Self-Terminating, Oxidative Radical Cyclizations


Abstract
:Introduction
Results and Discussion
1. Inorganic Radicals
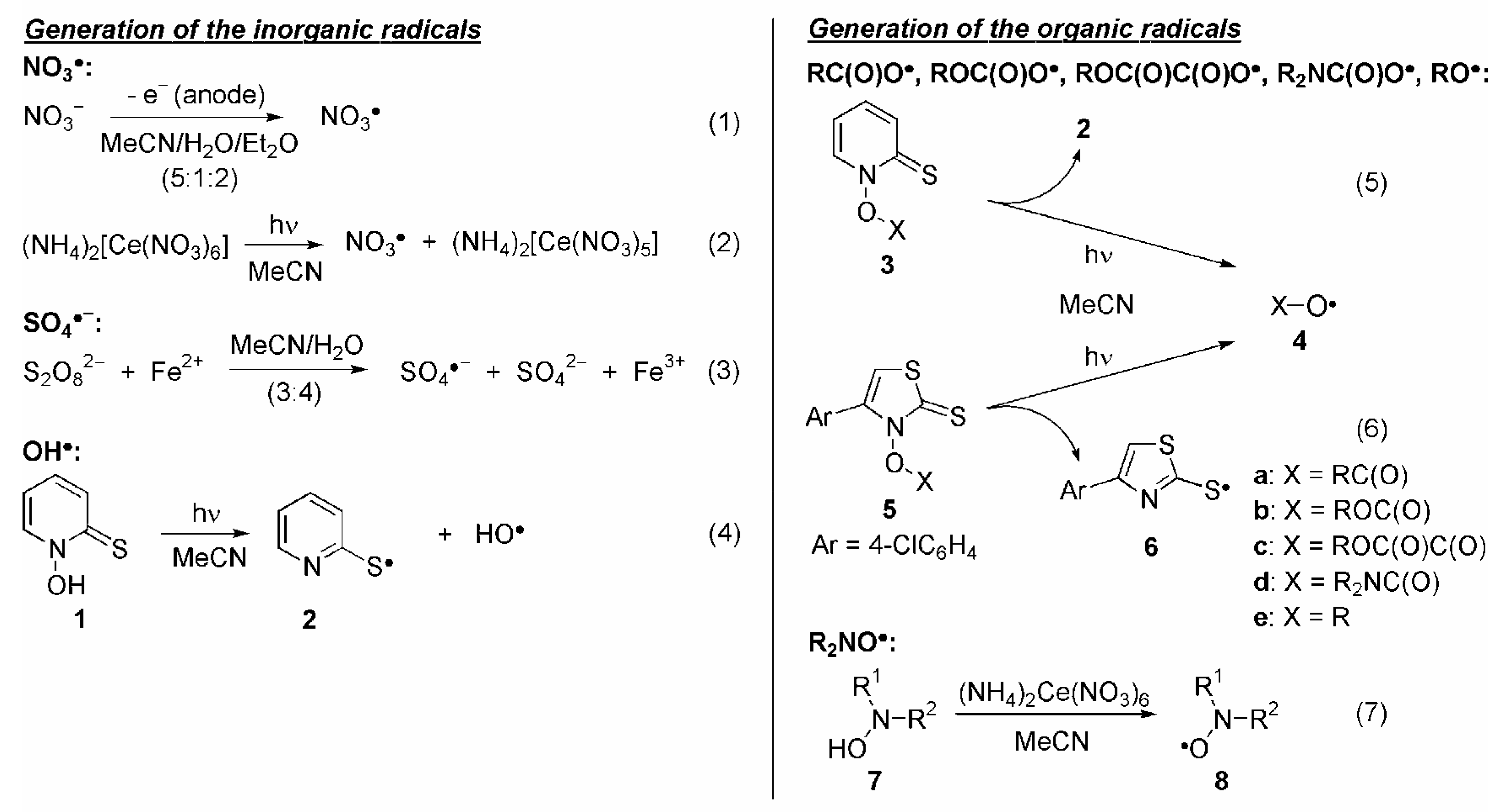

{kind=link}
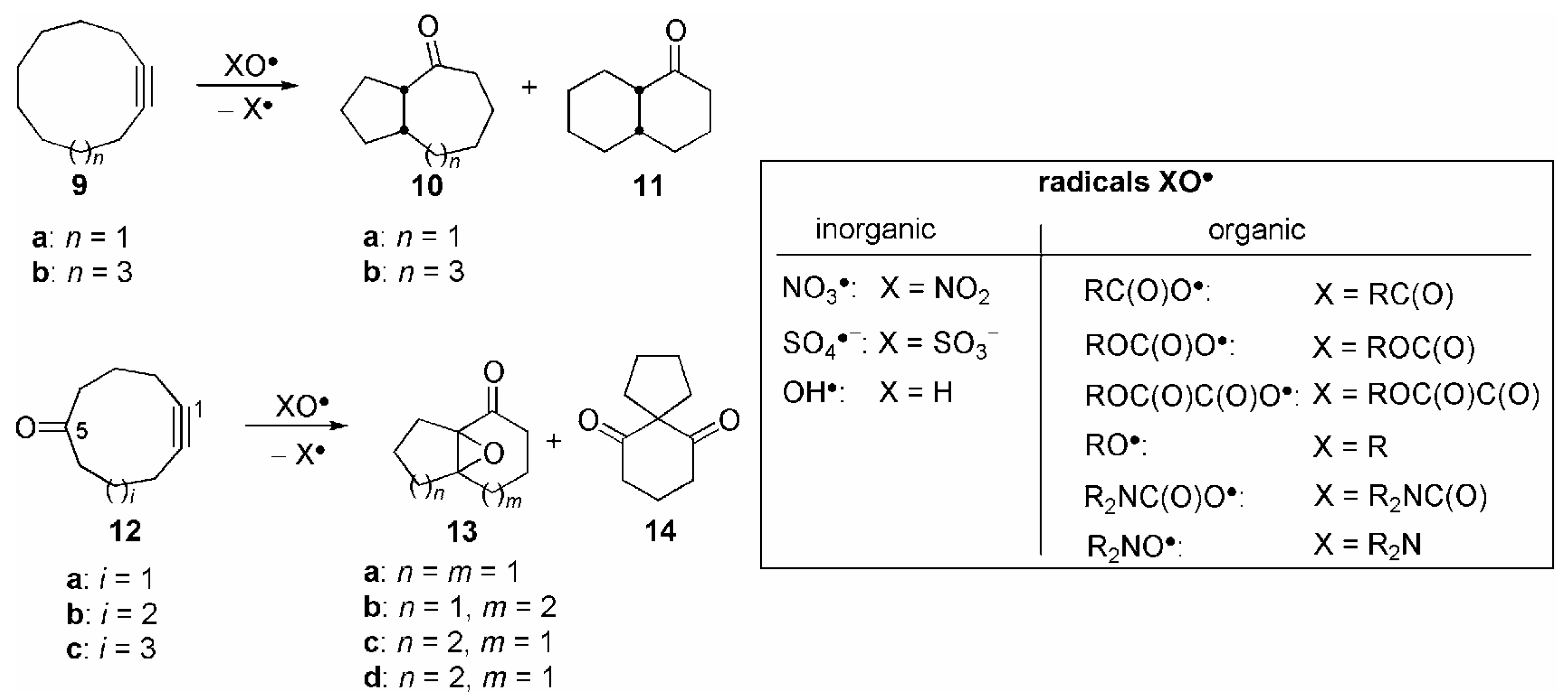
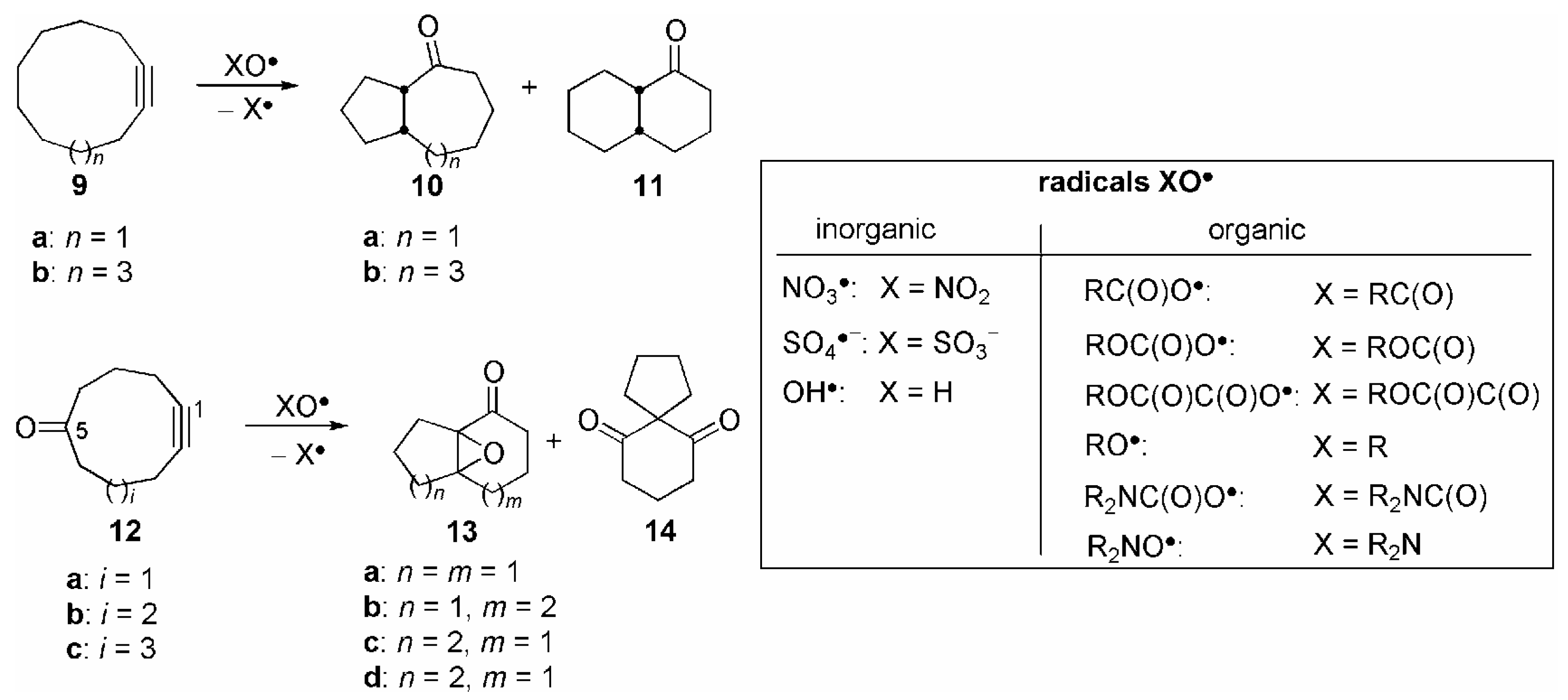{kind=link}
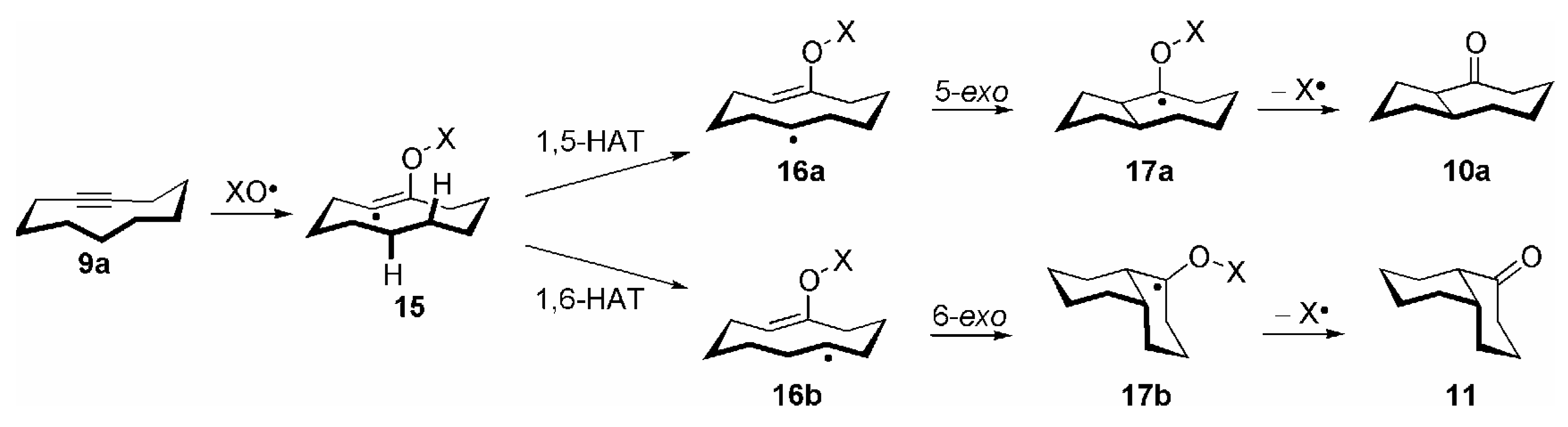{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Entry | 9 | X | Yield (%) |
|---|---|---|---|
| 1 | a | NO2[a] | 10a: 53, 11: 17[b] |
| 2 | b | NO2[a] | 10b: 35[b,c] |
| 3 | a | SO3− | 10a: 52, 11: 27[d] |
| 4 | a | H | 10a + 11: 63[d,e] |
| 5 | a | RC(O); R = Me[f] | 10a: 35, 11: 31[d,e] |
| 6 | a | RC(O); R = C6H5[f] | 10a: 42,11: 39[d,e] |
| 7 | a | RC(O); R = 4-MeOC6H4[f] | 10a: 47,11: 42[d,e] |
| 8 | a | ROC(O); R = Me[f] | 10a + 11: 94[d,e] |
| 9 | a | ROC(O); R = Me[f] | 10a + 11: 52[d,e] |
| 10 | a | ROC(O); R = Allyl[f] | 10a + 11: 89[d,e] |
| 11 | a | ROC(O); R = C6H5[f] | 10a + 11: 72[d,e] |
| 12 | a | ROC(O)C(O); R = Et[f] | 10a + 11: 82[d,e] |
| 13 | a | R2NC(O); R = Et[f] | 10a + 11: 89[d,e] |
| 14 | a | R2NC(O); R = Et[f] | 10a + 11: 69[d,e] |
| 15 | a | R2NC(O); R = Et, C6H5[f] | 10a + 11: 58[d,e] |
| 16 | a | R; R = Me[[g] | 10a + 11: 32[d,e] |
| 17 | a | R2N; R,R = –C(O)CH2CH2C(O)– | 10a + 11: 50[d,e,h] |
| 18 | a | R2N; R,R = –C(O)-2-C6H4-C(O)– | 10a + 11: 27[d,e,h] |

| Entry | 12 | X | Yield(%) (13+14) | Product ratio (%)[a] | ||
|---|---|---|---|---|---|---|
| 13b | 13c | 14 | ||||
| 1 | a | NO2[b] | 13a: 77[c] | |||
| 2 | b | NO2[b] | 74[c] | 43 | 57 | − |
| 3 | b | SO3− | 52[d] | 43 | 43 | 14 |
| 4 | c | NO2[b] | 13d: 11[c] | |||
| 5 | b | H | 82[d,e] | 6 | 12 | 82 |
| 6 | b | RC(O); R = Me[f] | 43[d,e,g] | 5 | 6 | 89 |
| 7 | b | RC(O); R = C6H5[f] | 57[d,e] | 4 | 8 | 88 |
| 8 | b | RC(O); R = 4-MeC6H4[f] | 93[d,e] | 5 | 5 | 90 |
| 9 | b | RC(O); R = 4-MeOC6H4[f] | 80[d,e] | 6 | 9 | 85 |
| 10 | b | RC(O); R = 3-NO2C6H4[f] | 74[d,e] | 10 | 29 | 61 |
| 11 | b | RC(O); R = 4-NO2C6H4[f] | 85[d,e] | 11 | 30 | 59 |
| 12 | b | RC(O); R = 4-FC6H4[f] | 88[d,e] | 7 | 9 | 84 |
| 13 | b | ROC(O); R = Me[f] | 84[d,e] | 9 | 12 | 79 |
| 14 | b | ROC(O); R = Allyl[f] | 75[d,e] | 10 | 18 | 72 |
| 15 | b | ROC(O); R = C6H5[f] | 63[d,e] | 15 | 34 | 51 |
| 16 | b | ROC(O)C(O); R = Et[f] | 48[d,e,g] | 10 | 17 | 73 |
| 17 | b | R2NC(O); R = Et[f] | 75[d,e] | 11 | 13 | 76 |
| 18 | b | R; R = Me[h] | 53[d,e] | 6 | 49 | 45 |
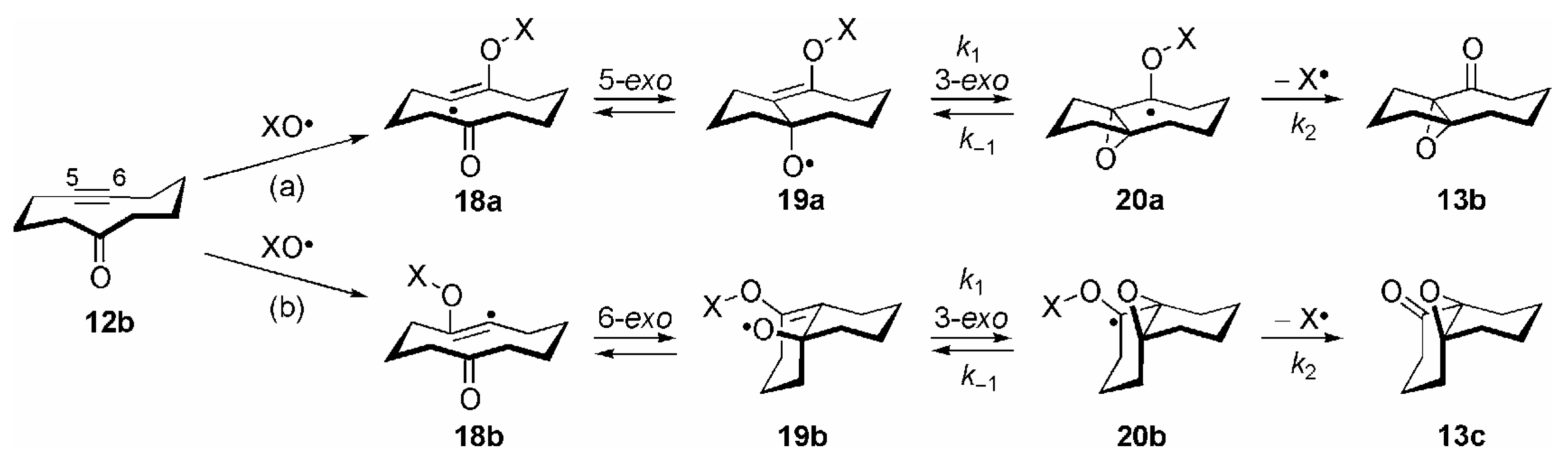

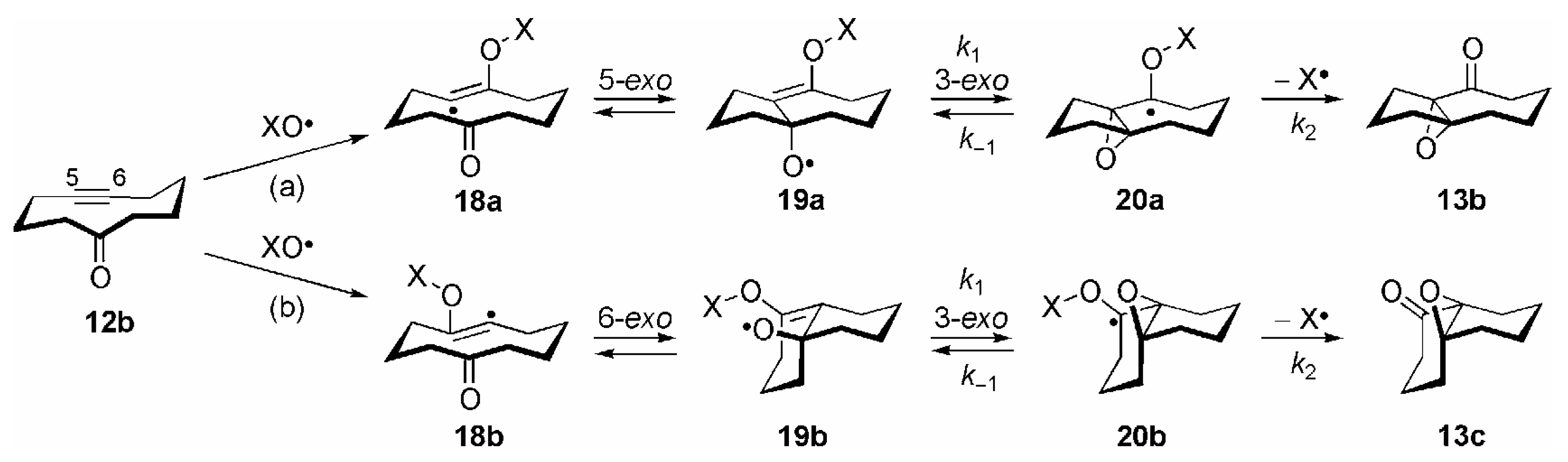
2. Other Oxygen-Centered Radicals
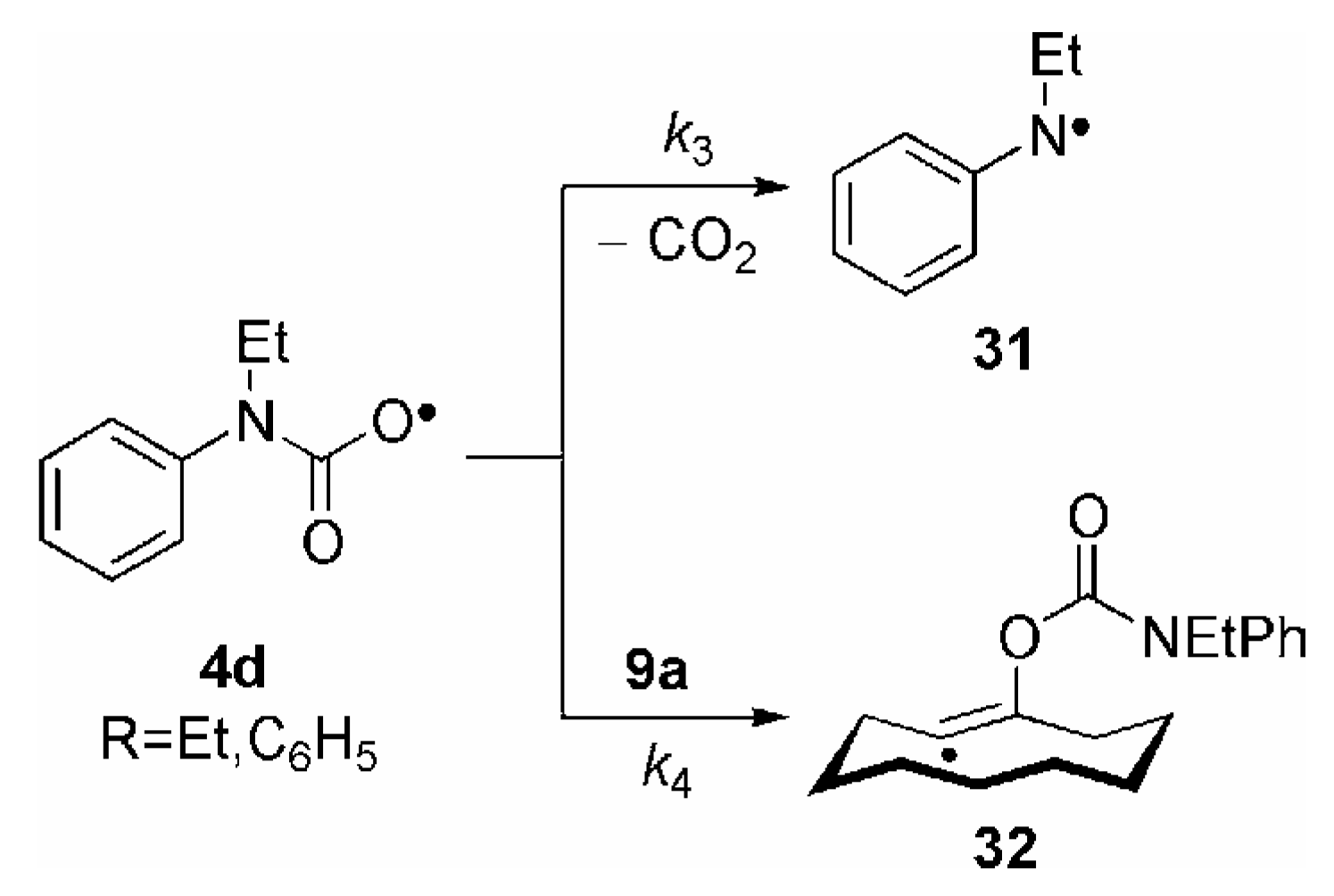
3. Some Mechanistic Considerations
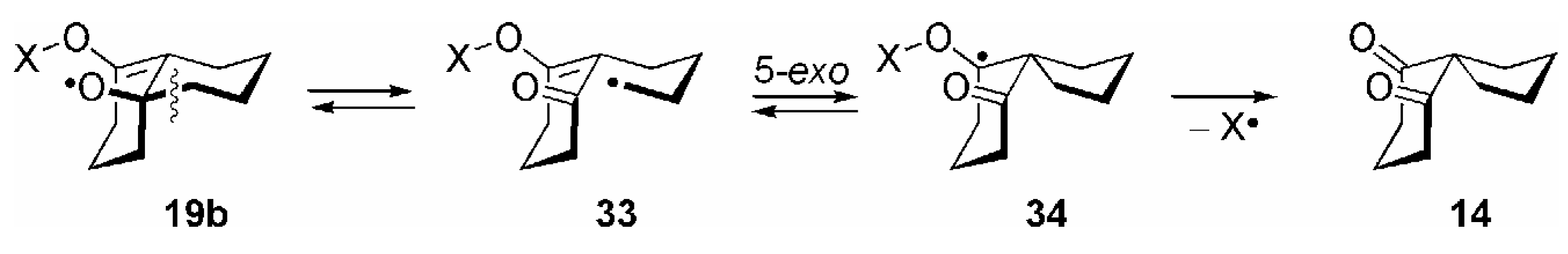
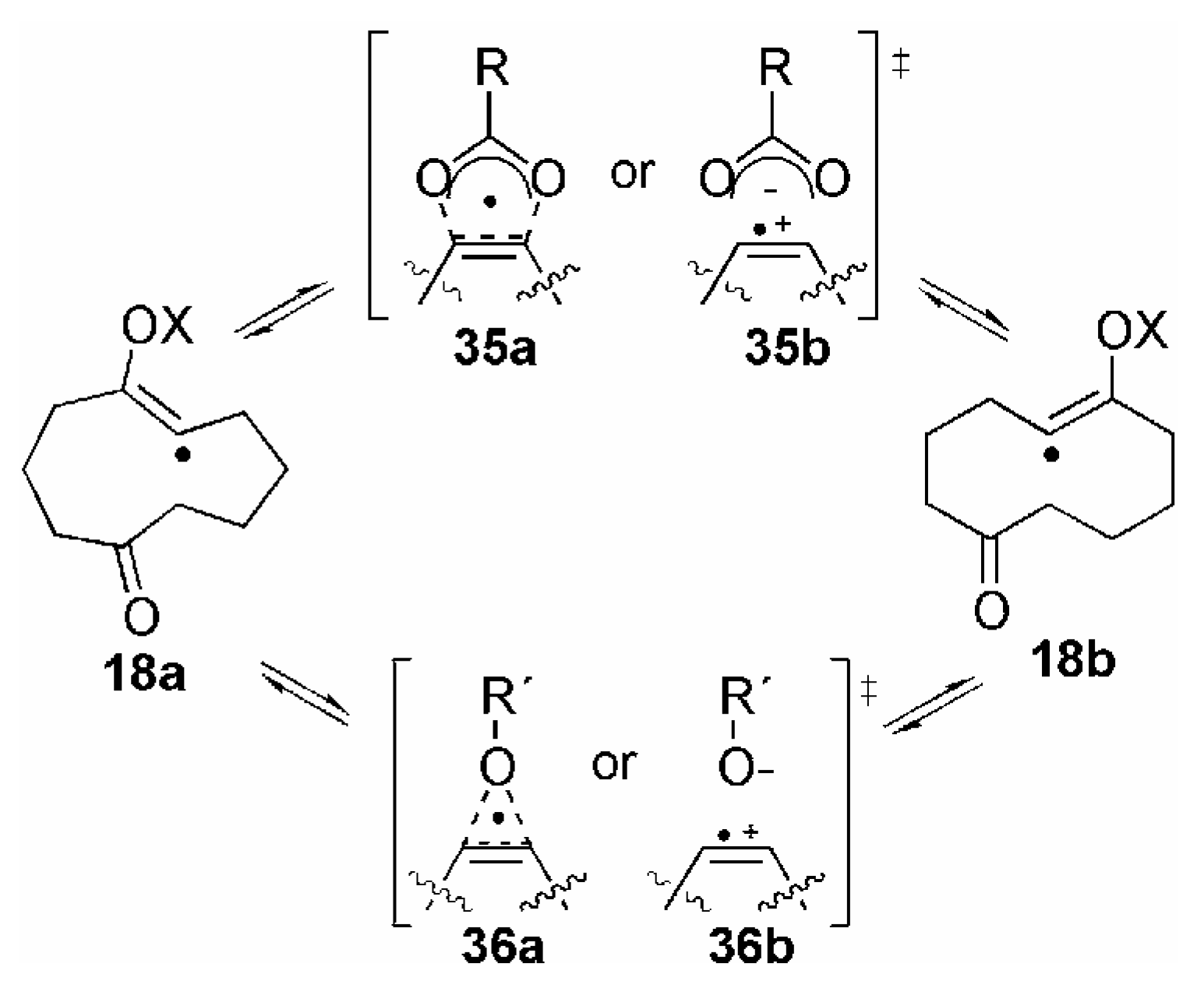
Conclusions and Future Outlook
Acknowledgments
References and Notes
- Curran, D. P.; Porter, N. A.; Giese, B. Stereochemistry of Radical Reactions; VCH: Weinheim, New York, Basel, Cambridge, Tokyo, 1996. [Google Scholar]
- A recent compilation is given in: Linker, T.; Schmittel, M. Radikale and Radikalionen in der Organischen Synthese; Wiley-VCH: Weinheim, New York, Chichester, Brisbane, Singapore, Toronto, 1998. [Google Scholar]
- Giese, B. Radicals in Organic Synthesis: Formation of Carbon-Carbon Bonds; Pergamon: Oxford, 1986. [Google Scholar]
- Baguley, P. A.; Walton, J. C. Angew. Chem. 1998, 110, 3272–3283.Angew. Chem. Int. Ed. Engl. 1998, 37, 3072–3082.
- Studer, A. Chem. Eur. J. 2001, 7, 1159–1164. [CrossRef]
- Olivier, C.; Renaud, P. Angew. Chem. 2000, 112, 946–949.Angew. Chem. Int. Ed. Engl. 2000, 39, 925–928.
- Hartung, J.; Kneuer, R.; Špehar, K. Chem. Commun. 2001, 799–800.
- Mikami, S.; Fujita, K.; Nakamura, T.; Yorimitsu, H.; Shinokubo, H.; Matsubara, S.; Oshima, K. Org. Lett. 2001, 3, 1853–1855.
- Hartung, J.; Gottwald, T.; Špehar, K. Synthesis 2002, 1469–1498. and cited refs.
- Hartung, J. Eur. J. Org. Chem. 2001, 619–632. and cited refs.
- Walling, C.; Clark, R. T. J. Am. Chem. Soc. 1974, 96, 4530–4534. [CrossRef]
- Walling, C.; El-Taliawi, G. J. Am. Chem. Soc. 1973, 95, 848–850. [CrossRef]
- Bottle, S.; Busfield, W. K.; Jenkins, I. D.; Skelton, B. W.; White, H. A.; Rizzardo, E.; Solomon, D. H. J. Chem. Soc., Perkin Trans. 2 1991, 1001–1007. [CrossRef]
- Fossey, J.; Lefort, D.; Sorba, J. Free Radicals in Organic Chemistry; Masson: Paris, 1995. [Google Scholar]
- Waters, W. A. The Chemistry of Free Radicals, 2nd ed.; Oxford University Press, 1950. [Google Scholar]
- Weissermel, K.; Arpe, H.-J. Industrial Organic Chemistry, 2nd ed.; VCH: Weinheim, 1993. [Google Scholar]
- Meier, H. Synthesis 1972, 235–253.
- Fokin, A. A.; Peleshanko, S. A.; Gunchenko, P. A.; Gusev, D. V.; Schreiner, P. R. Eur. J. Org. Chem. 2000, 3357–3362.
- Grossi, L.; Strazzari, S. J. Org. Chem. 1999, 64, 8076–8079. [CrossRef]
- Tomat, R.; Rigo, A. J. Appl. Electrochem. 1986, 16, 8–14. [CrossRef]
- Mella, M.; Freccero, M.; Soldi, T.; Fasani, E.; Albini, A. J. Org. Chem. 1996, 61, 1413–1422. [CrossRef]
- Sulpizio, A.; Mella, M.; Albini, A. Tetrahedron 1989, 45, 7545–7552.
- Shono, T.; Yamamoto, Y.; Takigawa, K.; Maekawa, H.; Ishifune, M.; Kashimura, S. Chem. Lett. 1994, 1045–1048.
- Shono, T.; Soejima, T.; Takigawa, K.; Yamaguchi, Y.; Maekawa, H.; Kashimura, S. Tetrahedron Lett. 1994, 35, 4161–4164. [CrossRef]
- Shono, T.; Chuankamnerdkarn, M.; Maekawa, H.; Ishifune, M.; Kashimura, S. Synthesis 1994, 895–897.
- Baciocchi, E.; Del Giacco, T.; Rol, C.; Sebastiani, G. V. Tetrahedron Lett. 1985, 26, 541–544.
- Baciocchi, E.; Del Giacco, T.; Rol, C.; Sebastiani, G. V. Tetrahedron Lett. 1985, 26, 3353–3356.
- Baciocchi, E.; Del Giacco, T.; Sebastiani, G. V. Tetrahedron Lett. 1987, 28, 1941–1944.
- Wille, U.; Plath, C. Liebigs Ann./Recueil 1997, 111–119. [CrossRef]
- Correctly, by taking both possible directions of enumeration in the carbocyclic ring into account, the transannular 1,5-HAT of 15 to 16a should be considered as a 1,5/1,7-HAT. By analogy, the reactions 16a →17a and 16b →17b should be termed as 5-exo/7-endo and 6-exo/6-endo cyclizations, respectively. For the sake of clarity the nomenclature of these transannular radical reactions is based on the most probable process; see ref. [14], p. 52.
- Due to the electron withdrawing character of the carbonyl oxygen, a C=O double bond in 12 is sufficiently electron deficient, so that addition of the electrophilic NO3• cannot occur. In the case of a C=C double bond competition experiments had revealed that at least two electron withdrawing substituents are required to direct the initial NO3• attack regioselectively at the alkyne site: Stademann, A. Diploma Thesis, Universität Kiel (Germany), 2000.
- Plath, C. Diploma Thesis; Universität Kiel (Germany), 1997. [Google Scholar]
- Wille, U.; Lietzau, L. Tetrahedron 1999, 55, 10119–10134.
- Estimated value for the redox couple NO3•/NO3−: E0 = 2.0 V vs. SCE (in MeCN): Baciocchi, E.; Del Giacco, T.; Murgia, S. M.; Sebastiani, G. V. Chem. Commun. 1987, 1246–1248. [CrossRef]
- Lietzau, L.; Wille, U. Heterocycles 2001, 55, 377–380.
- Stademann, A.; Wille, U. manuscript in preparation.
- Dreessen, T.; Wille, U. manuscript in preparation.
- Wille, U.; Lietzau, L. Tetrahedron 1999, 55, 11465–11474.
- Davies, M. J.; Gilbert, B. C. J. Chem. Soc., Perkin Trans. 2 1984, 1809–1815. [CrossRef]
- Niehaus, H.; Hildenbrand, K. J. Chem. Soc., Perkin Trans. 2 2000, 947–952. [CrossRef]
- Walling, C.; Zhao, C.; El-Taliawi, G. M. J. Org. Chem. 1983, 48, 4910–4914. [CrossRef]
- Deeble, D. J.; Schuchmann, M. N.; Steenken, S.; von Sonntag, C. J. Phys. Chem. 1990, 94, 8186–8192. [CrossRef]
- Lomoth, R.; Naumov, S.; Brede, O. J. Phys. Chem. A 1999, 103, 6571–6579. and cited refs.
- Minisci, F.; Citterio, A. Acc. Chem. Res. 1983, 16, 27–32. [CrossRef]
- Walling, C.; Camaioni, D. M. J. Am. Chem. Soc. 1975, 97, 1603–1604. [CrossRef]
- Walling, C.; Camaioni, D. M.; Kim, S. S. J. Am. Chem. Soc. 1978, 100, 4814–4818. [CrossRef]
- Effenberger, F.; Kottmann, H. Tetrahedron 1985, 41, 4171–4182.
- Wille, U. Org. Lett. 2000, 2, 3485–3488.
- Adam, W.; Ballmeier, D.; Epe, B.; Grimm, G. N.; Saha-Möller, C. R. Angew. Chem. 1995, 107, 2326–2328. Angew. Chem. Int. Ed. Engl. 1995, 34, 2156–2158.
- Wille, U. Tetrahedron Lett. 2002, 43, 1239–1242.
- Giese, B. Houben-Weyl, Methoden der Organischen Chemie, Band E 19a, C-Radikale; Regitz, M., Giese, B., Eds.; Georg Thieme Verlag: Stuttgart, New York, 1989; p. 140. [Google Scholar]
- Wille, U. J. Am. Chem. Soc. 2002, 124, 14–15. [CrossRef]
- Benzing, E. Angew. Chem. 1960, 72, 709. and cited refs.
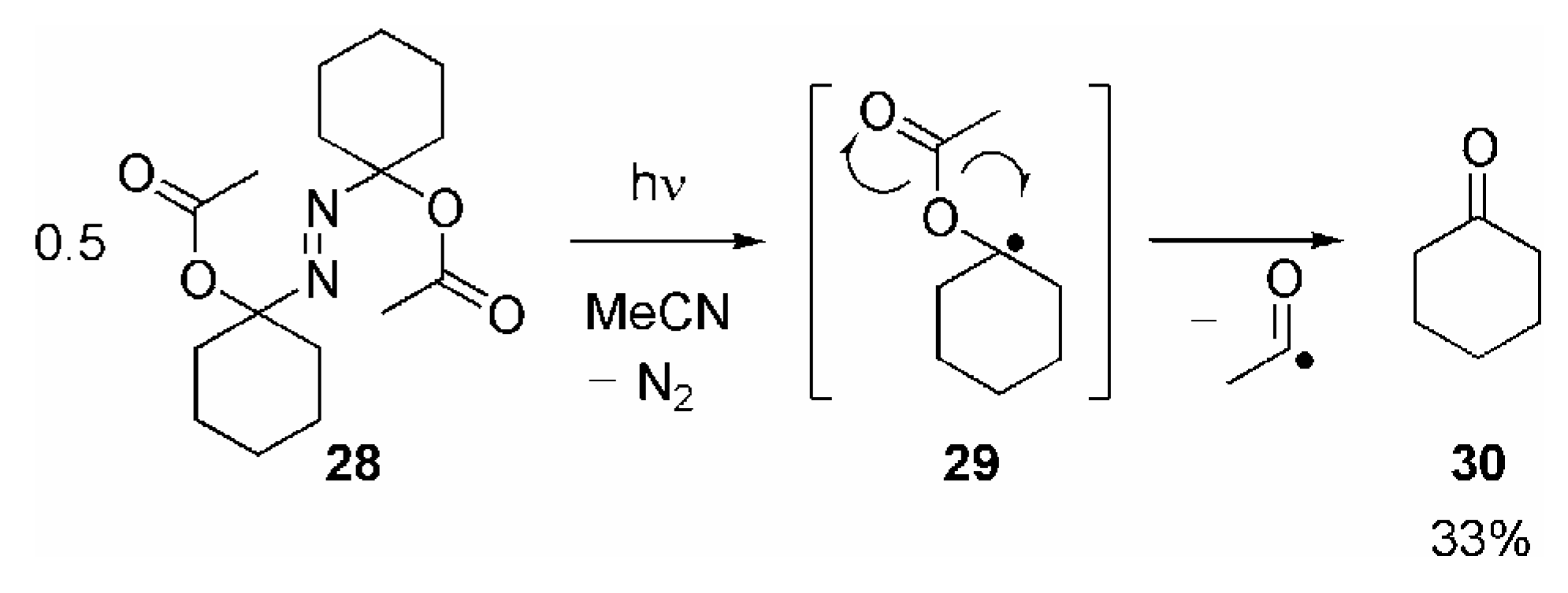
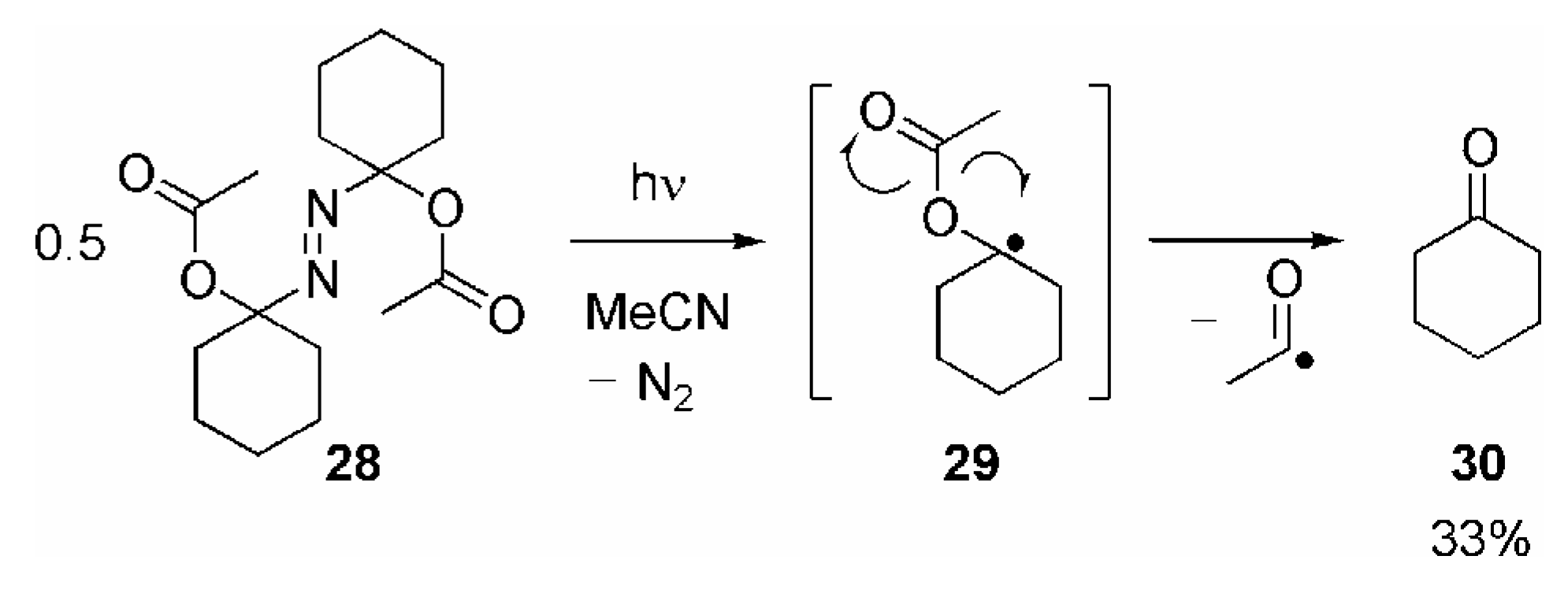
- Since the final fragmentation step is expected to be the same, the yield of 30 appears to be too low compared with the yields of 10a/11 obtained in the reaction of the various RC(O)O• with 9a. This discrepancy may be explained by the different reaction conditions.Whereas 10a/11 are generated by unimolecular fragmentation of highly diluted 17a/b, irradiation of 28 leads to formation of two molecules of 29 in close vicinity or even in the same solvent cage. Recombination 29 or other bimolecular reactions may be considered to compete with its fragmentation.
- Chateauneuf, J.; Lusztyk, J.; Maillard, B.; Ingold, K. U. J. Am. Chem. Soc. 1988, 110, 6727–6731. [CrossRef]
- Edge, D. J.; Kochi, J. K. J. Am. Chem. Soc. 1973, 95, 2635–2643. [CrossRef]
- Korth, H. G.; Chateauneuf, J.; Lusztyk, J.; Ingold, K. U. J. Am. Chem. Soc. 1988, 110, 5929–5931. [CrossRef]
- Beckwith, A. L. J.; Hay, B. P. J. Am. Chem. Soc. 1988, 110, 4415–4416. [CrossRef]
- Beckwith, A. L. J.; Hay, B. P. J. Am. Chem. Soc. 1989, 111, 230–234. [CrossRef]
- Van Sickle, D. E. J. Org. Chem. 1969, 34, 3446–3451. [CrossRef]
- To our knowledge, absolute rate data for these processes were not determined.
- Togo, H.; Fuji, M.; Yokoyama, M. Bull. Chem. Soc. Jpn. 1991, 64, 57–67. [CrossRef]
- Bucher, G.; Halupka, M.; Kolano, C.; Schade, O.; Sander, W. Eur. J. Org. Chem. 2001, 545–552.
- Barton, D. H. R.; Crich, D. J. Chem. Soc., Perkin Trans. 1 1986, 1603–1611. [CrossRef]
- Simakov, P. A.; Martinez, F. N.; Horner, J. H.; Newcomb, M. J. Org. Chem. 1998, 63, 1226–1232. [CrossRef]
- Newcomb, M.; Kumar, M. U.; Boivin, J.; Crépon, E.; Zard, S. Z. Tetrahedron Lett. 1991, 32, 45–48.
- Musa, O. M.; Horner, J. H.; Shahin, H.; Newcomb, M. J. Am. Chem. Soc. 1996, 118, 3862–3868. [CrossRef]
- Danen, W. C.; West, C. T.; Kensler, T. T. J. Am. Chem. Soc. 1973, 95, 5716–5724. [CrossRef]
- Esker, J. L.; Newcomb, M. J. Org. Chem. 1994, 59, 2779–2786. [CrossRef]
- Burdi, D.; Aveline, B. M.; Wood, P. D.; Stubbe, J.; Redmond, R. W. J. Am. Chem. Soc. 1997, 119, 6457–6460. [CrossRef]
- Hageman, H. J.; Oosterhoff, P.; Overeem, T.; Verbeek, J. J. Photochem. Photobiol. A: Chem. 1997, 110, 17–21. [CrossRef]
- Albéniz, A. C.; Espinet, P.; López-Fernández, R.; Sen, A. J. Am. Chem. Soc. 2002, 124, 11278–11279. [CrossRef]
- Benoit, D.; Chaplinski, V.; Braslau, R.; Hawker, C. J. J. Am. Chem. Soc. 1999, 121, 3904–3920. [CrossRef]
- Jargstorff, C.; Wille, U. Eur. J. Org. Chem. 2003, 3173–3178.
- The reason for the lower yield of 10a/11 obtained using thiazolethiones 5 as radical precursor is unclear. It was reported that the photoproducts arising from photolysis of thiazolethiones undergo a complicated chemistry: Adam, W.; Hartung, J.; Okamoto, H.; Marquardt, S.; Nau, W. M.; Pischel, U.; Saha-Möller, C. R.; Špehar, K. J. Org. Chem. 2002, 67, 6041–6049.
- Gregor, J.; Azhar, I.; Wille, U. to be published.
- GC-MS analysis revealed that in the reaction of the nitroxyl radicals with 9a small amounts of trans-10a and, presumably, trans-11 were also formed. It is believed that these compounds result from a subsequent acid-catalyzed isomerization of 10a/11, since the CAN mediated oxidation of hydroxylamines in acetonitrile is accompanied by production of equimolar amounts of protons.
- Absence of light was essential to avoid generation of NO3• from CAN through a photo-induced electron transfer (Scheme 1.2).
- Most of the radicals used in these investigations were also tested in their reaction with open-chain alkynes of type 21 (see Scheme 5).
- Laurie, D.; Nonhebel, D. C.; Suckling, C. J.; Walton, J. C. Tetrahedron 1993, 49, 5869–5872.
- Krishnamurthy, V.; Rawal, V. H. J. Org. Chem. 1997, 62, 1572–1573.
- The reversibility of allyloxyl radical formation was considered by: Ziegler, F. E.; Petersen, A. K. J. Org. Chem. 1994, 59, 2707–2714.
- Handbook of Chemistry and Physics, 63rd ed.; CRC Press: Boca Raton, Florida, 1982.
- Wille, U.; Jargstorff, C. J. Chem. Soc., Perkin Trans. 1 2002, 1036–1041. [CrossRef]
- Theoretically, further β-fragmentation pathways other than that shown in Scheme 8 are possible in both isomeric allyloxyl radicals 19a/b. However, subsequent cyclizations would involve kinetically unfavourable 5-endo, 6-endo or 4-exo steps (see refs. [1,2,14]), and no products arising from such reactions were observed; see [84].
- However, it might be expected that the energy required for cleavage of the O–X bond in a radical of type C•(O)-X is different from that in an even-electron system, but the general trend should be unaffected.
- Luft, N. W. Z. Elektrochem. 1956, 60, 94–100.
- Batt, L.; McCulloch, R. D. Int. J. Chem. Kinet. 1976, 8, 491–500.
- Orlov, Y. D.; Lebedev, Y. A. Bull. Acad. Sci. USSR Div. Chem. Sci. (Engl. Transl.) 1984, 33, 1227. [CrossRef]
- Gray, P.; Williams, A. Trans. Faraday Soc. 1959, 55, 760–777.
- Batt, L.; McCulloch, R. D. Int. J. Chem. Kinet. 1976, 8, 911–933.
- Mihalick, J. E.; Gatev, G. G.; Brauman, J. I. J. Am. Chem. Soc. 1996, 118, 12424–12431. [CrossRef]
- No literature data for the homolytic scission of an O−S bond in organic sulfates were available.
- A reversible addition of a tin-centered radical to the C≡C triple bond in 12b was proposed: Baldwin, J. E.; Adlington, R. M.; Robertson, J. Tetrahedron 1991, 47, 6795–6812. [CrossRef]
- Zipse, H.; Bootz, M. J. Chem. Soc., Perkin Trans. 2 2001, 1566–1572. [CrossRef] and cited refs.
© 2004 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Dreessen, T.; Jargstorff, C.; Lietzau, L.; Plath, C.; Stademann, A.; Wille, U. Self-Terminating, Oxidative Radical Cyclizations. Molecules 2004, 9, 480-497. https://doi.org/10.3390/90600480
Dreessen T, Jargstorff C, Lietzau L, Plath C, Stademann A, Wille U. Self-Terminating, Oxidative Radical Cyclizations. Molecules. 2004; 9(6):480-497. https://doi.org/10.3390/90600480
Chicago/Turabian StyleDreessen, Tim, Christian Jargstorff, Lars Lietzau, Christian Plath, Arne Stademann, and Uta Wille. 2004. "Self-Terminating, Oxidative Radical Cyclizations" Molecules 9, no. 6: 480-497. https://doi.org/10.3390/90600480
APA StyleDreessen, T., Jargstorff, C., Lietzau, L., Plath, C., Stademann, A., & Wille, U. (2004). Self-Terminating, Oxidative Radical Cyclizations. Molecules, 9(6), 480-497. https://doi.org/10.3390/90600480