Isolation and X-ray Crystal Structure of Tetrahydroisoquinoline Alkaloids from Calycotome Villosa Subsp. intermedias

Abstract
:Introduction
Results and Discussion
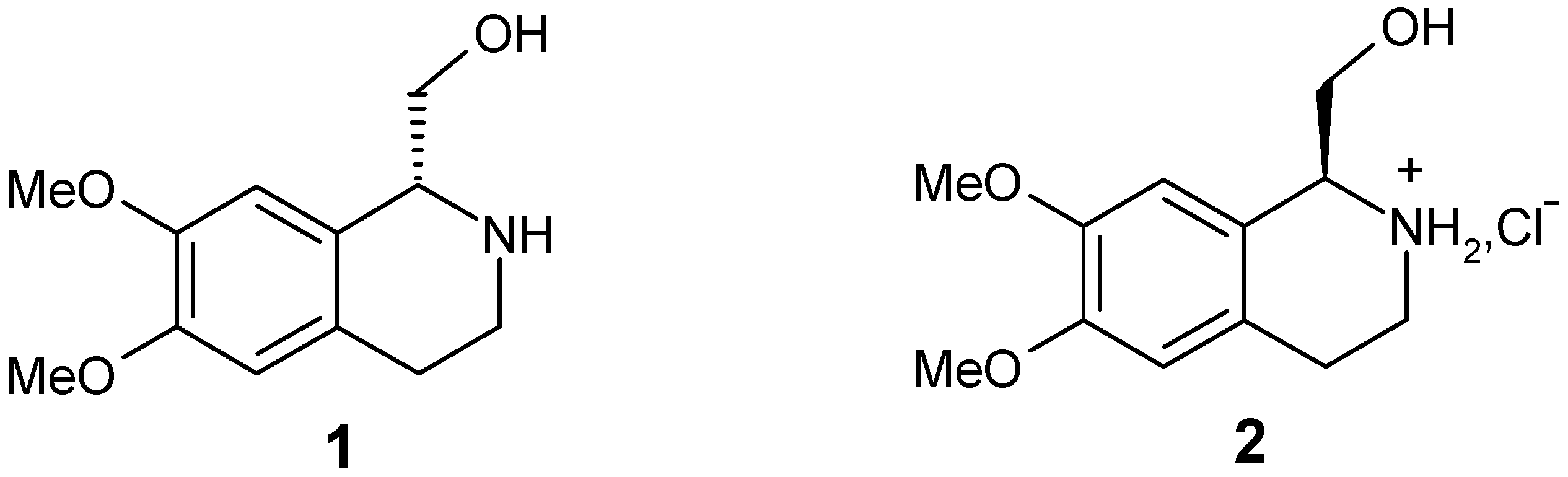

{kind=link}
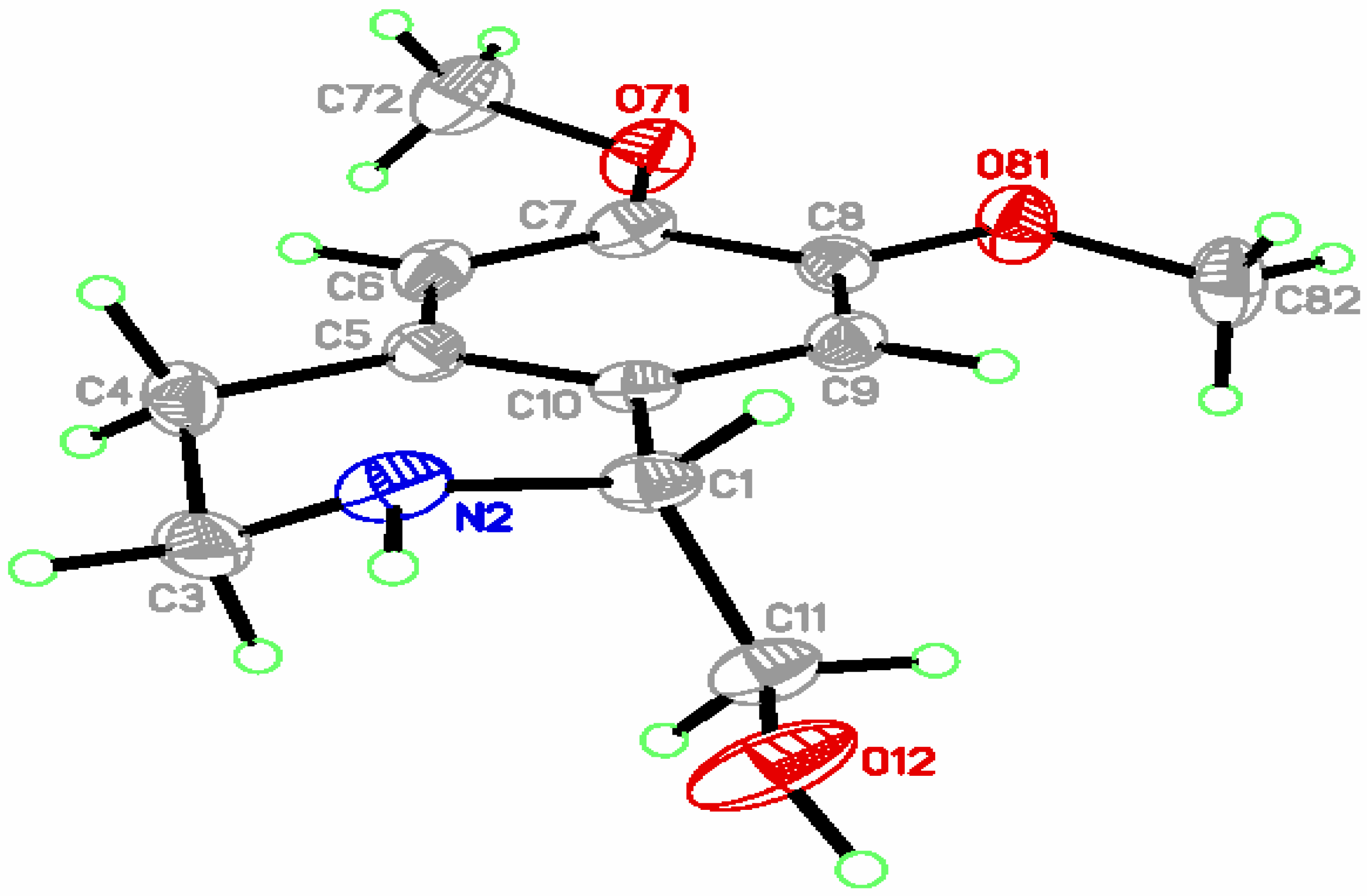
{kind=link}
{kind=link}
| Bond lengths [Å] | |||
| C1 C10 | 1.5200(16) | N2 C3 | 1.4712(19) |
| C1 C11 | 1.5306(17) | C3 C4 | 1.5206(19) |
| C1 N2 | 1.4761(16) | C4 C5 | 1.5170(16) |
| Bond angles [°] | |||
| N2 C1 C10 | 110.36(11) | C5 C4 C3 | 110.70(11) |
| N2 C1 C11 | 112.29(10) | C10 C5 C4 | 120.70(11) |
| C10 C1 C11 | 111.91(10) | N2 C3 C4 | 108.33(10) |
| C3 N2 C1 | 112.61(10) | C5 C10 C1 | 122.56(10) |
| Torsion angles [°] | |||
| C11 C1 N2 C3 | -77.74(14) | C1 N2 C3 C4 | -69.21(14) |
| N2 C3 C4 C5 | 51.84(14) | C3 C4 C5 C10 | -19.79(16) |
| C10 C1 N2 C3 | 47.86(14) | C4 C5 C10 C1 | 0.44(16) |
| N2 C1 C10 C5 | -13.40(15) | C11 C1 C10 C5 | 112.42(13) |
| Bond lengths [Å] | |||
| C6 C7 | 1.5200(12) | C7 N8 | 1.4983(11) |
| C7 C71 | 1.5275(12) | N8 C9 | 1.4883(12) |
| C5 C10 | 1.5123(12) | C9 C10 | 1.5195(13) |
| Bond angles [°] | |||
| N8 C7 C6 | 109.89(7) | N8 C9 C10 | 108.46(7) |
| N8 C7 C71 | 107.23(7) | C5 C10 C9 | 111.10(8) |
| C9 N8 C7 | 112.95(7) | C6 C7 C71 | 114.65(7) |
| C5 C6 C7 | 122.27(8) | C6 C5 C10 | 122.02(8) |
| Torsion angles [°] | |||
| C71 C7 N8 C9 | 173.06(7) | C6 C7 N8 C9 | 47.85(9) |
| C10 C5 C6 C7 | -1.13(13) | C5 C6 C7 N8 | -13.15(11) |
| C7 N8 C9 C10 | -68.14(10) | C5 C6 C7 C71 | -133.99(8) |
| N8 C9 C10 C5 | 49.53(10) | C6 C5 C10 C9 | -17.45(12) |
Conclusions
Experimental
General
Plant material
Extraction and isolation
X-ray Crystal Structure Determination [24]
| (1): C12H17NO3 | (2): C12H18ClNO3 | |
| Measurement Temp. (K) | 173(2) | 100(2) |
| M (g/mol) | 223.27 | 259.72 |
| Symmetry (S. G., Z) | Monoclinic (P21, 2) | Monoclinic (P21, 2) |
| Cell parameters (Å, °) | ||
| V = 575.55(10) Å3 | V = 652.37(9) Å3 | |
| dcal. (g·cm-3) | 1.288 | 1.322 |
| Crystal dimensions (mm) | 0.47*0.17*0.11 | 0.38*0.34*0.14 |
| F(000) | 240 | 276 |
| μ(Mo Kα)(mm-1) | 0.092 | 0.290 |
| Tmin./Tmax. | 0.9579 / 0.9899 | 0.8979 / 0.9606 |
| Total measured reflexions / Refined [I>2*σ(I)] | 9590 / 2341 | 14717 / 3870 |
| R / Rw (on F2) | 0.0303 / 0.0724 | 0.0234 / 0.0632 |
Acknowledgements
References and Notes
- Bently, K. W. The Isoquinoline Alkaloids; Harwood Academic Publishers: Amsterdam, 1998. [Google Scholar]
- Shamma, M. The Isoquinoline Alkaloids. Chemistry and Pharmacology; Academic Press: New York, 1972; Vol. 25. [Google Scholar]
- Herbert, R. B. The Chemistry and Biology of Isoquinoline Alkaloids; Philipson, J. D., Roberts, M. F., Zenk, M. H., Eds.; Springer Verlag: Berlin, Heidelberg, New York, Tokyo, 1985; p. 213. [Google Scholar]
- Shamma, M.; Moniot, J. L. Isoquinoline Alkaloid Research 1972-1977; Plenum Press: New York, 1978. [Google Scholar]
- Kitamura, M.; Hsiao, Y.; Ohta, M.; Tsukamota, M.; Ohta, T.; Takaya, H.; Noyori, R. J. Org. Chem. 1994, 59, 297.
- Locher, C.; Peerzada, N. J. Chem. Soc., Perkin Trans. 1 1999, 179.
- He, Y.; Nikulin, V. I.; Vansal, S. S.; Feller, D. R.; Miller, D. D. J. Med. Chem. 2000, 43, 591.
- Taniyama, D.; Hasegawa, M.; Tomioka, K. Tetrahedron: Asymmetry 1999, 10, 221.
- Gray, N. M.; Cheng, B. K.; Mick, S. J.; Lair, C. M.; Contreras, P. C. J. Med. Chem. 1989, 32, 1242.
- Wanner, K. Th.; Beer, H.; Höfner, G.; Ludwig, M. Eur. J. Org. Chem. 1998, 2019.
- El Antri, A.; Messouri, I.; Chendid Tlemcani, R.; Bouktaib, M.; El Alami, R.; El Bali, B.; Lachkar, M. Molecules 2004, 9, 568.
- Dalton, D. R. The Alkaloids - The Fundamental Chemistry, a Biogenetic Approach; Marcel Dekker: New York, 1979. [Google Scholar]
- Brossi, A.; Burkhardt, F. Helv. Chim. Acta 1961, 44, 1558.
- Tosun, F.; Tanker, M.; Ozden, T.; Tosun, A. Planta Med. 1987, 53, 499.
- White, E. P. New Zealand J. Sci. Tech. 1944, 25B, 137.
- Czarnocki, Z. J. Chem. Res. (S) 1992, 334.
- Czarnocki, Z. J. Chem. Res. (M) 1992, 2801.
- Morimoto, T.; Suzuki, N.; Achiva, K. Tetrahedron: Assymetry 1989, 9, 183.
- Pedrosa, R.; Andrés, C.; Iglesias, J. M. J. Org. Chem. 2001, 66, 243.
- Czarnocki, Z.; Maurin, J. K. Acta Cryst. 1993, C49, 1992.
- Czarnocki, Z.; Maurin, J. K.; Winnicka-Maurin, M. Acta Cryst. 1994, C50, 1779.
- Maurin, J. K.; Czarnocki, Z.; Paluchowska, B. Acta Cryst. 1996, C52, 953.
- Allen, F. M.; Kennard, O.; Watson, D. G.; Btammer, L.; Orpen, A. G.; Taylor, R. J. Chem. Soc., Perkin Trans. 2 1987, S1.
- CCDC 223477 and 223478 contain the supplementary crystallographic data for compounds 1 and 2. These data can be obtained free of charge via www.ccdc.cam.ac.uk/conts/retrieving.htlm (or from the CCDC, 12 Union Road Cambridge CB2 1EZ, UK; fax: +44 1223 336033; e-mail: deposit@ccdc.cam.ac.uk).
- Blessing, R. H. Acta Cryst. 1995, A51, 33.
- Sheldrick, G. M. Acta Cryst. 1990, A46, 467.
- Sheldrick, G. M. SHELXL-97, Program for the refinement of crystal structures. University of Göttingen: Göttingen, (Germany), 1997. [Google Scholar]
- Sample availability: Samples of compounds 1 and 2 are available from the authors and MDPI.
© 2004 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
El Antri, A.; Messouri, I.; Bouktaib, M.; El Alami, R.; Bolte, M.; El Bali, B.; Lachkar, M. Isolation and X-ray Crystal Structure of Tetrahydroisoquinoline Alkaloids from Calycotome Villosa Subsp. intermedias. Molecules 2004, 9, 650-657. https://doi.org/10.3390/90800650
El Antri A, Messouri I, Bouktaib M, El Alami R, Bolte M, El Bali B, Lachkar M. Isolation and X-ray Crystal Structure of Tetrahydroisoquinoline Alkaloids from Calycotome Villosa Subsp. intermedias. Molecules. 2004; 9(8):650-657. https://doi.org/10.3390/90800650
Chicago/Turabian StyleEl Antri, Ali, Ibtissam Messouri, Mohamed Bouktaib, Rachid El Alami, Michael Bolte, Brahim El Bali, and Mohammed Lachkar. 2004. "Isolation and X-ray Crystal Structure of Tetrahydroisoquinoline Alkaloids from Calycotome Villosa Subsp. intermedias" Molecules 9, no. 8: 650-657. https://doi.org/10.3390/90800650
APA StyleEl Antri, A., Messouri, I., Bouktaib, M., El Alami, R., Bolte, M., El Bali, B., & Lachkar, M. (2004). Isolation and X-ray Crystal Structure of Tetrahydroisoquinoline Alkaloids from Calycotome Villosa Subsp. intermedias. Molecules, 9(8), 650-657. https://doi.org/10.3390/90800650