, YG; Han, YS; Cho, S-J; Ahn, B-Y; Kim, S-H; Cho, Y. The crystal structure of a Fe-superoxide dismutase from the hyperthermophile Aquifex pyrophilus at 1.9 Å resolution: Structural basis for thermostability. J. Mol. Biol 1997, 270, 259–274. [Google Scholar]
- Horovitz, A; Serrano, L; Avron, B; Bycroft, M; Fersht, AR. Strength and co-operativity of contributions of surface salt bridges to protein stability. J. Mol. Biol 1990, 216, 1031–1044. [Google Scholar]
- Bycroft, M; Fersht, AR. Surface electrostatic interactions contribute little to stability of barnase. J. Mol. Biol 1991, 220, 779–788. [Google Scholar]
- Shirley, BA; Stanssens, P; Hahn, U; Pace, CN. Contribution of hydrogen bonding to the conformational stability of ribonuclease T1. Biochemistry 1992, 31, 725–732. [Google Scholar]
- Yip, KS; Stillman, TJ; Britton, KL; Artymiuk, PJ; Baker, PJ; Sedelnikova, SE; Engel, PC; Pasquo, A; Chiaraluce, R; Consalvi, V; Scandurra, R; Rice, DW. The structure of Pyrococcus furiosus glutamate dehydrogenase reveals a key role for ion-pair networks in maintaining enzyme stability at extreme temperatures. Structure 1995, 3, 1147–1158. [Google Scholar]
- Vogt, G; Woell, S; Argos, P. Protein thermal stability, hydrogen bonds, and ion pairs. J. Mol. Biol 1997, 269, 631–643. [Google Scholar]
- Karshikoff, A; Ladenstein, R. Ion pairs and the thermotolerance of proteins from hyperthermophiles: A “traffic rule” for hot roads. Trends Biochem. Sci 2001, 26, 550–556. [Google Scholar]
- Leemhuis, H; Rozeboom, HJ; Dijkstra, BW; Dijkhuizen, L. Improved thermostability of Bacillus circulans cyclodextrin glycosyltransferase by the introduction of a salt bridge. Proteins 2004, 54, 128–134. [Google Scholar]
- Bae, E; Phillips, GN, Jr. Identifying and engineering ion pairs in adenylate kinases. Insights from molecular dynamics simulations of thermophilic and mesophilic homologues. J. Biol. Chem 2005, 280, 30943–30948. [Google Scholar]
- Sadeghi, M; Naderi-Manesh, H; Zarrabi, M; Ranjbar, B. Effective factors in thermostability of thermophilic proteins. Biophys. Chem 2006, 119, 256–270. [Google Scholar]
- Lim, JH; Hwang, KY; Choi, J; Lee, DY; Ahn, BY; Cho, Y; Kim, KS; Han, YS. Mutational effects on thermostable superoxide dismutase from Aquifex pyrophilus: Understanding the molecular basis of protein thermostability. Biochem. Biophys. Res. Commun 2001, 288, 263–268. [Google Scholar]
- Schafer, G; Kardinahl, S. Iron superoxide dismutases: Structure and function of an archaic enzyme. Biochem. Soc. Trans 2003, 31, 1330–1334. [Google Scholar]
- Villeret, V; Clantin, B; Tricot, C; Legrain, C; Roovers, M; Stalon, V; Glansdorff, N; van Beeumen, J. The crystal structure of Pyrococcus furiosus ornithine carbamoyltransferase reveals a key role for oligomerization in enzyme stability at extremely high temperatures. Proc. Natl. Acad. Sci. USA 1998, 95, 2801–2806. [Google Scholar]
- Shima, S; Thauer, RK; Ermler, U; Durchschlag, H; Tziatzios, C; Schubert, D. A mutation affecting the association equilibrium of formyltransferase from the hyperthermophilic Methanopyrus kandleri and its influence on the enzyme’s activity and thermostability. Eur. J. Biochem 2000, 267, 6619–6623. [Google Scholar]
- Walden, H; Bell, GS; Russell, RJ; Siebers, B; Hensel, R; Taylor, GL. Tiny TIM: A small, tetrameric, hyperthermostable triosephosphate isomerase. J. Mol. Biol 2001, 306, 745–757. [Google Scholar]
- Ogasahara, K; Khechinashvili, NN; Nakamura, M; Yoshimoto, T; Yutani, K. Thermal stability of pyrrolidone carboxyl peptidases from the hyperthermophilic Archaeon, Pyrococcus furiosus. Eur. J. Biochem 2001, 268, 3233–3242. [Google Scholar]
- Feng, S; Zhao, T-J; Zhou, H-M; Yan, Y-B. Effects of the single point genetic mutation D54G on muscle creatine kinase activity, structure and stability. Int. J. Biochem. Cell Biol 2007, 39, 392–401. [Google Scholar]
- Marklund, S; Marklund, G. Involvement of the superoxide anion radical in the autoxidation of pyrogallol and a convenient assay for superoxide dismutase. Eur. J. Biochem 1974, 47, 469–474. [Google Scholar]
- Bradford, MM. Rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal. Biochem 1976, 73, 248–254. [Google Scholar]
- Tams, JW; Welinder, KG. Unfolding and refolding of Coprinus cinereus peroxidase at high pH, in urea, and at high temperature. Effect of organic and ionic additives on these processes. Biochemistry 1996, 35, 7573–7579. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substituted residue | Fe content per subunit | Specific activity (U/mg) | Elution volume (mL) | |
|---|---|---|---|---|
| WT | - | 0.47 | 1,383 ± 108 | 14.41 |
| M202 | A202→stop | 0.37 | 1,320 ± 37 | 14.45 |
| M211 | K211→stop | 0.47 | 1,434 ± 98 | 14.28 |
| (kJ/mol) | mNI (kJ/mol M) | (kJ/mol) | mIU (kJ/mol M) | (kJ/mol) | |
|---|---|---|---|---|---|
| WT | 170 ± 18 | 30 ± 6 | 46 ± 3 | 10.8 ± 0.7 | 355 ± 28 |
| M202 | 156 ± 5 | 30 ± 2 | 32 ± 2 | 9.3 ± 0.5 | 285 ± 12 |
| M211 | 166 ± 17 | 26 ± 5 | 42 ± 3 | 9.5 ± 0.7 | 332 ± 29 |
© 2009 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Wang, S.; Yan, Y.-B.; Dong, Z.-Y. Contributions of the C-Terminal Helix to the Structural Stability of a Hyperthermophilic Fe-Superoxide Dismutase (TcSOD). Int. J. Mol. Sci. 2009, 10, 5498-5512. https://doi.org/10.3390/ijms10125498
Wang S, Yan Y-B, Dong Z-Y. Contributions of the C-Terminal Helix to the Structural Stability of a Hyperthermophilic Fe-Superoxide Dismutase (TcSOD). International Journal of Molecular Sciences. 2009; 10(12):5498-5512. https://doi.org/10.3390/ijms10125498
Chicago/Turabian StyleWang, Sha, Yong-Bin Yan, and Zhi-Yang Dong. 2009. "Contributions of the C-Terminal Helix to the Structural Stability of a Hyperthermophilic Fe-Superoxide Dismutase (TcSOD)" International Journal of Molecular Sciences 10, no. 12: 5498-5512. https://doi.org/10.3390/ijms10125498
APA StyleWang, S., Yan, Y.-B., & Dong, Z.-Y. (2009). Contributions of the C-Terminal Helix to the Structural Stability of a Hyperthermophilic Fe-Superoxide Dismutase (TcSOD). International Journal of Molecular Sciences, 10(12), 5498-5512. https://doi.org/10.3390/ijms10125498