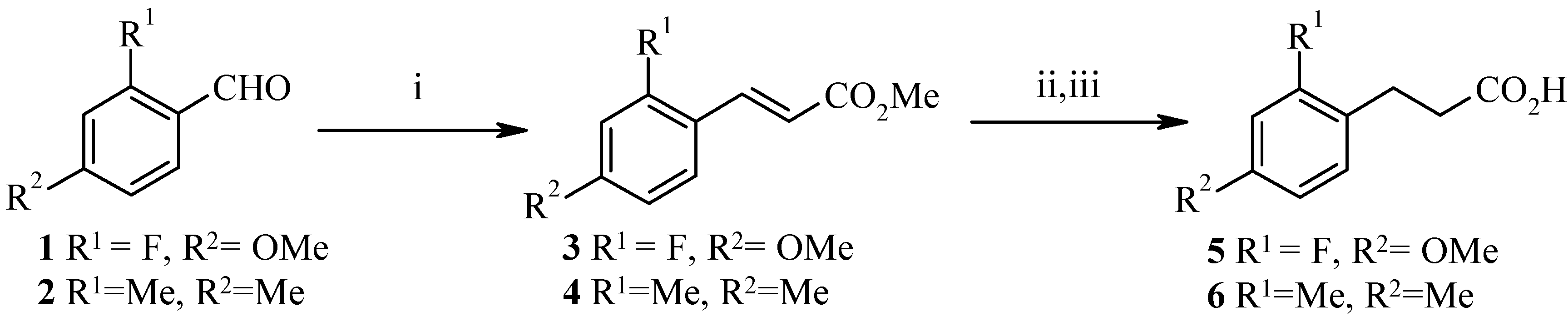Results and Discussion
As part of a collaborative medicinal chemistry effort, we needed to develop an enantioselective synthesis of (2
S)-2-(hydroxymethylphenyl) propionic acids for use as chiral building blocks. It was envisaged that an auxiliary-directed stereoselective alkylation of a phenylpropionamide should afford the target compounds after auxiliary removal. The synthesis of the starting phenylpropionic acids is detailed in
Scheme 1 and
Scheme 2. 3-(2-Fluoro-4-methoxyphenyl) propionic acid (
5) and 3-(2,4-dimethyl-phenyl)propionic acid (
6) were synthesised from the corresponding benzaldehydes
1 and
2 via Horner/Emmons reaction. Hydrogenation of the resulting α,β-unsaturated esters
3 and
4 followed by hydrolysis afforded the phenylpropionic acids
5 and
6 in high yield. The synthesis of 3-(2-fluoro-4-methylphenyl) propionic acid (
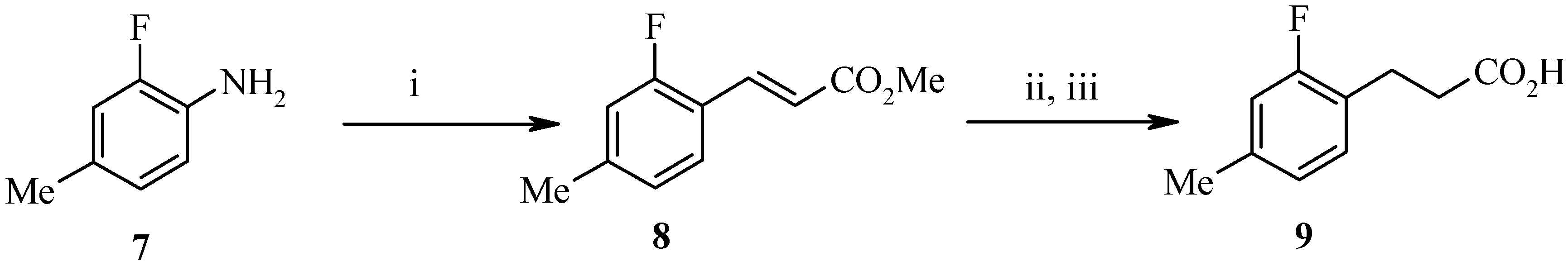
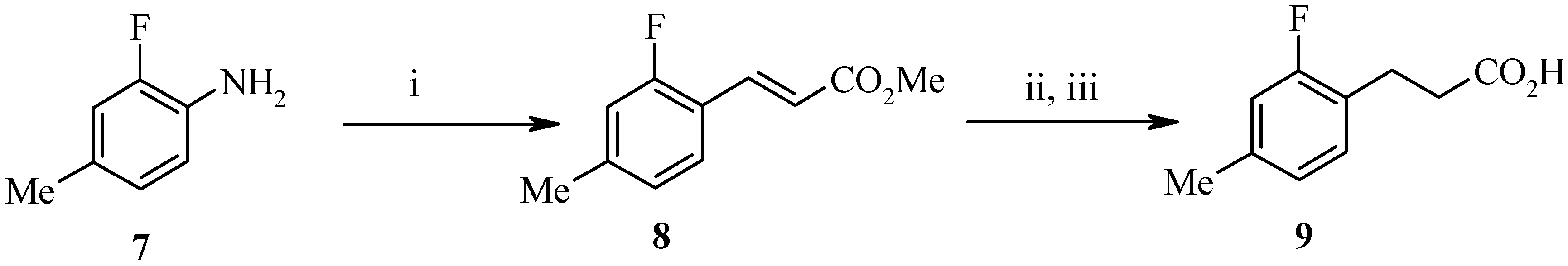
9) shown in
Scheme 2 began from 2-fluoro-4-methylaniline (
7) as the benzaldehyde was not commercially available. Diazonium salt formation from the aniline followed by an
in situ Heck type coupling with Pd(dba)
2 and methyl acrylate afforded the α,β−unsaturated ester
8 in high yield. Hydrogenation and hydrolysis gave the required acid
9.
Scheme 1.
Reagents and Conditions: (i) Methyl diethylphosphonoacetate, NaH, DMF; (ii) 10% Pd on C, H2; (iii) NaOH.
Scheme 2.
Reagents and Conditions: (i) NaNO2, H2SO4, Pd(dba)2, methyl acrylate; (ii) 10% Pd on C, H2; (iii) NaOH.
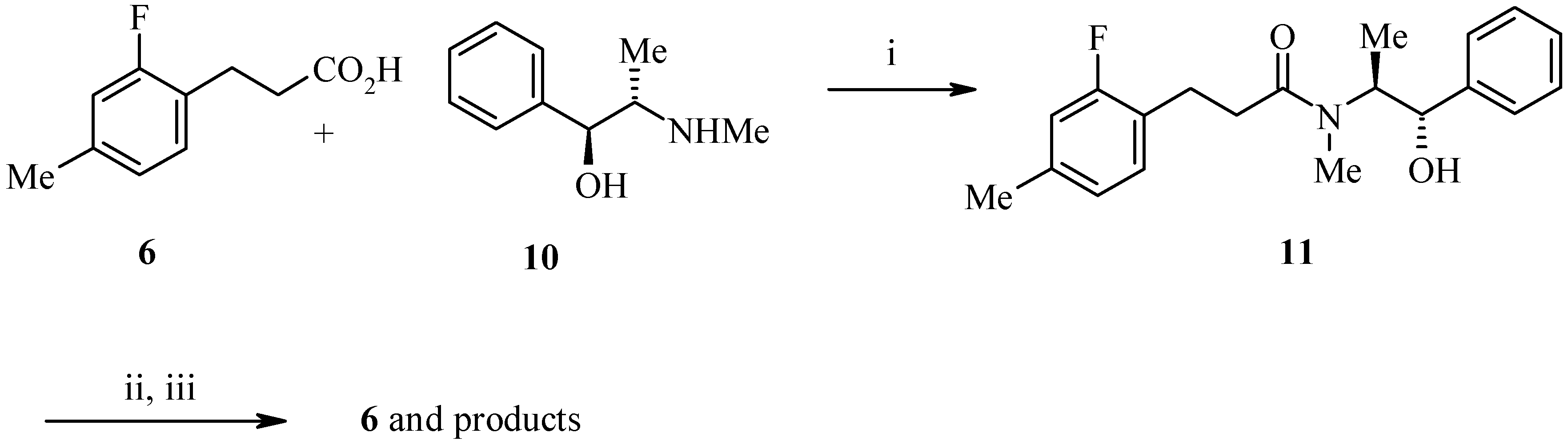
Our research plan was to couple a chiral auxiliary onto the required phenylpropionic acid and to direct alkylation from one face of the enolate. We initially used pseudoephedrine as a chiral auxiliary as Myers had reported excellent enantiomeric excesses for the alkylation of pseudoephedrine glycinamide [
2]. Adapting this method, we successfully coupled 3-(2-fluoro-4-methylphenyl) propionic acid (
9) to (
S,S)-(+)-pseudoephedrine (
10) in the presence of EDC, DIEA to give amide
11 (
Scheme 3). Alkylation of
11 with LDA and methoxymethyl chloride (MOMCl) or benzyloxymethyl chloride (BOMCl) gave a mixture of products with low conversion to the desired material. Further work on
11 was abandoned in favour of the approach outlined in
Scheme 4.
Scheme 3.
Reagents and Conditions: (i) HOBt.H2O, EDC, NMM, THF; (ii) LiCl, LDA, MOMCl, -78°C; (iii) 3N HCl reflux.
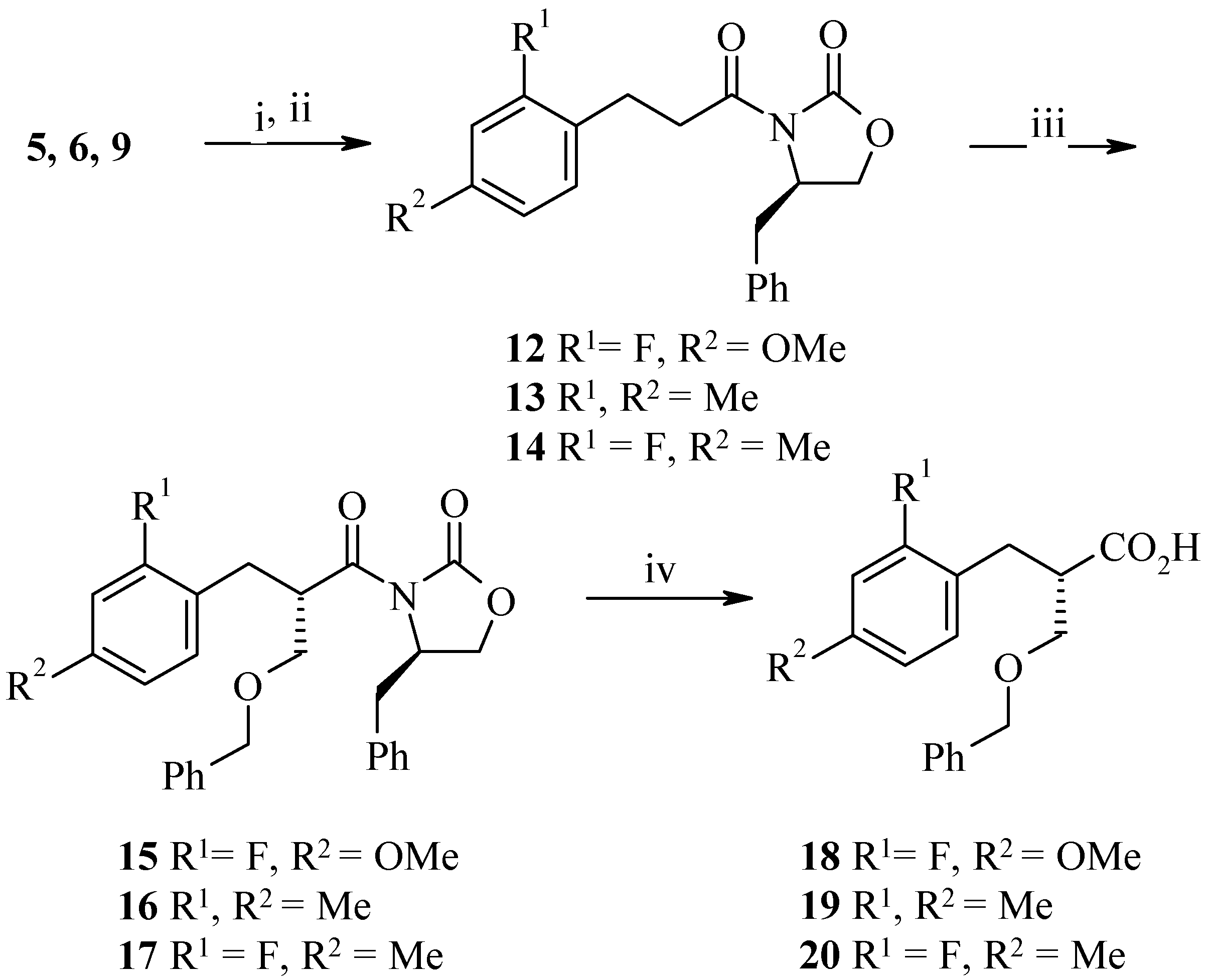
In 1990, Evans first reported the use of TiCl
4 as a pre-complexation agent with a series of 3-acylated-2-oxazolidinones [
3]. After deprotonation with an amine base, alkylation proceeded almost exclusively from one face of the enolate-titanium complex. Recently, Rawlings and co-workers reported a completely diastereoselective benzyloxymethylation of (4
R)-4-isopropyl-3-(3-phenyl-propionyl)-2-oxazolidinone as a key step in the asymmetric synthesis of A factor [
4]. The deployment of BOMCl in this case had the advantage of allowing for reductive removal of the benzyl protecting group after subsequent synthetic operations. Using this procedure we were able to access compounds
15–
17 via benzyloxymethylation of a series of 4-benzyl-3-[3-(2,4-disubstituted-phenyl)-propionyl]-2-oxazolidinones
12–
14 with complete diastereoselectivity (
Scheme 4).
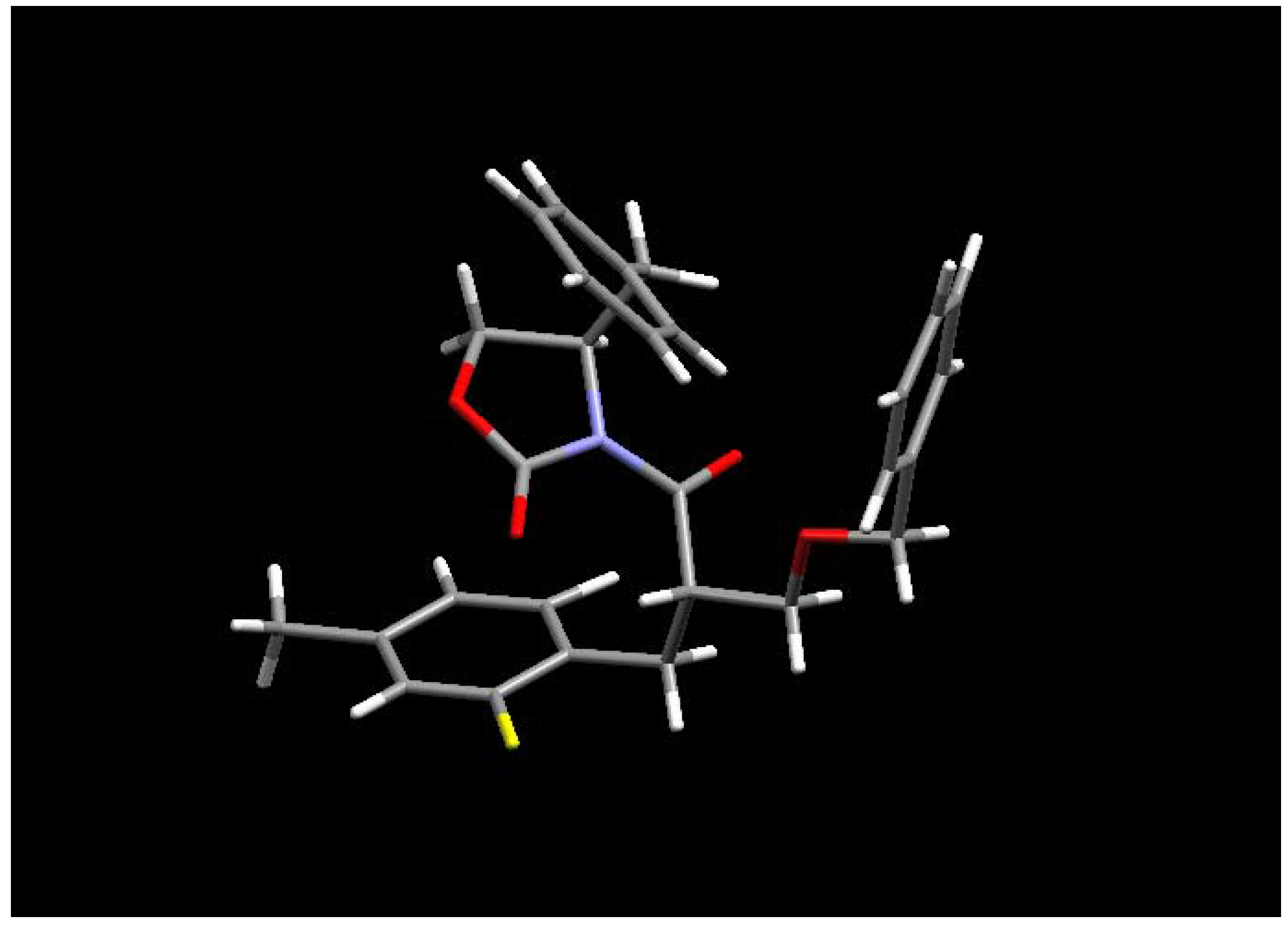
An X-ray crystal structure of (4
R)-4-benzyl-3-[(2
S)-2-benzyloxymethyl-3-(2-fluoro-4-methyl-phenyl)propionyl]-2-oxazolidinone (
17) revealed that alkylation had indeed occurred from the least hindered face of the enolate delivering the required
S stereochemistry at C2 (
Figure 1). Furthermore, the respective minor diastereomers were not observed by
1H-NMR, HPLC nor LC/MS. Removal of the auxiliary, to give
18–
20, was achieved using LiOH·H
2O and 35% aqueous hydrogen peroxide in degassed tetrahydrofuran/water in good yield [
4,
5]. It should be noted that when 1.0 equivalent of LiOH·H
2O was used we observed an impurity resulting from attack of the lithium peroxide anion onto the oxazolidinone carbonyl. This by-product was eliminated when 0.9 equivalent of LiOH·H
2O was used.
Scheme 4.
Reagents and Conditions: (i) SOCl2, DMF; (ii) n-BuLi, (4R)-4-benzyl-2-oxazolidinone; (iii) TiCl4, DIEA, BOMCl; (iv) LiOH·H2O, 35% aq H2O2, THF/H2O.
Figure 1.
X-ray crystal structure of
17 [
6].
Figure 1.
X-ray crystal structure of
17 [
6].
Experimental
General
1H-NMR (400 MHz) and 13C-NMR (100 MHz) were recorded on a VarianUNITY INOVA spectrometer in d-CHCl3 solutions. LCMS were run on an Applied Biosystems/MDS Sciex API-2000 LC/MS/MS system. HPLC analysis was performed on a Millenium 100 HPLC System and retention times (tR) are reported at 214 nm. X-ray crystallography was carried out on a Bruker Smart Apex X-ray diffractometer. The images were generated using Mercury 1.2.1 from pdb files. Analytical thin layer chromatograms were visualised under UV or with 20% w/v solution of phosphomolybdic acid in ethanol. Flash chromatography was performed with Merck silica gel No. 9385. Anhydrous solvents were purchased from Aldrich Chemical Co. in Sure/SealTM bottles. The chiral auxiliary was also purchased from Aldrich.
Representative procedure for the synthesis of 3-arylacrylic acid methyl esters: preparation of 3-(2-fluoro-4-methoxyphenyl) acrylic acid methyl ester (3)
To a suspension of sodium hydride (3.46g, 86.9 mmol, 60% wt in oil, petrol washed) in anhydrous N,N-dimethylformamide (35 mL) at 0°C was added neat methyl diethylphosphonoacetate (16.0 mL, 86.9 mmol) over a 20 min period. The resulting pale solution was warmed to room temperature over 15 min then re-cooled to 0°C. A solution of 2-fluoro-4-methoxybenzaldehyde 1 (12.18 g, 79.0 mmol) in anhydrous N,N-dimethylformamide (25 mL) was added via cannula to the reaction. The resulting orange solution was warmed to room temperature and stirred for 20 h. The reaction was cooled in an ice/water bath and quenched with water (200 mL). A white solid precipitated out of solution and was collected by filtration. The product was washed with water (x4) and dried at the pump to afford 3-(2-fluoro-4-methoxyphenyl) acrylic acid methyl ester (3, 12.45 g, 75%) as a white solid, mp 46-48°C; 1H-NMR δ: 3.77 (s, 3H), 3.78 (s, 3H), 6.37 (d, J = 16.4 Hz, 1H), 6.60 (dd, J = 12.8, 2.4 Hz, 1H), 6.67 (dd, J = 8.8, 2.4 Hz, 1H), 7.40 (t, J = 8.4Hz, 1H), 7.71 (d, J = 16.4 Hz, 1H); 13C-NMR δ: 51.2, 55.3, 101.5 (d, J2CF = 25.8 Hz), 110.5, 114.7 (d, J3CF = 12.2 Hz), 117.3, 129.5, 136.9, 162.1 (d, J1CF = 252.2 Hz), 162.3 (d, J3CF = 11.4 Hz), 167.2.
In situ diazotisation-Heck type coupling: synthesis of 3-(2-fluoro-4-methylphenyl) acrylic acid methyl ester (8)
2-Fluoro-4-methylaniline (7, 5.0 g, 7.99 mmol) was dissolved in acetic acid (40 mL), cooled to 15° C and treated with the dropwise addition of concentrated sulfuric acid (4.26 mL). A solution of sodium nitrite (3.03 g, 44.0 mmol) in water (7.5 mL) was then added dropwise to the reaction mixture while keeping the reaction temperature below 15° C. The reaction was warmed to 45° C and the resulting red solution was treated with Pd(dba)2 (207 mg, 0.36 mmol), followed by the dropwise addition of methyl acrylate (3.96 mL, 8.79 mmol). The reaction temperature was increased to 70° C after the addition was complete. After stirring at room temperature overnight the reaction was quenched with ice and extracted into ether three times. The combined organic layers were washed with ice cold water three times, saturated sodium hydrogen carbonate, dried over sodium sulfate, filtered and concentrated to give 3-(2-fluoro-4-methylphenyl) acrylic acid methyl ester (8) as a brown oil (6.10 g, 85% yield); 1H-NMR δ: 2.33 (s, 3H), 3.77 (s, 3H), 6.49 (d, J = 16.0 Hz, 1H), 6.91 (d, J = 11.6 Hz, 1H), 6.95 (d, J = 8.4 Hz, 1H), 7.40 (t, J = 8.0 Hz, 1H), 7.78 (d, J = 16.0 Hz, 1H); 13C-NMR δ: 23.7, 34.4, 55.5, 101.7 (d, J2CF = 25.1 Hz), 109.7, 118.8 (d, J3CF = 16.0 Hz), 130.7, 130.8, 159.5, 161.6 (d, J1CF = 243.8 Hz), 179.0.
Representative procedure for the syntheses of 3-arylpropionic acids: synthesis of 3-(2-fluoro-4-methoxyphenyl)propionic acid (5)
Methyl ester 3 (12.0 g, 57.1 mmol) was dissolved in methanol (150 mL), then 10% Pd on C (700 mg) was added and the mixture was hydrogenated for 20 h. The reaction was evacuated, filtered and concentrated to afford the saturated ester (11.0g, 91%). The reduced ester was heated to reflux in 2N aqueous sodium hydroxide (150 mL) for 3 h. The reaction was cooled and extracted with ethyl acetate (x2). The aqueous layer was acidified with concentrated hydrochloric acid and the resulting white solid was collected by filtration, washing with water. The product was dried at the pump to give 3-(2-fluoro-4-methoxyphenyl)propionic acid (5, 8.2 g, 80%), mp 86-87°C; 1H-NMR: δ 2.65 (t, J = 7.6 Hz, 2H), 2.91 (t, J = 7.6 Hz, 2H), 3.77 (s, 3H), 6.57-6.63 (m, 2H), 7.10 (t, J = 8.4 Hz, 1H); 13C-NMR δ: 23.7, 34.4, 55.5, 101.7 (d, J2CF = 25.1 Hz), 109.7, 118.8 (d, J3CF = 16Hz), 130.7, 130.8, 159.5, 161.6 (d, J1CF = 243.8 Hz), 179.0.
Representative procedure for the synthesis of acyloxazolidinones [7]: preparation of (4R)-4-benzyl-3-[3-(2-fluoro-4-methoxyphenyl)propionyl]-2-oxazolidinone (12)
Acid 5 (3.99 g, 20.1 mmol) was heated to reflux in thionyl chloride (30 mL) and 2 drops of N,N-dimethylformamide for 1 h. The reaction was cooled and volatiles were removed in vacuo. In a separate flask, (4R)-(+)-4-benzyl-2-oxazolidinone (3.57 g, 20.1 mmol) was dissolved in tetrahydrofuran (30 mL) and 1,10-phenanthroline (40 mg) was added as indicator. The solution was cooled to -78°C and n-butyllithium (9.0 mL, 2.23 M solution in hexanes) was added until a brown colour persisted. A solution of the above prepared acid chloride in tetrahydrofuran (10 mL) was added via a cannula to the reaction. After addition was complete, the reaction was warmed to 0°C for 30 min. The reaction was quenched with saturated aqueous sodium hydrogen carbonate and the aqueous layer was extracted into dichloromethane (x3). The combined organic layers were dried over anhydrous sodium sulfate, filtered, concentrated and recrystallized from ethyl acetate (15 mL)/petroleum ether (20 mL) to afford (4R)-4-benzyl-3-[3-(2-fluoro-4-methoxyphenyl)-propionyl]-2-oxazolidinone (12, 5.06 g, 70%) as a white powder, mp 67-69°C; [α]D = - 29.4° (c = 0.11, CHCl3); 1H-NMR δ: 2.75 (dd, J = 17.4, 9.2 Hz, 1H), 2.97 (t, J = 7.2 Hz, 2H), 3.12-3.27 (m, 3H), 3.72 (s, 3H), 4.10-4.13 (m, 2H), 4.61-4.65 (m, 1H), 6.56-6.63 (m, 2H), 7.12-7.30 (m, 6H); 13C-NMR δ 22.8, 35.7, 37.5, 54.8, 55.2, 65.9, 101.3 (d, J2CF = 25.9 Hz), 109.5, 118.8 (d, J3CF = 15.9 Hz), 127.0, 128.6, 129.1, 130.7, 130.8, 135.1, 153.1, 159.2, 159.3, 161.3 (d, J1CF = 243.8 Hz), 171.9; Anal. Calcd. C20H20FNO4 requires C, 67.22; H, 5.64; N, 3.92. Found C, 67.23; H, 5.55; N, 4.06%.
Characterization data for (4R)-4-benzyl-3-[3-(2-fluoro-2,4-dimethylphenyl)propionyl]-2-oxazoli-dinone (13): HPLC tR = 10.94 min (73%); LCMS tR = 10.25 min (m/z 338.0 [M+H]+); 1H-NMR δ: 2.28 (s, 3H), 2.33 (s, 3H), 2.74-2.80 (m, 1H), 2.95-3.00 (m, 2H), 3.16-3.29 (m, 3H), 4.13-4.15 (m, 2H), 4.64-4.68 (m, 1H), 6.93-6.98 (m, 2H), 7.09-7.11 (m, 1H), 7.17-7.19 (m, 2H), 7.26-7.36 (m, 3H); 13C-NMR δ: 19.45, 21.11, 36.14, 38.09, 55.36, 66.40, 126.92, 127.92, 129.06, 129.16, 129.65, 131.34, 135.51, 135.66, 136.09, 136.16, 153.61, 172.85.
Characterization data for (4R)-4-benzyl-3-[3-(2-fluoro-4-methylphenyl)propionyl]-2-oxazolidinone (14): HPLC tR = 10.42 min (67%); 1H-NMR δ: 2.29 (s, 3H), 2.61-2.77 (m, 1H), 2.91 (t, J = 7.7Hz, 1H), 2.99 (t, J = 7.5 Hz, 1H), 3.10-3.30 (m, 3H), 4.10-4.18 (m, 2H), 4.62-4.67 (m, 1H), 6.80-6.86 (m, 2H), 7.03-7.32 (m, 6H).
Synthesis of (4R)-4-benzyl-3-[(2S)-2-benzyloxymethyl-3-(2-fluoro-4-methoxyphenyl)propionyl]-2-oxazolidinone (15)
A solution of 12 in anhydrous dichloromethane (75 mL) was cooled to between –5° and 0°C and treated with neat TiCl4 (1.19 mL, 10.9 mmol). After stirring for 10 min, diisopropylethylamine (2.04 mL, 11.72 mmol) was added dropwise and the resulting blood-red solution was stirred at 0°C for 1 h. Benzyloxychloromethyl ether (2.56 mL, 18.4 mmol) was added dropwise and the reaction was warmed to room temperature for 2 h. The reaction was quenched with saturated aqueous ammonium chloride then extracted into dichloromethane (x3). The combined organic layers were dried with anhydrous sodium sulfate, filtered, concentrated and purified by flash chromatography eluting with 80% dichloromethane/petrol to afford (4R)-4-benzyl-3-[(2S)-2-benzyloxymethyl-3-(2-fluoro-4-methoxy-phenyl)propionyl]-2-oxazolidinone (15, 3.22 g, 81%); HPLC tR = 11.32 min (82%); LCMS tR = 10.81 min (m/z 478.4 [M+H]+); 1H-NMR δ: 2.61-2.68 (m, 1H), 2.87-2.93 (m, 2H), 3.14-3.22 (m, 1H), 3.62-3.66 (m, 1H), 3.73 (s, 3H), 3.99-4.06 (m, 2H), 4.53-4.86 (m, 5H), 6.52-6.57 (m, 1H), 7.13-7.33 (m, 12H); 13C-NMR δ 27.3, 37.7, 44.1, 55.2, 55.5, 65.9, 70.2, 73.1, 101.4 (d, J2CF = 25.8 Hz), 109.7, 117.0 (d, J3CF = 15.9 Hz), 127.2, 127.6, 127.7, 128.3, 128.8, 129.4, 131.68, 131.74, 135.3, 138.1, 153.0, 159.6, 161.7 (d, J1CF = 243.0 Hz), 173.8.
Characterization data for (4R)-4-benzyl-3-[(2S)-2-benzyloxymethyl-3-(2-fluoro-2,4-dimethylphenyl)-propionyl]-2-oxazolidinone (16): HPLC tR = 12.13 min (70%); LCMS tR = 11.35 min (m/z 458.4 [M+H]+); 1H-NMR δ: 2.30 (s, 3H), 2.37 (s, 3H), 2.68-2.74 (m, 1H), 2.96-2.98 (m, 2H), 3.20 (dd, J = 3.1, 13.5 Hz, 1H), 3.70-3.74 (m, 1H), 3.79-3.84 (m, 2H), 3.91-4.00 (m, 2H), 4.52-4.56 (m, 1H), 4.59 (d, J = 2.0 Hz, 1H), 4.64-4.70 (m, 2H), 4.82 (s, 1H), 6.92-6.99 (m, 2H), 7.06-7.08 (m, 1H), 7.19-7.22 (m, 2H), 7.26-7.32 (m, 3H), 7.34-7.39 (m, 5H).
Characterization data for (4R)-4-benzyl-3-[(2S)-2-benzyloxymethyl-3-(2-fluoro-4-methylphenyl)-propionyl]-2-oxazolidinone (17): HPLC tR = 11.84 min (81%); LCMS tR = 10.77 min (m/z 462.3 [M+H]+); 1H-NMR δ: 2.29 (s, 3H), 2.67 (dd, J = 13.6, 9.6Hz, 1H), 2.92-3.02 (m, 2H), 3.20 (dd, J = 13.6, 3.2Hz, 1H), 3.67 (dd, J = 9.6Hz, J = 5.2Hz, 1H), 3.81 (dd, J = 9.2, 7.2 Hz, 1H), 4.01-4.09 (m, 2H), 4.40-4.50 (m, 1H), 4.54 (s, 2H), 4.59-4.63 (m, 1H), 6.79-6.84 (m, 2H), 7.07 (t, J = 7.6Hz, 1H), 7.16-7.18 (m, 2H), 7.23-7.29 (m, 4H), 7.31-7.32 (m, 4H); 13C-NMR δ: 21.0, 27.6, 37.8, 44.0, 55.3, 65.9, 70.3, 73.2, 115.8 (d, J2CF = 21.3Hz), 124.6, 127.2, 127.6, 127.7, 128.3, 128.9, 129.5, 130.1, 131.1, 135.3, 138.1, 161.1 (d, J1CF = 243.0 Hz ), 173.8.
Representative procedure for the synthesis of (2S)-2-benzyloxymethyl-3-phenylpropionic acids: synthesis of (2S)-2-benzyloxymethyl-3-(2-fluoro-4-methoxyphenyl)propionic acid (18)
35% Aqueous hydrogen peroxide (3.7 mL, 38 mmol) was added under nitrogen to a solution of 15 (2.50 g, 5.24 mmol) in a degassed mixture of tetrahydrofuran (50 mL) and water (13 mL). After 5 min, lithium hydroxide monohydrate (198 mg, 4.72 mmol) was added and the reaction was stirred at room temperature for 17 h. Tetrahydrofuran was removed in vacuo and the residue was diluted with water (50 mL) and additional lithium hydroxide monohydrate (50 mg) was added to keep the solution basic. The aqueous phase was extracted with ethyl acetate (x 3). The aqueous phase was then acidified to pH 2 with concentrated hydrochloric acid and the solution was extracted into dichloromethane (x 3). The combined organic layers were washed with water (x 2), dried over anhydrous sodium sulfate, filtered and concentrated to give (2S)-2-benzyloxymethyl-3-(2-fluoro-4-methoxyphenyl)propionic acid (18, 1.3 g, 78%) as a clear colourless oil; [α]D = +7.8° (c 0.42, CHCl3); HPLC tR = 9.11 min (92%); LCMS tR = 8.51 min (m/z 301.1 [M+H-H2O] +, 319.4 [M+H]+); 1H-NMR δ: 2.82-3.02 (m, 3H), 3.62 (d, J =5.6 Hz, 2H), 3.75 (s, 3H), 4.52 (d, J = 4.0 Hz, 2H), 6.59 (d, J = 10.0 Hz, 2H), 7.07 (t, J = 8.8 Hz, 1H), 7.25-7.33 (m, 5H); 13C-NMR δ: 27.2, 46.2, 55.4, 69.5, 73.1, 101.5 (d, J2CF = 25.8 Hz), 109.7, 117.1 (d, J3CF = 15.9 Hz), 127.6, 128.3, 131.50, 131.56, 137.7, 159.6, 159.7, 161.6 (d, J1CF = 243.8 Hz), 179.0.
Characterization data for (2S)-2-benzyloxymethyl-3-(2,4-dimethylphenyl)propionic acid (19): HPLC tR = 9.88 min (93%); LCMS tR = 9.39 min (m/z 299.4 [M+H]+); 1H-NMR δ: 2.28 (s, 6H), 2.83-2.94 (m, 1H), 2.95-3.08 (m, 2H), 3.62-3.67 (m, 2H), 4.52 (s, 2H), 6.89-6.92 (m, 1H), 6.96-7.01 (m, 2H), 7.27-7.35 (m, 5H); 13C-NMR δ: 19.45, 21.23, 31.42, 46.71, 69.94, 73.46, 126.87, 127.89, 128.61, 129.75, 131.49, 133.85, 136.29, 136.34, 138.06, 180.03.
Characterization data for (2S)-2-benzyloxymethyl-3-(2-fluoro-4-methylphenyl)propionic acid (20): HPLC tR = 9.77 min (85%); LCMS tR = 8.89 min (m/z 303.1 [M+H]+); 1H-NMR δ: 2.28 (s, 3H), 2.80-3.00 (m, 3H), 3.61 (d, J = 5.2Hz, 2H), 4.48 (d, J = 2.8Hz, 2H), 6.78 (d, J = 2.8Hz, 1H), 6.81 (s, 1H), 7.02 (t, J = 8.4Hz, 1H), 7.12-7.14 (m, 1H), 7.22-7.32 (m, 4H); 13C-NMR δ 21.1, 27.8, 41.6, 46.5, 54.1, 69.9, 73.4, 116.1 (d, J2CF = 21.3Hz), 122.5 (d, J2CF = 15.2Hz), 124.9, 127.5, 127.9, 128.6, 129.2, 131.3 (d, J3CF = 5.3Hz), 136.1, 138.1, 138.9 (d, J3CF = 7.5Hz), 161.3 (d, J1CF = 243.8Hz), 178.9

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}