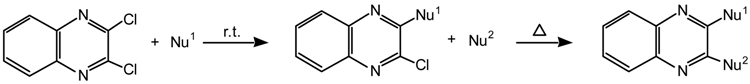
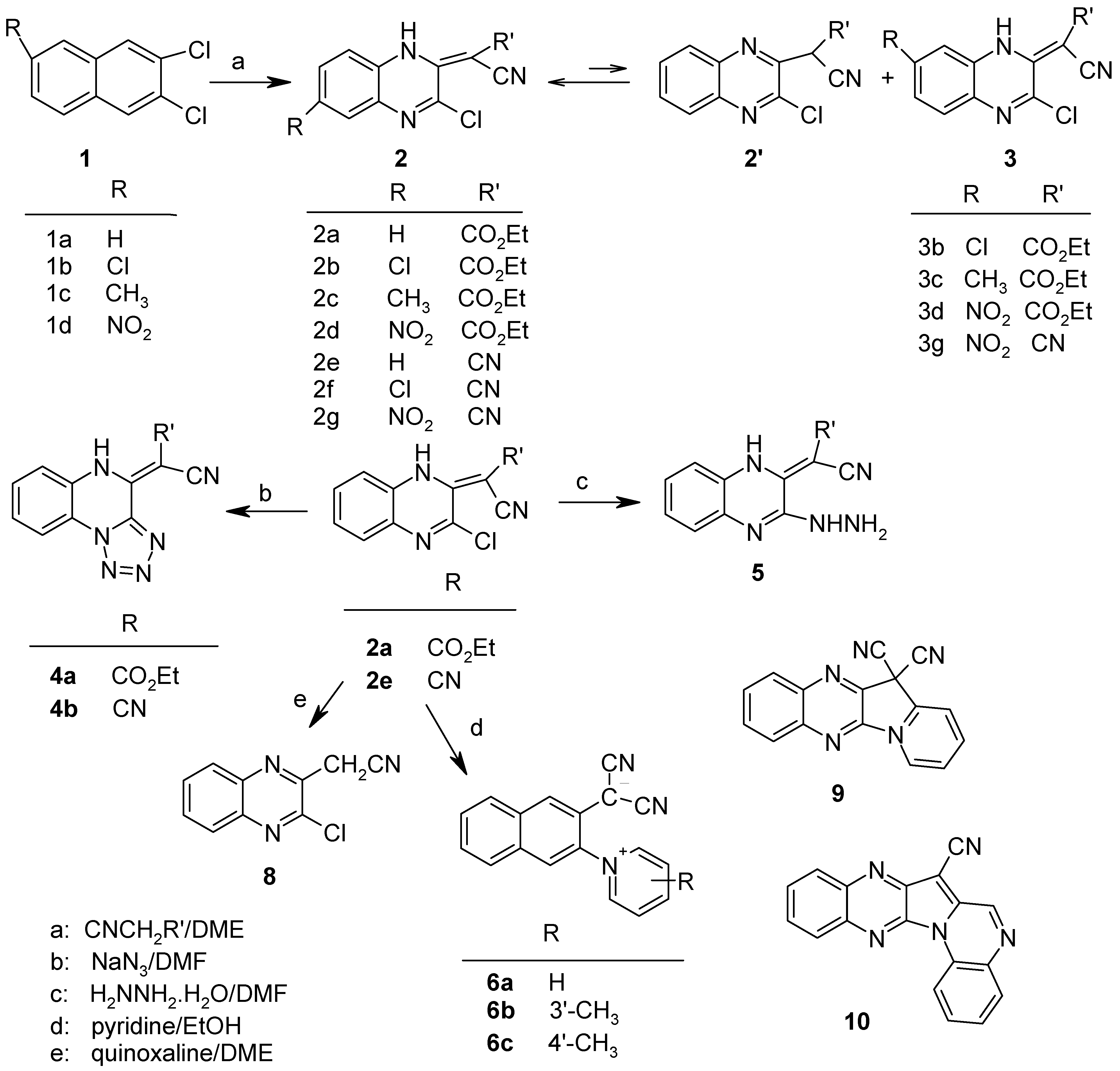
The 3-chloro-2-(cyanomethylene)-1,2-dihydroquinoxalines were prepared by reacting 2,3-dichloro-quinoxaline derivatives 1a-d with the appropriate active methylene compound in the presence of sodium hydride. This is exemplified by the preparation of compound 2a as follows: ethyl cyanoacetate (1.1 mL, 10 mmoles) was added dropwise with stirring to a suspension of sodium hydride (0.25 g, 10.4 mmoles) in dimethoxyethane (20 mL). After the addition, the stirring was continued for 30 min. and then 2,3-dichloroquinoxaline (1.0 g, 5 mmoles) was added. The reaction mixture was stirred at room temperature for 3 h and then heated under reflux for 1 h. The dimethoxyethane was removed on a rotatory evaporator in vacuo and the resulting residue was treated with cold aqueous hydrochloric acid to give a yellow product. This was filtered, washed with cold water, dried and then recrystallized from ethanol to give yellow crystals of 3-chloro-2-(ethoxycarbonylcyanomethylene)-1,2-dihydroquinoxaline (2a, yield: 85%); m.p. 175–177°C (lit. [3] 174–175°C); IR (cm-1) (KBr): 2200 (νCN), 1630 (sh) (νC=O); 1H-NMR: 14.49 (br s, NH, D2O exchangeable), 7.78 (d, IH, ArH), 7.63 (t, IH, ArH), 7.37-7.50 (m, 2H, ArH), 4.37 (q, 2H, CH2), 1.41 (t, 3H, CH3 ); 13C-NMR: 170.7 (C=O), 146.0, 143.6, 134.2, 132.3, 128.6, 126.6, 116.5 (CN), 116.4, 69.3 (=C(CN)2), 62.1 (OCH2), 14.2 (CH3); MS: 275 (19.4, M+), 240 (2.2, [M–Cl]+), 231 (7.3, [M-CO2]+), 212 (10.5, [M-Cl–C2H4]+), 203 (100, [M–CO2–C2H4]+), 167 (62.4, [M–CO2C2H5–Cl]+), 114 (14.2), 102 (53.5), 76 (23.9), 75 (21.0).
Compounds 2b-2g were obtained similarly:
3,6-Dichloro-2-(ethoxycarbonylcyanomethylene)-1,2-dihydroquinoxaline (2b): golden yellow crystals (yield: 82%); m.p. 162-164oC (dec.); IR (cm-1): 2200 (νCN), 1640 (νCO); 1H-NMR: 13.84 (br s, NH, D2O exchangeable), 8.01 (s, 1H, H-5), 7.72 (d, 1H, H-8), 7.46 (d, 1H, H-7), 4.28 (q, 2H, CH2), 1.30 (t, 3H, CH3); Analysis: Calc. For C13H9Cl2N3O2: C, 50.34; H, 2.93; N, 13.55. Found: C, 50.15; H, 2.85; N, 13.39.
3,7-Dichloro-2-(ethoxycarbonylcyanomethylene)-1,2-dihydroquinoxaline (3b): 1H-NMR: 13.85 (br s, NH, D2O exchangeable), 8.10 (s, 1H, H-8), 7.80 (d, 1H, H-5), 7.66 (d, 1H, H-6), 4.28 (q, 2H, CH2), 1.21 (t, 3H, CH3).
3-Chloro-6-methyl-2-(ethoxycarbonylcyanomethylene)-1,2-dihydroquinoxaline (2c): golden yellow crystals (yield: 79%); m.p. 159-161oC (dec.); IR (cm-1): 2190 (ν CN), 1640 (ν C=O); 1H‑NMR: 14.46 (br s, NH, D2O exchangeable), 7.51 (s, 1H, H-5), 7.45 (d, 1H, H-7), 7.31 (d, 1H, H-8), 4.35 (q, 2H, CH2), 2.48 (s, 3H, CH3), 1.40 (t, 3H, CH3); 13C-NMR: 170.8 (C=O), 145.5, 143.2, 137.2, 134.2, 133.8, 127.9, 125.9, 116.5 (CN), 68,3 (=C–(CN)2), 62.5 (OCH2) 21.0 (CH3), 14.0 (CH3); Analysis: Calc. For C14H12ClN3O2: C, 58.04; H, 4.17; N, 14.5. Found: C, 58.00; H, 4.10; N, 14.26.
3-Chloro-7-methyl-2-(ethoxycarbonylcyanomethylene)-1,2-dihydroquinoxaline (3c): 1H-NMR: 14.39 (br s, NH, D2O exchangeable), 7.59 (d, 1H, H-5), 7.26 (d, 1H, H-6), 7.15 (s, 1H, H-8), 4.35 (q,2H, CH2), 2.51 (s, 3H, CH3). 1.35 (t, 3H, CH3); 13C-NMR: 170.7 (C=O), 145.8, 143.8, 133.8, 132.5, 128.3, 128.1, 128.0, 116.0 (CN), 68.6 (=C(CN)2), 61.9 (OCH2), 21.8 (CH3), 14.1 (CH3).
3-chloro-2-(ethoxycarbonylcyanomethylene)-6-nitro-1,2-dihydroquinoxaline (2d): golden yellow crystals (yield: 88%); m.p 191-1993oC (dec.); IR (cm-1): 2210 (υCN), 1650 (υCO); 1H‑NMR: 11.42 (br s, NH, D2O exchangeable), 8.48 (s, 1H, H-5), 8.27 (d, 1H, H-7), 7.48 (d, 1H, H-8), 4.37 (q, 2H, CH2) 1.39 (t, 3H, CH3); Analysis: Calc. for C13H9ClN4O4: C 48.69, H 2.83, N 17.47. Found C, 48.51; H. 2.80; N, 17.28.
3-Chloro-2-(dicyanomethylene)-1,2-dihydroquiinoxaline (2e): golden brown crystals (yield: 78%); m.p. 240-243o C (dec.) (lit. [3] 217o C); IR (cm-1): 2210 (υCN); 1H-NMR: 10.99 (br s, NH, D2O exchangeable), 7.85 (d, 1H, Ar-H), 7.63-7.75 (m, 2H, Ar-H), 7.46 (t, 1H, Ar-H). 13C-NMR: 147.3, 142.3, 134.1, 132.0, 130.8, 127.7, 126.1, 118.0, 116.7 (CN), 47.3 (=C(CN)2); UV (MeOH): 206 (4.18), 222 (4.43), 290 (4.32), 315(sh) (3.95), 435 (4.03); MS:228 (79.8, M+), 192 (20.7, [M–HCl]+), 163 (100, [M–CH (CN)2]+ ), 114 (8.3), 102 (83.8), 76 (37.7), 75 (38.3).
3,6-Dichloro-2-(dicyanomethylene)-1,2-dihydroquinoxaline (2f): brown crystals (yield: 75%); m.p. 190-191o C (dec.); IR (cm-1): 2200 (υCN); 1H-NMR: 10.14 (br s, NH, D2O exchangeable), 8.27 (s, 1H, H-5), 7.63 (d, 1H, H-8), 7.30 (d, 1H, H-7); 13C-NMR: 150.0, 142.3, 135.3, 133.6, 129.1, 125.5, 119.7, 118.5 (CN), 45.6 (=C(CN)2). MS: 264 (54.8, [M+2]+), 262 (79.5, M+), 226 (23.4, [M–HCl]+), 197 (100, [M–CH(CN) 2]+), 162 (12.4, [M–CH(CN)2–Cl]+), 136(52.5), 110 (9.1), 100 (42.4), 75 (28.9). Analysis : Calc. for C11H4Cl2N4: C, 50.22; H, 1.53; N, 21.30. Found: C, 50.41; H, 1.50; N, 21.18.
3-Chloro-2-(dicyanomethylene)-6-nitro-1,2-dihdroquinoxaline (2g): brown crystals (yield: 80%); m.p 255-257oC (dec.); IR (cm-1): 2208 (υCN); 1H-NMR: 8.33(s, 1H, H-5), 8.19 (d, 1H, H-7), 7.50 (d, 1H, H-8); 13C-NMR: 155.5, 145.6, 144.5, 143.4, 142.0, 134.3, 125.8, 123.9, 123.3 (CN?), 44.5 (-C(CN)2); MS: 273 (41.9, M+), 243 (27.6, [M–NO]+), 237 (10.8, [M–HCl]+), 227 (25.2 [M–NO2]+), 215 (24.21), 208 (12.7, [M–CH(CN)2]+), 162 (16.0), 117 (13.0), 101 (30.0), 75 (48.5); Analysis: Calc. for C11H4ClN5O2: C, 48.28; H, 1.47; N, 25.59. Found: C, 48.20; H, 1.44; N, 25.40.

{kind=link}