Masked ω-Lithio Ester Enolates: Synthetic Applications

Abstract
:Introduction
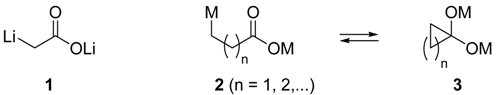
Results and Discussion
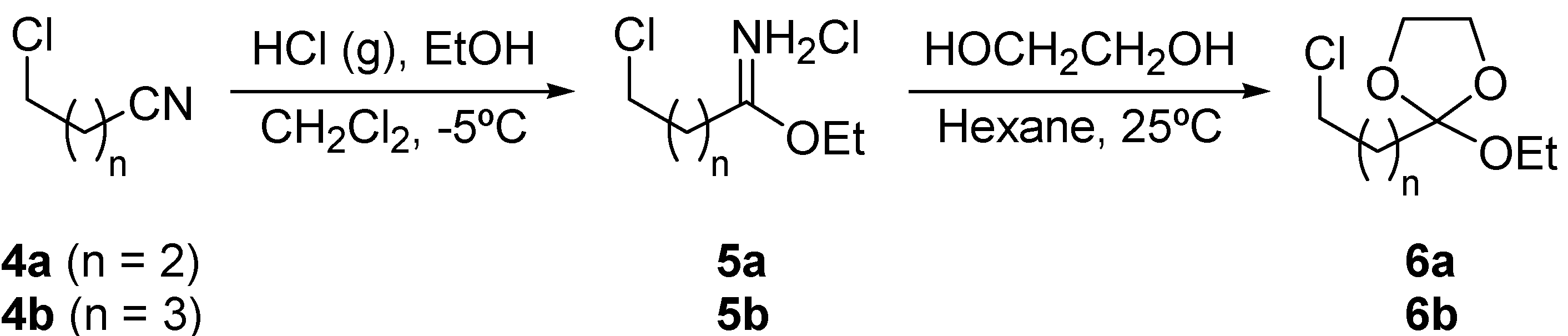
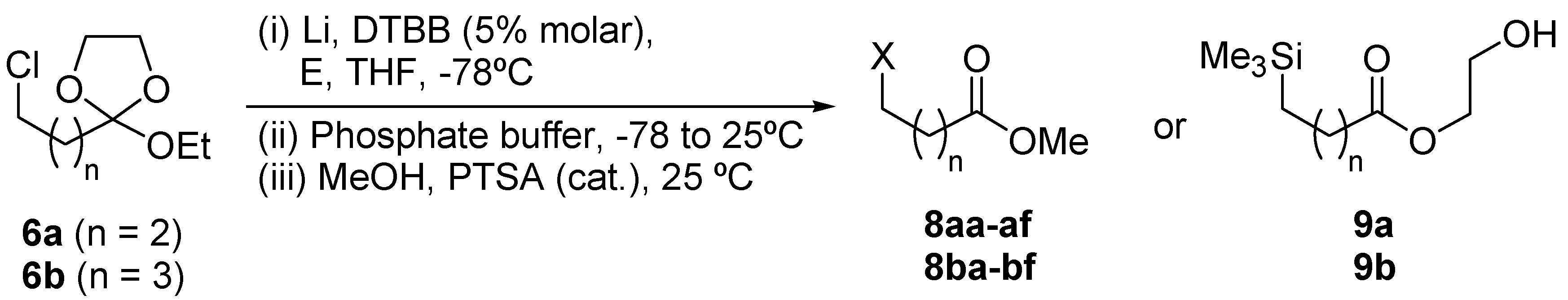


{kind=link}
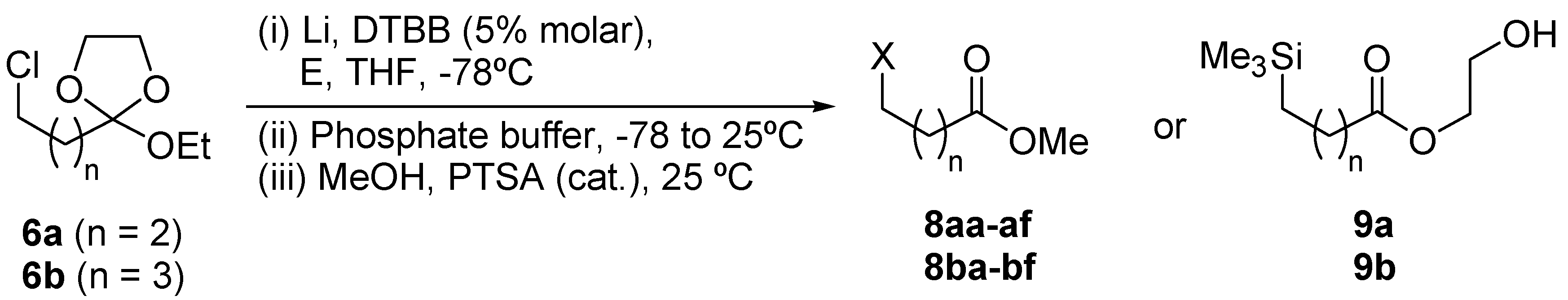{kind=link}
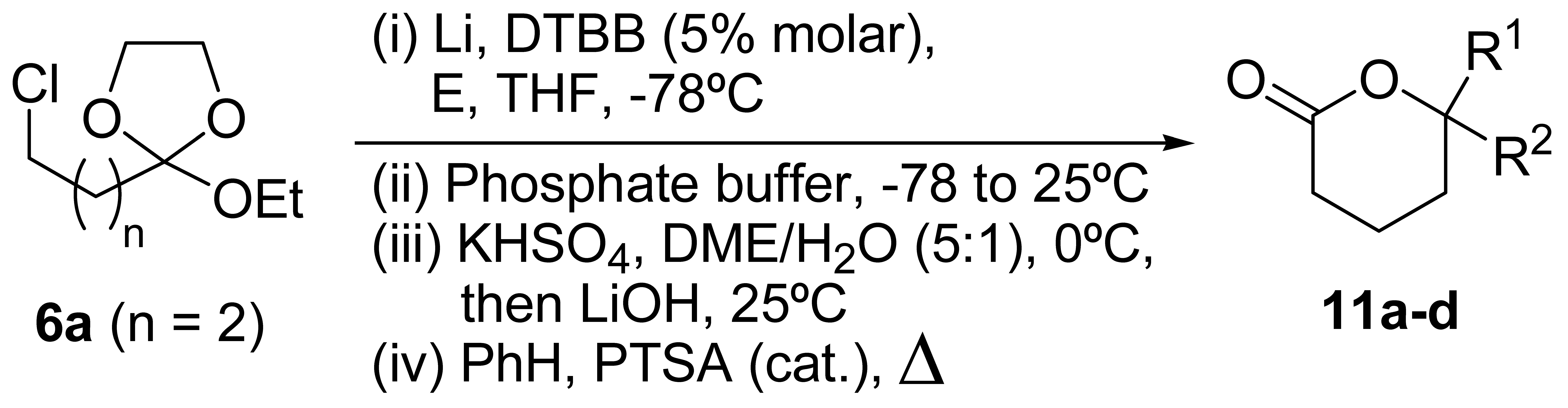{kind=link}
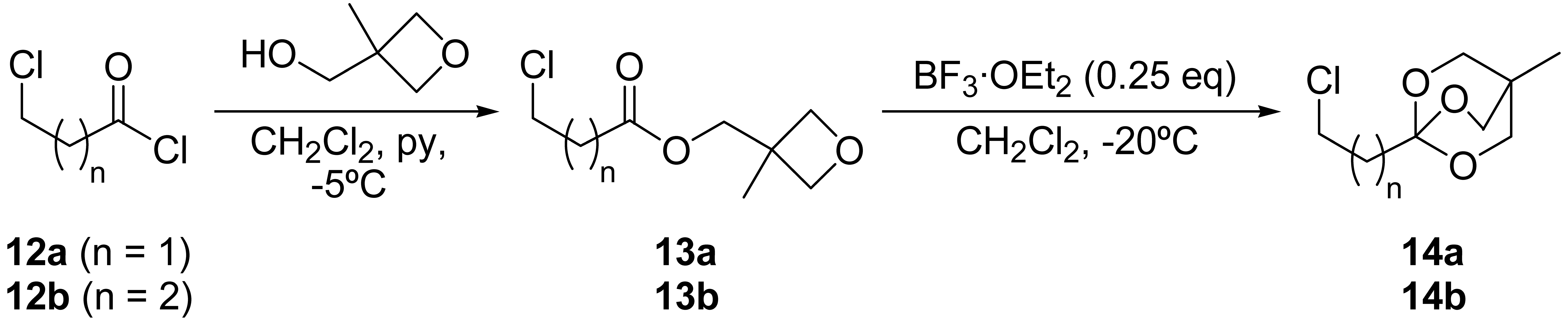{kind=link}
{kind=link}
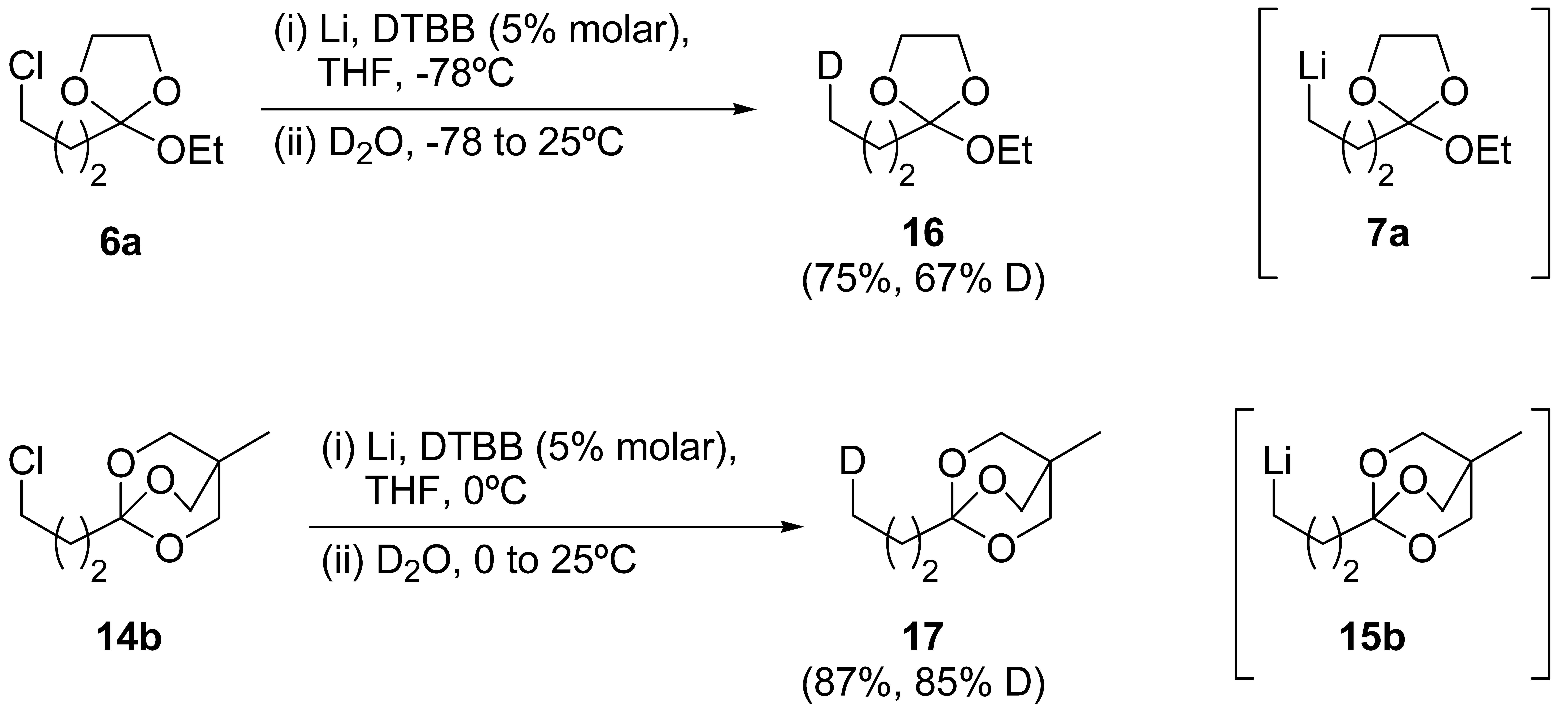{kind=link}
| Entry | Starting material | Electrophile (E) | Product 8 or 9a | |||
| No. | n | X | Yield (%)b | |||
| 1 | 6a | ButCHO | 8aa | 2 | ButCHOH | 58 |
| 2 | 6a | PhCHO | 8ab | 2 | PhCHOH | 63 (54)c (47)d |
| 3 | 6a | (CH2)5CO | 8ac | 2 | (CH2)5COH | 59 |
| 4 | 6a | Et2CO | 8ad | 2 | Et2COH | 51 |
| 5 | 6a | PhCOMe | 8ae | 2 | PhC(OH)Me | 52 |
| 6 | 6a | PhCH=NPh | 8af | 2 | PhCHNHPh | 57 |
| 7 | 6a | Me3SiCl | 9ae | 2 | Me3Si | 40 |
| 8 | 6b | ButCHO | 8ba | 3 | ButCHOH | 62 |
| 9 | 6b | PhCHO | 8bb | 3 | PhCHOH | 66 |
| 10 | 6b | (CH2)5CO | 8bc | 3 | (CH2)5COH | 56 |
| 11 | 6b | Et2CO | 8bd | 3 | Et2COH | 52 |
| 12 | 6b | PhCOMe | 8be | 3 | PhC(OH)Me | 49 |
| 13 | 6b | PhCH=NPh | 8bf | 3 | PhCHNHPh | 54 |
| 14 | 6b | Me3SiCl | 9be | 3 | Me3Si | 47 |
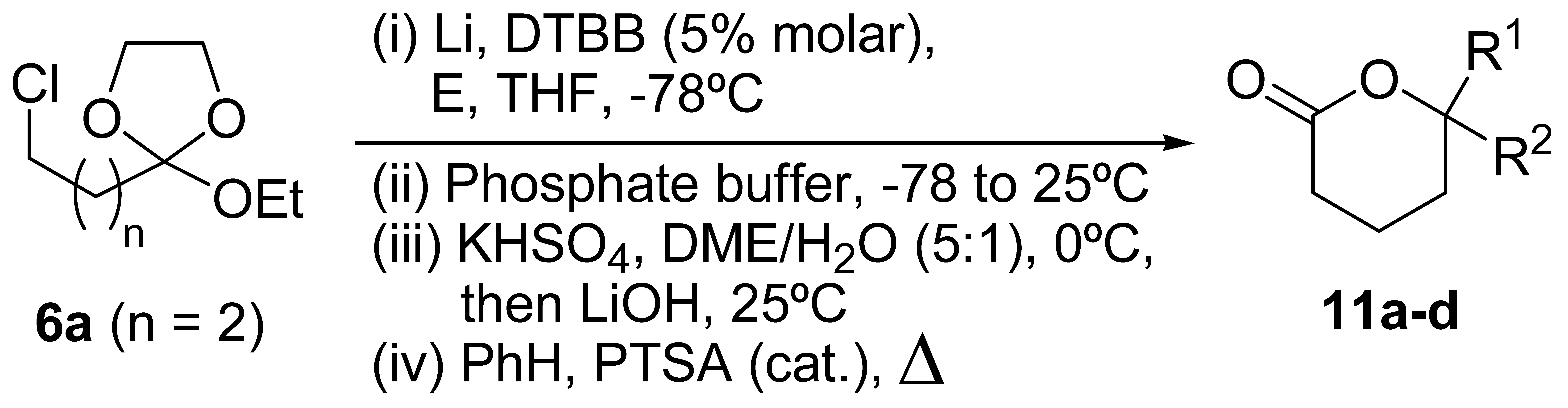
| Entry | Carbonyl compound (E) | Product 11a | |||
| No. | R1 | R2 | Yield (%)b | ||
| 1 | ButCHO | 11a | But | H | 47 |
| 2 | PhCHO | 11b | Ph | H | 33 |
| 3 | (CH2)5CO | 11c | (CH2)5 | 37 | |
| 4 | Et2CO | 11d | Et | Et | 35 |

| Entry | Carbonyl compound (E) | Product 15a | |||
| No. | R1 | R2 | Yield (%)b | ||
| 1 | ButCHO | 15a | But | H | 45 |
| 2 | PhCHO | 15b | Ph | H | 43 |
| 3 | (CH2)5CO | 15c | (CH2)5 | 39 | |
| 4 | Et2CO | 15d | Et | Et | 38 |
| 5 | PhCOMe | 15e | Ph | Me | 37 |
Conclusions
Acknowledgements
Experimental
General
Preparation of ortho esters 6.
DTBB-catalyzed lithiation of ortho esters 6: Preparation of esters 8 and 9.
Preparation of δ-lactones 11.
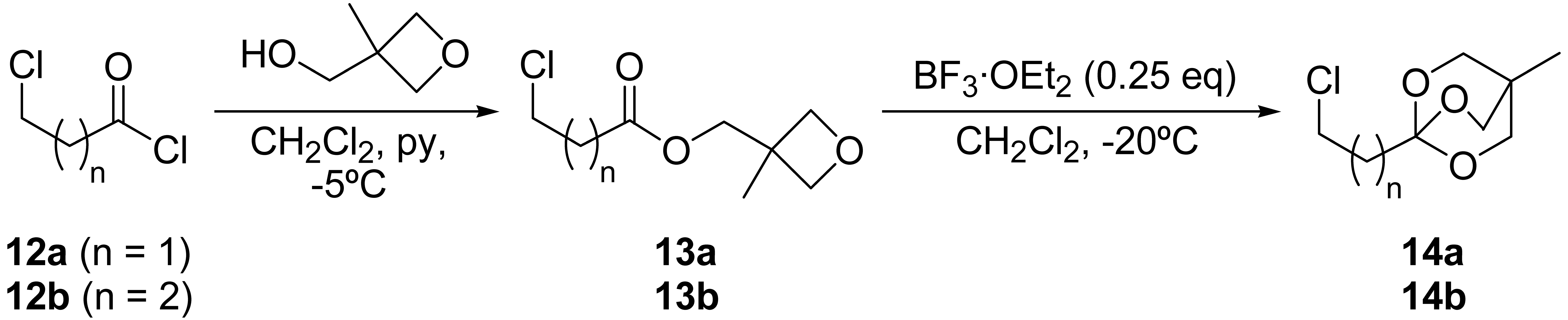
Preparation of ortho esters 14: Preparation of esters 13.
Rearrangement of esters 13: Preparation of ortho esters 14.
DTBB-catalyzed lithiation of ortho esters 14: Preparation of γ-lactones 15.
Preparation of esters 8aa, 8ab and 8ae.
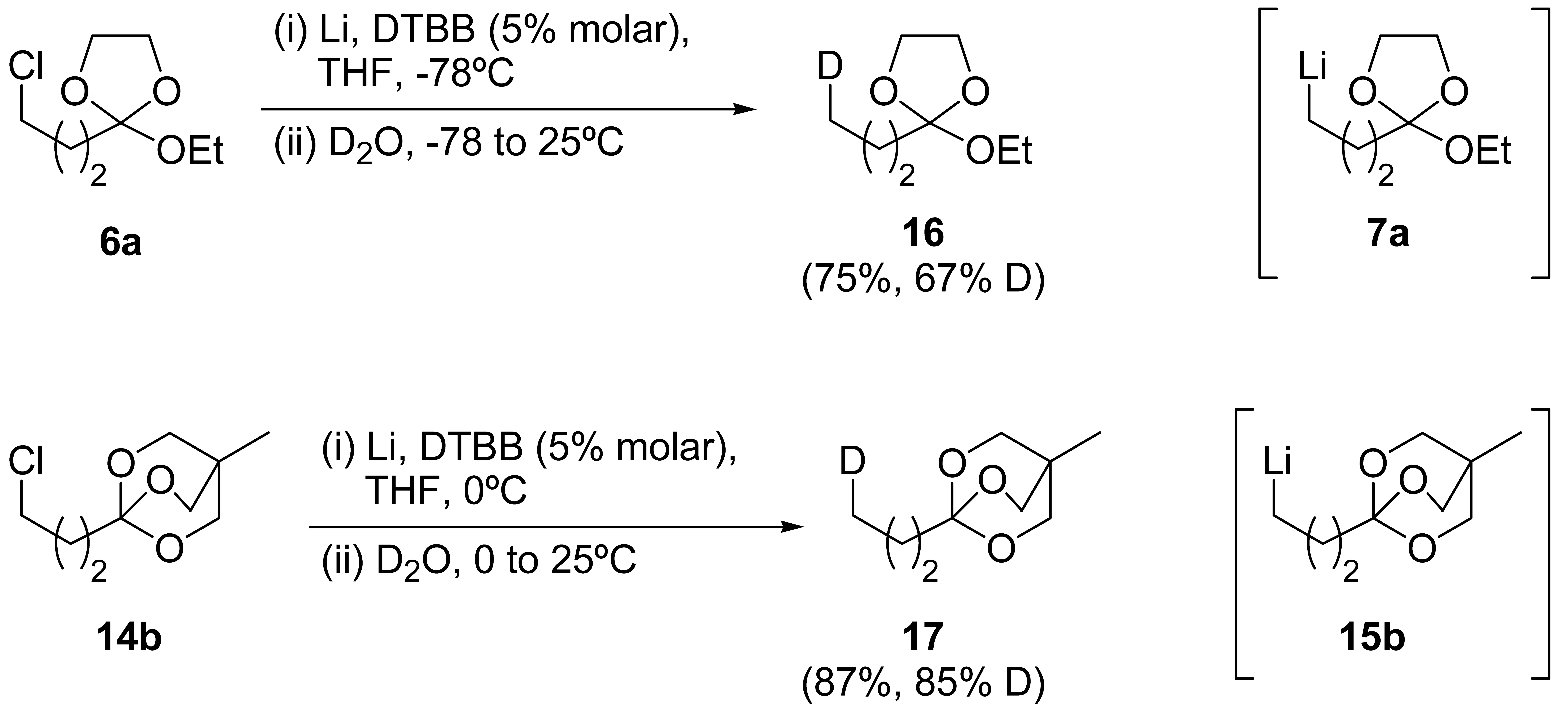
DTBB-catalyzed lithiation of ortho esters 6a and 14b.
Deuterolysis of lithiated intermediates. Preparation of compounds 16 and 17.
References and Notes
- Negishi, E. Organometallics in Organic Chemistry; Wiley: New York, 1980. [Google Scholar] Boudier, A.; Bromm, L. O.; Lotz, M.; Knochel, P. New applications of polyfunctional organometallic compounds in organic synthesis. Angew. Chem. Int. Ed. 2000, 39, 4414–4435. [Google Scholar]
- Review: Petragnani, N.; Yonashiro, M. The reactions of dianions of carboxylic acids and ester enolates. Synthesis 1982, 521–578. [Google Scholar]
- Review: Gil, S.; Parra, M. Dienediolates of carboxylic acids in synthesis. Recent advances. Curr. Org. Chem. 2002, 6, 283–302. [Google Scholar]
- Mekelburger, H. B.; Wilcos, C. S. Formation of enolates. In Comprehensive Organic Synthesis; Trost, B. M., Fleming, I., Eds.; Pergamon Press: Oxford, 1991; Vol. 2, pp. 99–131. [Google Scholar]
- Parra, M.; Sotoca, E.; Gil, S. A convenient generation of acetic acid dianion. Eur. J. Org. Chem. 2003, 1386–1388. [Google Scholar]
- Pastor, I. M.; Yus, M. Lithium α-lithioacetate and β-lithiopropionate: useful intermediates in organic synthesis. Tetrahedron Lett. 2000, 41, 5335–5339. [Google Scholar]
- Kuwajima, I.; Nakamura, E. Metal homoenolates. In Comprehensive Organic Synthesis; Trost, B. M., Fleming, I., Eds.; Pergamon Press: Oxford, 1991; Vol. 2, pp. 441–454. [Google Scholar] Ahlbrecht, H.; Beyer, U. Stereoselective of chiral homoenolate equivalents. Synthesis 1999, 365–390. [Google Scholar] Beak, P.; Gallagher, D. J.; Park, J. S.; Thayumanavan, S. Regioselective, diastereoselective, and enantioselective lithiation-substitution sequences: reactions pathways and synthetic applications. Acc. Chem. Res. 1999, 29, 552–560. [Google Scholar]
- Caine, D.; Frobese, A. S. A synthesis of γ-lactones by reaction of lithium β-lithiopropionate with aldehydes and ketones. Tetrahedron Lett. 1978, 883–886. [Google Scholar]
- Usually a sulfur-containing moiety, see: Thompson, C. M.; Green, D. L. C. Recent advances in dianion chemistry. Tetrahedron 1991, 47, 4223–4285. [Google Scholar] Thompson, C. M.; Frick, J. A. 4-(Phenylsulfonyl)butanoic acid. Preparation, dianion generation, and application to four-carbon chain extension. J. Org. Chem. 1989, 54, 890–896. [Google Scholar] Green, D. L. C.; Kiddle, J. J.; Thompson, C. M. Stereochemistry of remote dianion addition to imines. application to the synthesis of (1S, 8aS)-1-hydroxyindolizidine. Tetrahedron 1995, 51, 2865–2874. [Google Scholar] Thompson, C. M. “Remote” dianion-I. Utility of 4-phenylsulfonylbutanoic acid in the mild conversion of aldehydes and ketones to lactones. Tetrahedron Lett. 1987, 28, 4243–4246. [Google Scholar]
- For reviews see: Nájera, C.; Yus, M. Functionalized organolithium compounds in synthetic organic chemistry. Trends Org. Chem. 1991, 2, 155–181. [Google Scholar] Nájera, C.; Yus, M. Recent developments in the chemistry of functionalized organolithium compounds. Recent Res. Devel. Org. Chem. 1997, 1, 67–96. [Google Scholar] Nájera, C.; Yus, M. Functionalized organolithium compounds: New synthetic adventures. Curr. Org. Chem. 2003, 7, 867–926. [Google Scholar] Nájera, C.; Sansano, J. M.; Yus, M. Recent synthetic uses of functionalised aromatic and heteroaromatic organolithium reagents prepared by non-deprotonating methods. Tetrahedron 2003, 59, 9255–9303. [Google Scholar] Chinchilla, R.; Nájera, C.; Yus, M. Metallated heterocycles and their applications in synthetic organic chemistry. Chem. Rev. 2004, in press (cr020101a). [Google Scholar]
- Yus, M.; Foubelo, F. Reductive opening of saturated oxa-, aza- and thia-cycles by means of an arene-promoted lithiation: synthetic applications. Rev. Heteroatom Chem. 1997, 17, 73–107. [Google Scholar] Yus, M.; Foubelo, F. Reductive opening of heterocycles with lithium metal as a source of functionalised organolithium compounds: synthetic applications. In Targets in Heterocyclic Systems; The Royal Society of Chemistry: Cambridge, U.K., 2002; Vol. 6, pp. 136–171. [Google Scholar]
- Reviews on arene-catalyzed lithiation: Yus, M. Arene-catalyzed lithiation reactions. Chem. Soc. Rev. 1996, 25, 155–161. [Google Scholar] Ramón, D. J.; Yus, M. New methodologies based on arene-catalyzed lithiation reactions and their application to synthetic organic chemistry. Eur. J. Org. Chem. 2000, 225–237. [Google Scholar] Yus, M. From arene-catalyzed lithiation to other synthetic adventures. Synlett 2001, 1197–1205. [Google Scholar] Yus, M. Arene-catalyzed lithiation. In The Chemistry of Organolithium Compounds; Rapopport, Z., Marek, I., Eds.; Wiley: Chichester, 2003; chapter 11. [Google Scholar]
- For polymer supported arene-catalyzed version of this methodology, see: Gómez, C.; Ruiz, S.; Yus, M. Polymer supported arene-catalyzed lithiation reactions. Tetrahedron 1999, 55, 7017–7026. [Google Scholar] Yus, M.; Gómez, C.; Candela, P. Polyphenylene as an electron transfer catalyst in lithiation processes. Tetrahedron 2002, 58, 6207–6210. [Google Scholar] Arnauld, T.; Barrett, A. G. M.; Hopkins, B. T. ROMPgel-supported biphenyl and naphthalene: reagents for lithiation reactions with minimal purification. Tetrahedron Lett. 2002, 43, 1081–1083. [Google Scholar]
- For mechanistic studies, see: Herrera, R. P.; Guijarro, A.; Yus, M. Primary alkyl fluorides as regioselective alkylating reagents of lithium arene Dianions. Easy prediction of regioselectivity by MO calculations on the dianion. Tetrahedron Lett. 2003, 44, 1313–1316. [Google Scholar] Herrera, R. P.; Guijarro, A.; Yus, M. On the dichotomy of the SN2/ET reaction pathways: an apparent SN2 reactivity in the reaction of naphthalene dianion with alkyl fluorides. Tetrahedron Lett. 2003, 44, 1309–1312. [Google Scholar] Yus, M.; Herrera, R. P.; Guijarro, A. On the mechanism of arene-catalyzed lithiation: the role of arene dianions-naphthalene radical anion versus naphthalene dianion. Chem. Eur. J. 2002, 8, 2574–2584. [Google Scholar]
- Gil, J. F.; Ramón, D. J.; Yus, M. Intramolecular 1,6-hydride transfer in acyclic 1,6-diols: a mechanistic study. Tetrahedron 1994, 50, 7307–7314. [Google Scholar] Gil, J. F.; Ramón, D. J.; Yus, M. New masked δ-lithio carbonyl compounds: preparation and synthetic applications. Tetrahedron 1993, 49, 4923–4938. [Google Scholar] Ramón, D. J.; Yus, M. One-step synthesis of substituted 6,8-dioxabicyclo[3.2.1]octanes: easy preparation of racemic rontalin, brevicomins, and related systems. J. Org. Chem. 1992, 57, 750–751. [Google Scholar] Ramón, D. J.; Yus, M. Masked lithium bishomoenolates: useful intermediates in organic synthesis. J. Org. Chem. 1991, 56, 3825–3831. [Google Scholar]
- Preliminary communication: Pastor, I. M.; Yus, M. Masked β-, γ- and δ-lithium ester enolates: useful reagents in organic synthesis. Tetrahedron Lett. 2001, 42, 1029–1032. [Google Scholar]
- (a) For a review, see: DeWolfe, R. H. Synthesis of carboxylic and carbonic ortho esters. Synthesis 1974, 153–172. [Google Scholar] (b) See also: Casy, G.; Patterson, J. W.; Taylor, R. J. K. Methyl 7-hydroxyhept-5-ynoate. Org. Synth. 1988, 67, 193–201. [Google Scholar]
- Blomberg, C. The Barbier Reaction and Related One-Step Processes; Spring-Verlag: Berlin, 1993. [Google Scholar] Alonso, F.; Yus, M. Recent developments in Barbier-type reactions. Recent Res. Dev. Org. Chem. 1997, 1, 397–436. [Google Scholar]
- See, for example: Ramachandran, P. V.; Pitre, S.; Brown, H. C. Selective reduction 59. Effective intramolecular asymmetric reductions of α-, β-, and γ-keto acids with diisopino-campheylborane and intermolecular asymmetric reductions of the corresponding esters with β-chlorodiisopinocampheylborane. J. Org. Chem. 2002, 67, 5315–5319. [Google Scholar] Downham, R.; Edwards, P. J.; Entwistle, D. A.; Hughes, A. B.; Kim, K. S.; Ley, S. V. Tetrahedron: Asymmetr 1995, 6, 2403–2440.
- Greene, T. W.; Wuts, P. G. M. Protective Groups in Organic Synthesis; John Wiley & Sons: New York, 1999; pp. 437–441. [Google Scholar]
- Yus, M.; Martínez, P.; Guijarro, D. DTBB-catalysed dilithiation of styrene and its methyl-derivatives: introduction of two electrophilic reagents. Tetrahedron 2001, 57, 10119–10124. [Google Scholar]
- Borer, B. C.; Taylor, R. J. K. Trimethyl 4-lithioorthobutanoate: preparation and synthetic applications. Synlett 1990, 601–602. [Google Scholar]
- Zahorszhy, U. I. Bifunctional even-electron. III. Fragmentation behavior of aliphatic hydroxonium ions containing an additional carbomethoxy group. Org. Mass Spectrom. 1988, 23, 63–69. [Google Scholar]
- Babcock, B. W.; Dimmel, D. R.; Graves, D. P., Jr.; Mckelvey, R. D. Light-induced free-radical reactions of 2-methoxy-6-methyltetrahydropyran: irreversible ring opening and multisite hydrogen abstraction. J. Org. Chem. 1981, 46, 736–742. [Google Scholar]
- Henri, C.; Plenat, F.; Reliaud, C. Acid-catalyzed oxidation of spirocyclanones using hydrogen peroxide. Bull. Soc. Chim. Fr. 1968, 1566–1571. [Google Scholar]
- Hsu, J. -L.; Chen, C. -T.; Fang, J. -M. Cooperative catalysis of samarium diiodide and mercaptan in a stereoselective one-pot transformation of 5-oxopentanals into δ-lactones. Org. Lett. 1999, 1, 1989–1991. [Google Scholar]
- Baldwin, J. E.; Adlington, R. M.; Robertson, J. Carboxyclic ring expansion reactions via radical chain processes. Part II. Tetrahedron 1991, 47, 6795–6812. [Google Scholar]
- Corey, E. J.; Shimoji, K. Total synthesis of the major human urinary metabolite of prostaglandin D2, a key diagnostic indicator. J. Am. Chem. Soc. 1983, 105, 1662–1664. [Google Scholar]
- Atkins, M. P.; Golding, B. T.; Howes, D. A.; Sellars, P. J. Masking the carboxy group as a 2,6,7-trioxabicyclo[2.2.2]octane: application to the synthesis of alkylcobaloximes containing ester and carboxy groups. J. Chem. Soc., Chem. Commun. 1980, 207–208. [Google Scholar]
- Trost, B. M.; Rhee, Y. H. Ruthenium-catalyzed cycloisomerization-oxidation of homopropargyl alcohols. A new access to γ-butyrolactones. J. Am. Chem. Soc. 1999, 121, 11680–11683. [Google Scholar]
- Machrouhi, F.; Parlea, E.; Namy, J. -L. Barbier-type reactions of cyclic acid anhydrides and keto acids mediated by an SmI2/(NiI2-catalytic) system. Eur. J. Org. Chem. 1998, 2431–2436. [Google Scholar]
- Sample Availability: Not available.
© 2004 by MDPI (http:www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Yus, M.; Torregrosa, R.; Pastor, I.M. Masked ω-Lithio Ester Enolates: Synthetic Applications. Molecules 2004, 9, 330-348. https://doi.org/10.3390/90500330
Yus M, Torregrosa R, Pastor IM. Masked ω-Lithio Ester Enolates: Synthetic Applications. Molecules. 2004; 9(5):330-348. https://doi.org/10.3390/90500330
Chicago/Turabian StyleYus, Miguel, Rosario Torregrosa, and Isidro M. Pastor. 2004. "Masked ω-Lithio Ester Enolates: Synthetic Applications" Molecules 9, no. 5: 330-348. https://doi.org/10.3390/90500330
APA StyleYus, M., Torregrosa, R., & Pastor, I. M. (2004). Masked ω-Lithio Ester Enolates: Synthetic Applications. Molecules, 9(5), 330-348. https://doi.org/10.3390/90500330