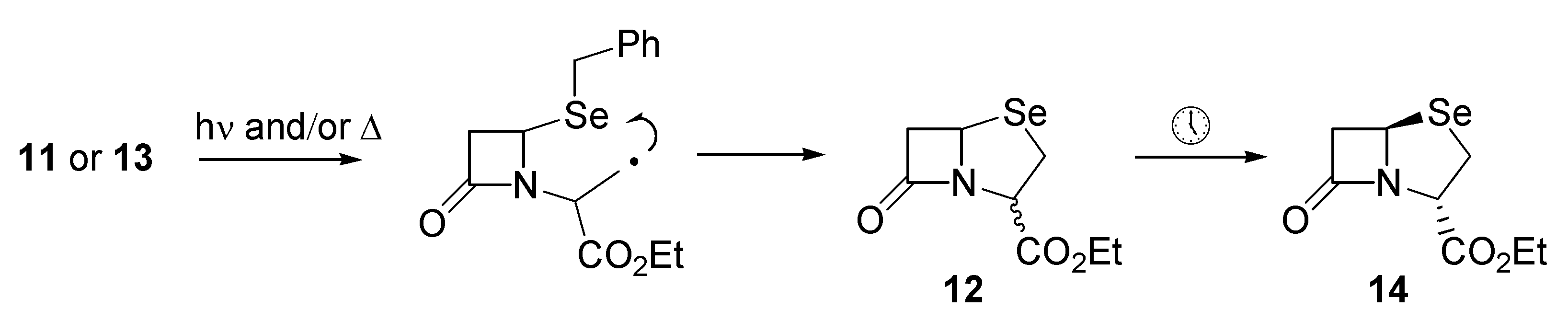Introduction
Free-radical homolytic substitution chemistry is rapidly gaining acceptance as a versatile synthetic method [
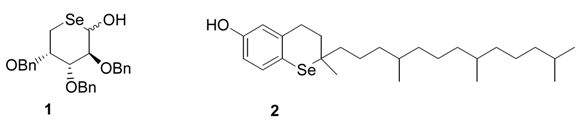
1]. Over the past few years, we have demonstrated the effectiveness of this chemistry for the preparation of selenium and tellurium-containing higher heterocycles. Indeed, so versatile is the free-radical approach that many classes of compound including some of hitherto unknown structure have been successfully prepared. Among these are included 5-deoxy-5-seleno pyranose sugars (eg.
1) [
2], and novel selenium and tellurium analogues (eg.
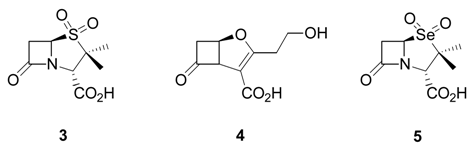
2) [
3] of the important antioxidant,
α-tocopherol.
It is generally appreciated that β-lactam based antibiotics have a limited future given increased resistance demonstrated by many strains of bacteria [
4]. There is an urgent need for the development of new classes of antibiotic and many laboratories have made significant progress in this area, especially with the introduction of peptide-based agents [
5].
A common mechanism of resistance in bacteria to the β-lactam class of antibiotics is through the β-lactamase enzymes [
6,
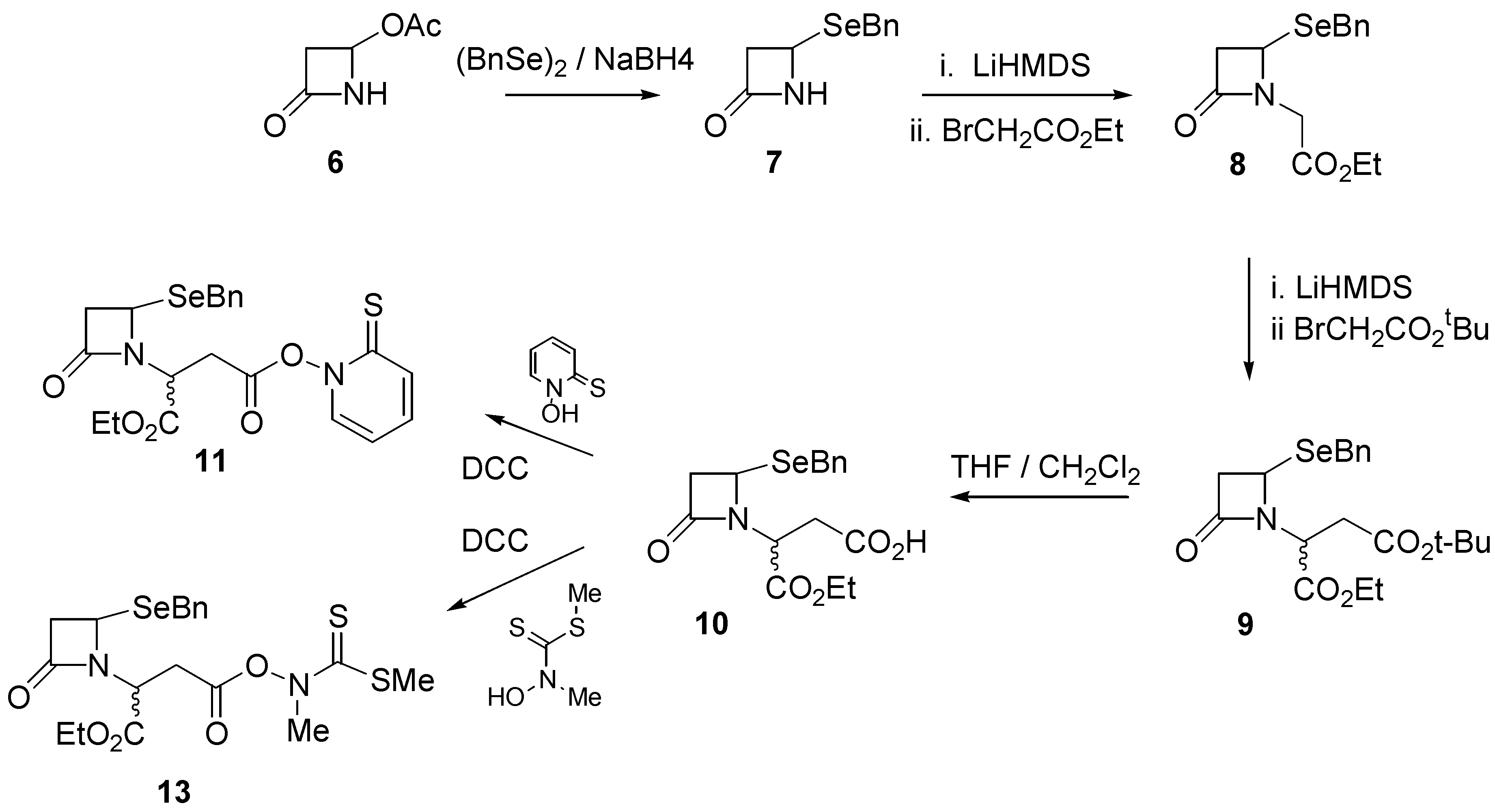
7]. These enzymes hydrolyse β-lactam antibiotics, rendering them impotent. Several β-lactamase inhibitors, such as sulbactam (
3) and clavulanic acid (
4) are known, are able to be administered in conjunction with antibiotics susceptible to β-lactamase activity [
8].
In an attempt to extend the variety of available β-lactamase inhibitors, we considered that substitution of the sulfur atom in the bicyclic core with selenium may provide an analogue (5) of sulbactam that preserves its shape and electronic properties, but which may differ in reactivity, and thus potency.
Selenium analogues of β-lactam antibiotics have been reported on previous occasions together with limited biological testing data [
9,
10]. In each report the yields of the target molecules are either poor, or the compounds prepared bear functionality that compromises biological activity. New methods for the preparation of these interesting systems are therefore important objectives. We now report that selenium analogues of β-lactam antibiotic nuclei are conveniently prepared by extension of our previously established radical methodologies.
Results and Discussion
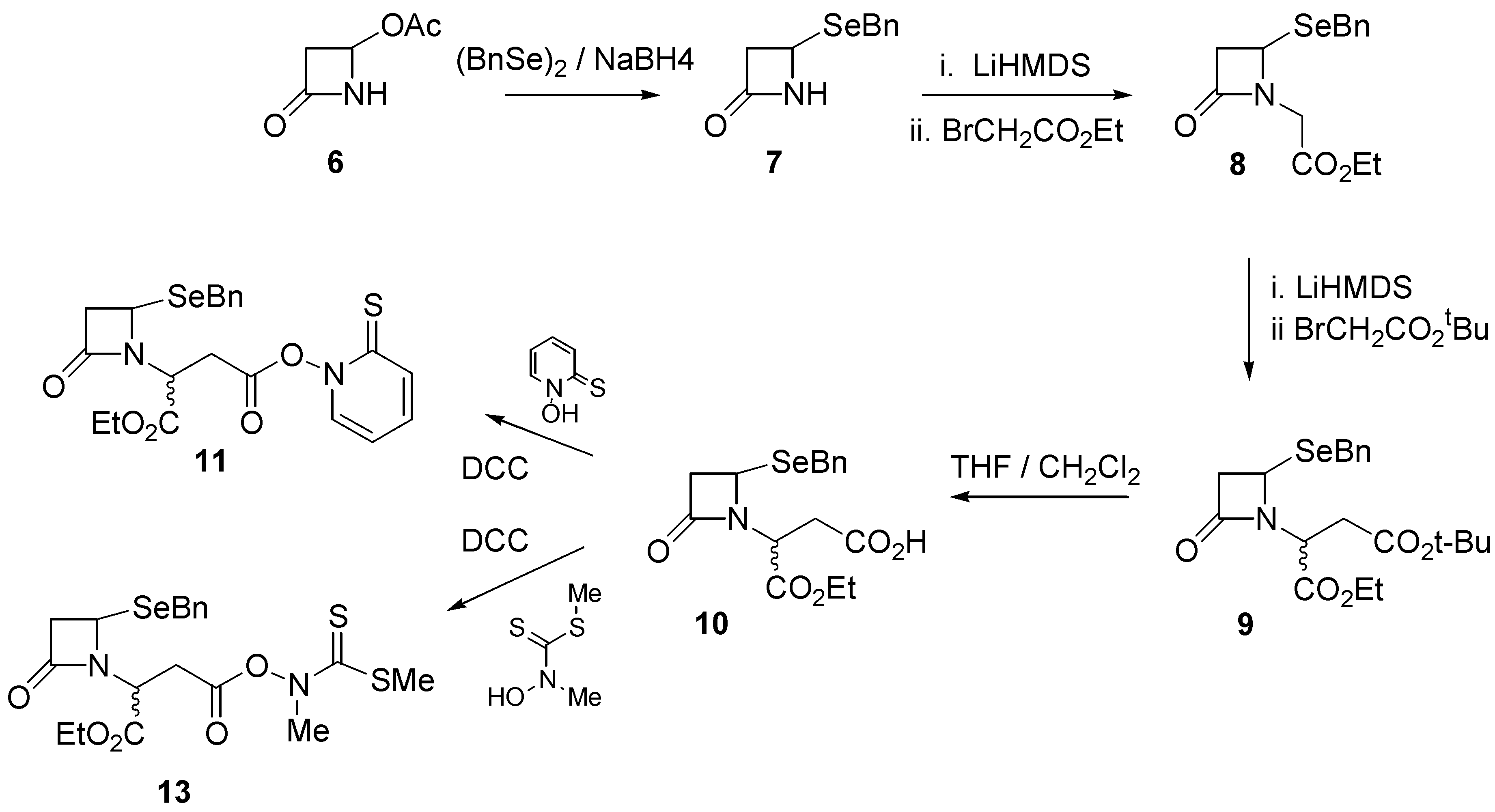
Our synthetic route began with commercially available 4-acetoxy-2-azetidinone (
6) which was reacted with, sodium benzylselenoate, prepared by treatment of dibenzyldiselenide and sodium borohydride, to give 4-(benzylseleno)-2-azetidinone (
7) in quantitative yield. Further reaction with ethyl bromoacetate under alkaline conditions (LiHMDS) afforded ester
8 in good (88%) yield. Ester
8 was further reacted with several equivalents of
t-butyl bromoacetate under alkaline conditions to provide the mixed diester
9 in 74% yield. Selective hydrolysis of the
t-butyl ester in a 1:1 mixture of trifluoroacetic acid and dichloromethane afforded the monoacid
10 in quantitative yield. Further reaction of
10 with N-hydroxypyridine-2-thione in the presence of dicyclohexylcarbodiimide (DCC) afforded the pyridinethioneoxycarbonyl (PTOC, Barton)[
11] ester free radical precursor
11 (
Scheme 1).
The Barton ester proved to be quite labile and was not purified. Rather, a concentrated solution of
11 was transferred into benzene, after which irradiation with a tungsten lamp for about two hours resulted in a distinct colour change and the appearance of several components by TLC. The dominant TLC spot was isolated and characterized and proved to be the required ethyl 1-aza-7-oxo- 4-selenabicyclo[3.2.0]heptane-2-carboxylate (
12) which was isolated in 35% yield (based on
10) as an inseparable mixture of diastereoisomers (
Scheme 2). Attempts to improve the yield through irradiation at 0° or at reflux, or thermolysis in the dark did not result in any significant improvement. Presumably
12 arises through a mechanism involving intramolecular homolytic substitution at selenium (
Scheme 2).
We were pleasantly surprised, however, to find significant improvements through the use of the thiohydroximate ester derivative
13 of compound
10 as described recently by Kim [
12]. The thiohydroximate ester
13 was prepared by the action of N-methylhydroxydithiocarbamate and dicyclohexylcarbodiimide (DCC) on the carboxylic acid
10. It was stable to purification by column chromatography, and was isolated as a thick yellow oil. When the radical precursor
13 was irradiated in benzene as described above for
11, the required selenapenam
12 was isolated in 71% yield.
Interestingly, the diastereomeric ratios ranged from 1:1 to 8:1 depending upon conditions, however, upon standing, epimerization to a single diastereoisomer
14 was observed within 72 hours (
Scheme 2). Selenapenam
14 displayed a signal at δ 301.0 in its
77Se NMR spectrum, as well as a doublet of doublets at δ 3.69 for the proton at position-2 in
14 in the 1H-NMR spectrum. Coupling constants of 3.9 and 15.9 Hz for this proton are indicative of the relative stereochemistry depicted in
14.
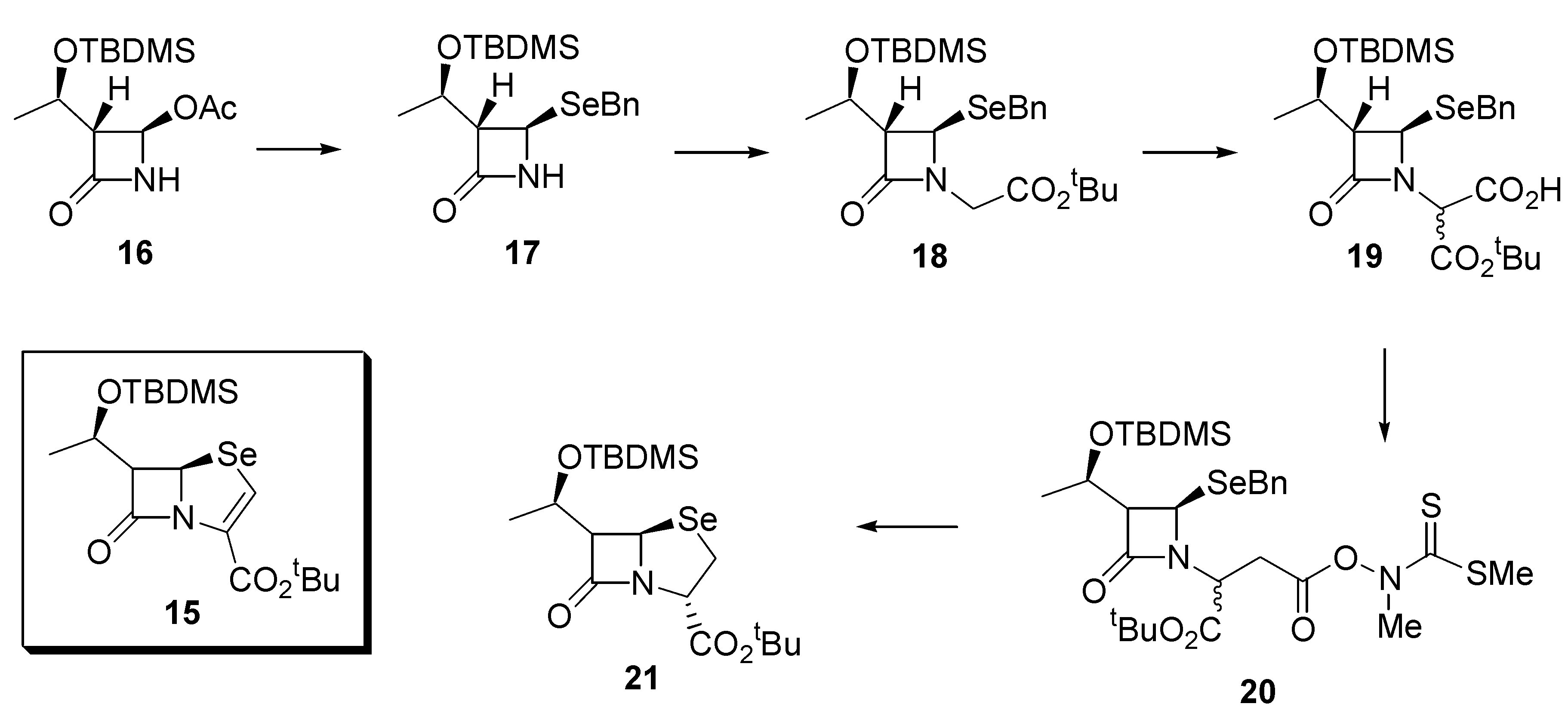
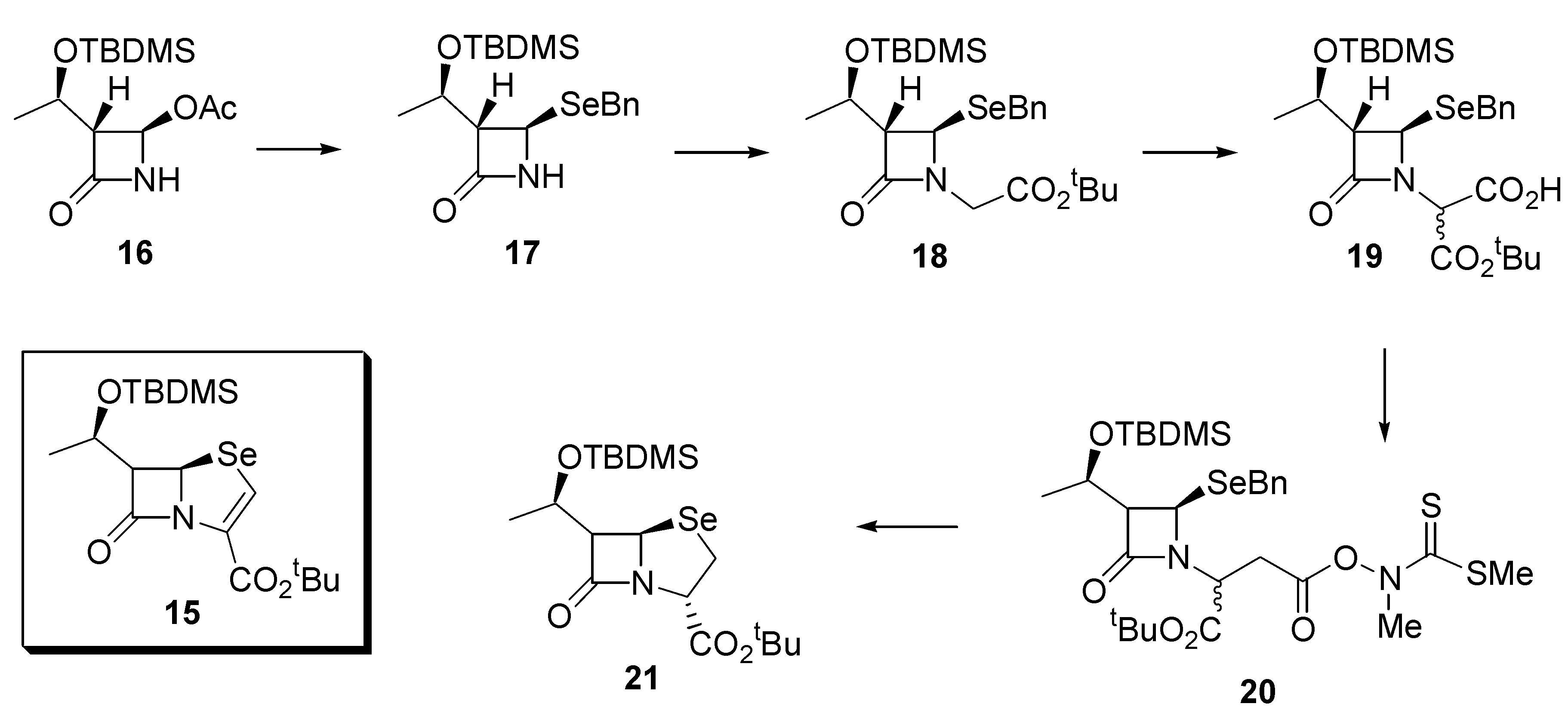
While the synthesis described above affords the general core structure of a β-lactamase inhibitor, we were also interested in the preparation of selenium-containing analogues of penem β-lactam antibiotics (eg.
15). To that end, the commercially available, optically pure, 4-acetoxy- azetidinone
16 was converted into the benzylseleno derivative
17 as described above for the preparation of
7, with complete retention of configuration (de > 98%). Because of the susceptibility of the protecting group in
17 to acidic workup, it was necessary to subtly change the order in which the subsequent steps were executed to that described in
Scheme 1. The selenide
17 was reacted with
t-butyl bromoacetate under alkaline conditions (LiHMDS) to afford the ester
18 in 86% yield. Subsequent reaction with ethyl bromoacetate and LiHMDS, followed by careful barium hydroxide hydrolysis provided the acid
19 as a 7:1 mixture of diastereoisomers in 68% yield. Further conversion of
19 into the thiohydroximate ester
20 and photolysis as described above afforded the required selenapenam
21 in 35% yield (based on
19) (
Scheme 3). Once again, NMR spectroscopy provided evidence for the formation of
21, in particular, the penam displayed a signal at δ 279.8 in its
77Se-NMR spectrum.
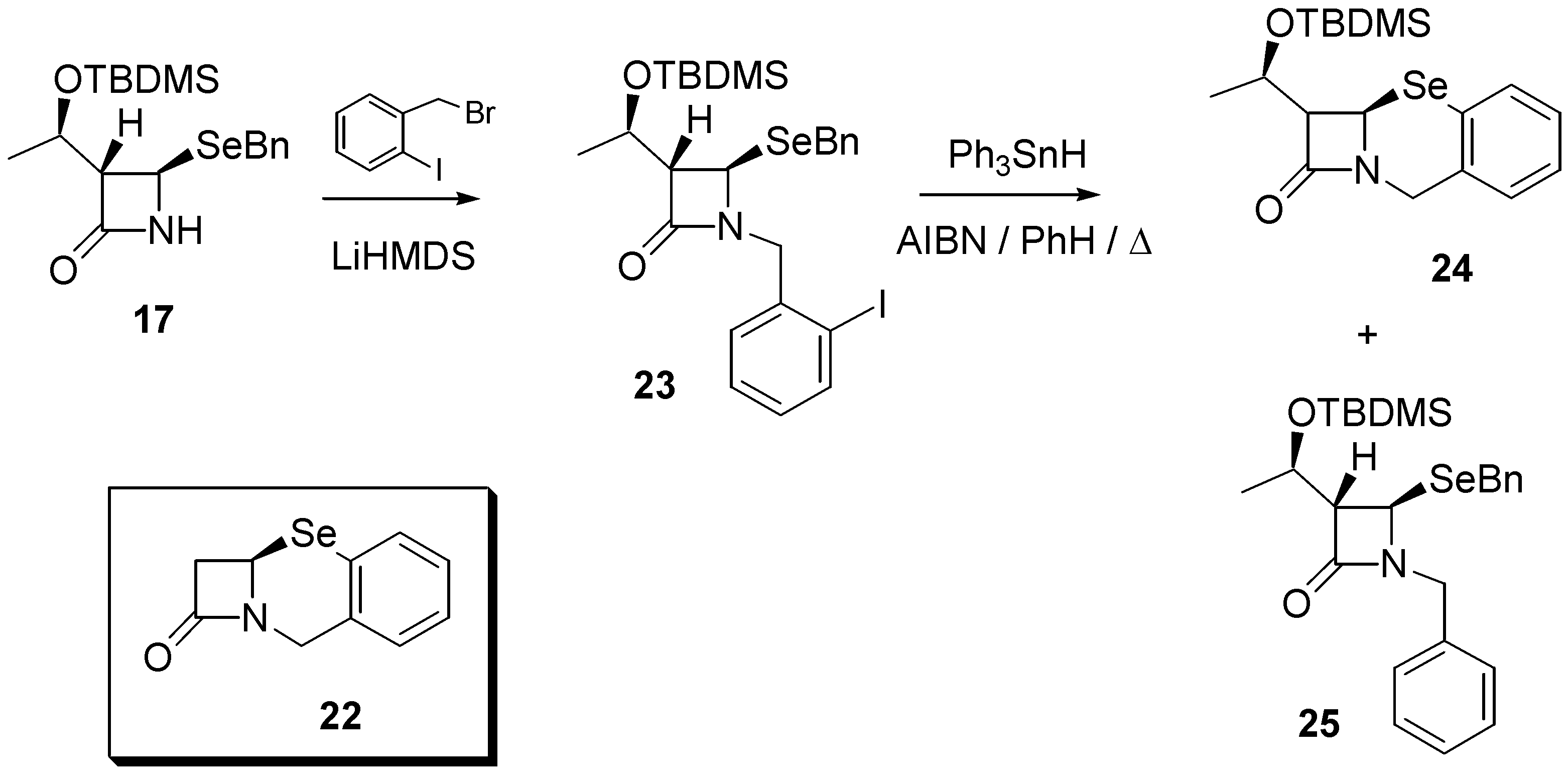
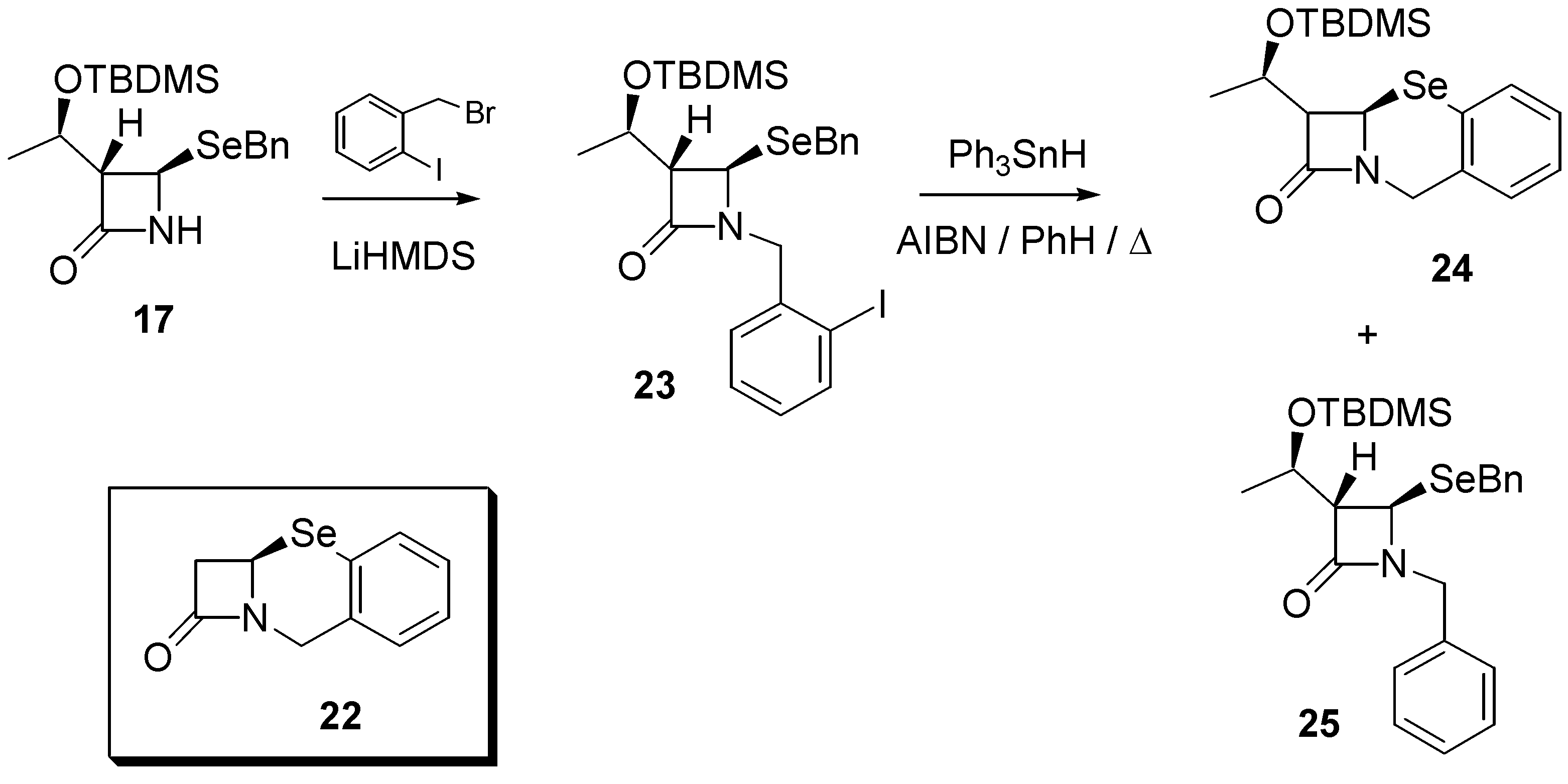
Finally, with the aim of exploring other biologically important selenium heterocycles, we considered the feasibility of preparing benzo-fused systems such as that found in 12,2a-dihydro- 1H,8H-azeto[2,1-b][1,3]benzoselenazin-1-one (
22). Therefore,
17 was reacted with 2-(bromo- methyl)iodobenzene under alkaline conditions to afford the aromatic iodide
23 in 65% yield. Reaction with triphenyltin hydride (0.015M) under standard radical conditions (AIBN, refluxing benzene) afforded the required product
24 in 59% yield, together with the uncyclized (directly reduced) byproduct
25 demonstrating, once again, that intramolecular homolytic substitution chemistry is a versatile method for the construction of selenium-containing rings (
Scheme 4). Selenide
24 exhibited a signal at δ 311.8 in its
77Se-NMR spectrum.

{kind=link}
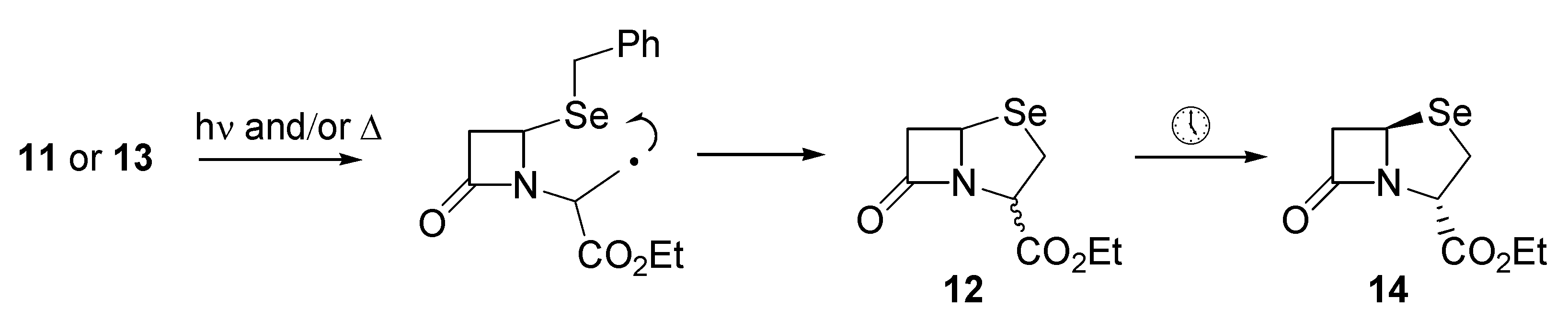{kind=link}
{kind=link}
{kind=link}