QSPR Calculation of Normal Boiling Points of Organic Molecules Based on the Use of Correlation Weighting of Atomic Orbitals with Extended Connectivity of Zero- and First-Order Graphs of Atomic Orbitals

Abstract
:Introduction
Molecular Descriptors
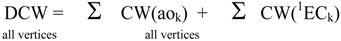

{kind=link}
| 1s1 | -0.246 |
| 1s2 | 0.165 |
| 2s2 | -0.556 |
| 2p2 | 1.780 |
| 2p3 | 3.738 |
| 2p4 | 2.722 |
| 2p5 | -4.591 |
| 2p6 | -0.726 |
| 3s2 | -0.437 |
| 3p2 | 1.760 |
| 3p3 | -2.030 |
| 3p4 | 5.491 |
| 3p5 | 4.532 |
| 3p6 | 0.093 |
| 3d10 | 0.551 |
| 4s2 | 2.873 |
| 4p5 | 0.193 |
| 0003 | 0.626 |
| 0004 | 1.648 |
| 0005 | 0.475 |
| 0006 | 0.175 |
| 0007 | 1.159 |
| 0008 | 0.623 |
| 0009 | 1.758 |
| 0010 | 0.546 |
| 0011 | 1.198 |
| 0012 | 0.463 |
| 0013 | 1.247 |
| 0014 | 3.437 |
| 0015 | 1.877 |
| 0016 | -0.404 |
| 1s1 | 0.939 |
| 1s2 | 0.155 |
| 2s2 | 0.104 |
| 2p2 | 0.704 |
| 2p3 | 4.943 |
| 2p4 | 0.748 |
| 2p5 | -2.191 |
| 2p6 | 0.222 |
| 3s2 | -0.183 |
| 3p2 | 0.827 |
| 3p3 | 4.546 |
| 3p4 | 5.322 |
| 3p5 | 0.939 |
| 3p6 | 8.663 |
| 3d10 | 9.470 |
| 4s2 | 8.444 |
| 4p5 | 8.422 |
| 0012 | 5.903 |
| 0015 | -2.827 |
| 0018 | 0.150 |
| 0020 | 0.376 |
| 0021 | 1.669 |
| 0024 | -0.381 |
| 0027 | 2.112 |
| 0030 | 1.574 |
| 0033 | 2.507 |
| 0035 | 0.685 |
| 0036 | 1.462 |
| 0038 | 1.577 |
| 0039 | 0.219 |
| 0042 | 0.224 |
| 0045 | 0.033 |
| 0048 | 1.204 |
| 0050 | 0.071 |
| 0051 | 1.528 |
| 0053 | 1.086 |
| 0054 | 1.323 |
| 0057 | 1.983 |
| 0059 | 0.574 |
| 0060 | 0.469 |
| 0062 | 0.669 |
| 0063 | -0.236 |
| 0066 | -0.161 |
| 0069 | 0.737 |
| 0070 | -2.190 |
| 0075 | 3.355 |
| 0078 | 3.944 |
| 0079 | 0.582 |
| 0080 | 0.582 |
| 0081 | 2.970 |
| 0082 | 0.904 |
| 0084 | 0.646 |
| 0086 | -0.466 |
| 0087 | -0.007 |
| 0089 | 0.376 |
| 0090 | 2.254 |
| 0091 | 4.903 |
| 0094 | -0.955 |
| 0096 | 2.028 |
| 0097 | -1.506 |
| 0098 | 4.564 |
| 0099 | 1.506 |
| 0100 | 5.589 |
| 0101 | 3.285 |
| 0102 | -5.967 |
| 0103 | 1.738 |
| 0105 | 1.969 |
| 0108 | 0.273 |
| 0109 | 4.121 |
| 0110 | 2.223 |
| 0111 | 2.796 |
| 0112 | 1.653 |
| 0116 | 4.641 |
| 0120 | -2.254 |
| 0122 | 0.616 |
| 0124 | 1.832 |
| 0134 | 1.828 |
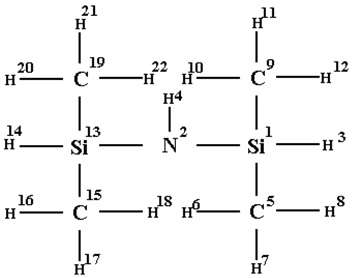
| atom | Nat | EC1 | ao | Nao | EC1 | CW(V) | CW(LI) |
|---|---|---|---|---|---|---|---|
| Si | 1 | 12 | 1s2 | 1 | 86 | 0.155 | -0.466 |
| 2s2 | 2 | 86 | 0.104 | -0.466 | |||
| 2p6 | 3 | 86 | 0.222 | -0.466 | |||
| 3s2 | 4 | 86 | -0.183 | -0.466 | |||
| 3p2 | 5 | 86 | 0.827 | -0.466 | |||
| N | 2 | 9 | 1s2 | 6 | 103 | 0.155 | 1.738 |
| 2s2 | 7 | 103 | 0.104 | 1.738 | |||
| 2p3 | 8 | 103 | 4.943 | 1.738 | |||
| H | 3 | 4 | 1s1 | 9 | 50 | 0.939 | 0.071 |
| H | 4 | 3 | 1s1 | 10 | 33 | 0.939 | 2.507 |
| C | 5 | 7 | 1s2 | 11 | 59 | 0.155 | 0.574 |
| 2s2 | 12 | 59 | 0.104 | 0.574 | |||
| 2p2 | 13 | 59 | 0.704 | 0.574 | |||
| H | 6 | 4 | 1s1 | 14 | 24 | 0.939 | -0.381 |
| H | 7 | 4 | 1s1 | 15 | 24 | 0.939 | -0.381 |
| H | 8 | 4 | 1s1 | 16 | 24 | 0.939 | -0.381 |
| C | 9 | 7 | 1s2 | 17 | 59 | 0.155 | 0.574 |
| 2s2 | 18 | 59 | 0.104 | 0.574 | |||
| 2p2 | 19 | 59 | 0.704 | 0.574 | |||
| H | 10 | 4 | 1s1 | 20 | 24 | 0.939 | -0.381 |
| H | 11 | 4 | 1s1 | 21 | 24 | 0.939 | -0.381 |
| H | 12 | 4 | 1s1 | 22 | 24 | 0.939 | -0.381 |
| Si | 13 | 12 | 1s2 | 23 | 86 | 0.155 | -0.466 |
| 2s2 | 24 | 86 | 0.104 | -0.466 | |||
| 2p6 | 25 | 86 | 0.222 | -0.466 | |||
| 3s2 | 26 | 86 | -0.183 | -0.466 | |||
| 3p2 | 27 | 86 | 0.827 | -0.466 | |||
| H | 14 | 4 | 1s1 | 28 | 50 | 0.939 | 0.071 |
| C | 15 | 7 | 1s2 | 29 | 59 | 0.155 | 0.574 |
| 2s2 | 30 | 59 | 0.104 | 0.574 | |||
| 2p2 | 31 | 59 | 0.704 | 0.574 | |||
| H | 16 | 4 | 1s1 | 32 | 24 | 0.939 | -0.381 |
| H | 17 | 4 | 1s1 | 33 | 24 | 0.939 | -0.381 |
| H | 18 | 4 | 1s1 | 34 | 24 | 0.939 | -0.381 |
| C | 19 | 7 | 1s2 | 35 | 59 | 0.155 | 0.574 |
| 2s2 | 36 | 59 | 0.104 | 0.574 | |||
| 2p2 | 37 | 59 | 0.704 | 0.574 | |||
| H | 20 | 4 | 1s1 | 38 | 24 | 0.939 | -0.381 |
| H | 21 | 4 | 1s1 | 39 | 24 | 0.939 | -0.381 |
| H | 22 | 4 | 1s1 | 40 | 24 | 0.939 | -0.381 |
Results and Discussion
| n | CAS | Molecule | DCW0 | DCW1 | NBPexp |
|---|---|---|---|---|---|
| 1 | 15933-59-2 | 1,1,3,3-Tetramethyldisilalazane | 5.460 | 8.398 | 99.000 |
| 2 | 105-54-4 | Butanoic acid, ethyl ester | 5.543 | 11.949 | 120.000 |
| 3 | 623-27-8 | 1,4-Benzenedicarboxaldehyde | 16.537 | 18.618 | 245.000 |
| 4 | 7212-44-4 | 1,6,10-Dodecatrien-3-ol, 3,7,11-trimethyl | 14.389 | 17.084 | 68.000 |
| 5 | 705-86-2 | 5-Hydroxydecanoic acid lactone | 8.596 | 13.405 | 117.000 |
| 6 | 620-22-4 | Benzonitrile, 3-methyl- | 12.826 | 16.075 | 99.000 |
| 7 | 621-33-0 | 3-Ethoxyaniline | 12.735 | 25.122 | 248.000 |
| 8 | 150-76-5 | Mequinol | 14.584 | 23.498 | 243.000 |
| 9 | 109-52-4 | Pentanoic acid | 9.042 | 14.320 | 185.000 |
| 10 | 75-55-8 | Aziridine, 2-methyl- | 1.529 | 9.325 | 66.000 |
| 11 | 586-39-0 | 3-Nitrosostyrene | 21.317 | 14.385 | 56.000 |
| 12 | 224-41-9 | Dibenz[a,j]anthracene | 39.917 | 53.421 | 531.000 |
| 13 | 105-05-5 | Benzene, 1,4-diethyl- | 11.384 | 19.030 | 184.000 |
| 14 | 110-42-9 | Decanoic acid, methyl ester | 8.330 | 13.914 | 108.000 |
| 15 | 111-69-3 | Hexanedinitrile | 6.906 | 8.394 | 295.000 |
| 16 | 1112-55-6 | Silane , tetraethyl- | 10.654 | 11.853 | 130.000 |
| 17 | 1719-57-9 | Silane, chloro(chloromethyl)dimethyl- | 5.129 | 10.177 | 115.000 |
| 18 | 123-31-9 | Hydroquinoline | 18.082 | 28.959 | 285.000 |
| 19 | 100-02-7 | Phenol, 4-nitro- | 23.197 | 25.692 | 279.000 |
| 20 | 2548-87-0 | 2-Octenal, (E)- | 7.837 | 15.141 | 84.000 |
| 21 | 6166-86-5 | 2,4,6,8,10-Pentamethylcyclopentasiloxane | 10.191 | 15.986 | 168.000 |
| 22 | 2031-79-0 | 1,1,3,3,5,5-Hexaethylcyclotrisiloxane | 5.493 | 10.852 | 117.000 |
| 23 | 3862-73-5 | Trifluoroaniline | 4.605 | 2.796 | 92.000 |
| 24 | 15980-15-1 | 1,4-Oxathiane | 3.112 | 11.780 | 147.000 |
| 25 | 108-41-8 | Benzene, 1-chloro-3-methyl- | 11.575 | 16.747 | 160.000 |
| 26 | 78-81-9 | 1-Propanamine, 2-methyl | 2.006 | 6.152 | 64.000 |
| 27 | 7087-68-5 | Diisopropylethylamine | 6.556 | 12.133 | 127.000 |
| 28 | 17477-29-1 | Propyldimethylchlorosilane | 3.718 | 9.954 | 113.000 |
| 29 | 75-35-4 | Ethylene, 1,1-dichloro- | 5.812 | -4.704 | 30.000 |
| 30 | 91-64-5 | Coumarin | 17.928 | 20.526 | 298.000 |
| 31 | 328-87-0 | 4-Chloro-3-cyanobenzotrifluoride | 6.405 | 11.573 | 210.000 |
| 32 | 616-25-1 | 1-Penten-3-ol | 6.438 | 12.844 | 114.000 |
| 33 | 75-85-4 | 2-Butanol, 2-methyl- | 4.231 | 9.165 | 102.000 |
| 34 | 138-86-3 | Limonene | 10.367 | 10.009 | 170.000 |
| 35 | 333-41-5 | Diazinon | 4.486 | 6.708 | 83.000 |
| 36 | 15570-12-4 | meta-Methoxybenzenethiol | 16.490 | 22.256 | 223.000 |
| 37 | 198-55-0 | Perylene | 37.005 | 50.768 | 495.000 |
| 38 | 192-97-2 | Benzo[e]pyrene | 37.005 | 50.768 | 492.000 |
| 39 | 205-99-2 | Benzo[b]fluoranthene | 37.005 | 52.659 | 481.000 |
| 40 | 218-01-9 | Chrysene | 32.121 | 45.048 | 448.000 |
| 41 | 217-59-4 | Triphenylene | 32.121 | 47.211 | 425.000 |
| 42 | 611-32-5 | Quinoline, 8-methyl- | 16.427 | 22.665 | 143.000 |
| 43 | 76783-59-0 | Ethyl-3-trifluoromethylbenzoate | 6.641 | 15.738 | 101.000 |
| 44 | 76-86-8 | Triphenylchlorosilane | 28.953 | 39.379 | 378.000 |
| 45 | 1241-94-7 | Phosphoric acid, 2-ethylhexyldiphenylester | 26.200 | 33.929 | 375.000 |
| 46 | 2943-75-1 | N-octyltriethoxysilane | 9.043 | 15.458 | 98.000 |
| 47 | 594-72-9 | Ethane,1,1-dichloro-1-nitro- | 11.751 | 11.958 | 124.000 |
| 48 | 62-73-7 | Dimethyl-2,2-dichlorovinyl phosphate | 10.283 | 20.471 | 140.000 |
| 49 | 123-15-9 | Pentanol, 2-methyl- | 4.196 | 8.437 | 119.000 |
| 50 | 6640-27-3 | Phenol, 2-chloro-4- methyl- | 16.248 | 19.546 | 195.000 |
| 51 | 537-92-8 | N-(3-tolyl)acetic acid amide | 17.896 | 30.207 | 303.000 |
| 52 | 105-99-7 | Hexanedioic acid, dibutyl ester | 13.754 | 22.346 | 305.000 |
| 53 | 77-35-2 | Phenanthrene, 9,10-dihydro- | 22.529 | 27.241 | 168.000 |
| 54 | 2713-33-9 | 3,4-Difluorophenol | 9.143 | 13.615 | 85.000 |
| 55 | 111-83-1 | Octane, 1-bromo- | 5.899 | 14.924 | 201.000 |
| 56 | 101-68-8 | Benzene, 1,1'-methylene bis(4-isocyanato)- | 26.548 | 29.352 | 200.000 |
| 57 | 597-49-9 | 3-Ethyl-3-pentanol | 5.346 | 14.104 | 141.000 |
| 58 | 18395-90-9 | di-tert-Butyldichlorosilane | 10.980 | 18.381 | 190.000 |
| 59 | 107-12-0 | Propanenitrile | 2.677 | 5.531 | 97.000 |
| 60 | 1825-62-3 | Silane, ethoxytrimethyl | 3.096 | 5.122 | 75.000 |
| 61 | 56-55-3 | Benz[a]anthracene | 32.121 | 40.882 | 438.000 |
| 62 | 243-17-4 | 2,3-Benzofluorene | 30.666 | 38.282 | 402.000 |
| 63 | 57-11-4 | Octadecanoic acid | 16.287 | 21.581 | 183.000 |
| 64 | 98-03-3 | Thiophenecarboxaldehyde | 10.468 | 15.608 | 198.000 |
| 65 | 605-39-0 | 2,2'-Dimethylbiphenyl | 20.976 | 28.363 | 258.000 |
| 66 | 831-91-4 | Benzene, [(phenylmethyl)thio] | 19.804 | 23.867 | 197.000 |
| 67 | 761-65-9 | Formamide, N,N-dibutyl- | 11.705 | 17.359 | 120.000 |
| 68 | 348-54-9 | Benzeneamine, 2-fluoro- | 8.870 | 14.610 | 182.000 |
| 69 | 136-77-6 | Hexylresorcinol | 21.636 | 31.606 | 333.000 |
| 70 | 100-53-8 | Benzenemethanethiol | 16.543 | 18.999 | 194.000 |
| 71 | 191-30-0 | 1,2,9,10-Dibenzopyrene | 44.801 | 59.414 | 595.000 |
| 72 | 109-73-9 | 1-Butanamine | 3.292 | 6.593 | 78.000 |
| 73 | 100-69-6 | Pyridine, 2-ethenyl- | 10.659 | 13.058 | 79.000 |
| 74 | 1712-70-5 | 1-Chloro-4-isopropenylbenzene | 14.368 | 18.139 | 214.500 |
| 75 | 95-56-7 | Phenol, 2-bromo- | 18.076 | 25.834 | 195.000 |
| 76 | 2984-50-1 | Oxirane, hexyl- | 4.137 | 8.771 | 63.000 |
| 77 | 100-43-6 | Pyridine, 4-ethenyl- | 10.659 | 8.905 | 62.000 |
| 78 | 919-31-3 | Propanenitrile, 3-(triethoxysilyl)- | 8.813 | 13.961 | 224.000 |
| 79 | 874-60-2 | 4-Methylbenzoic acid chloride | 15.476 | 24.765 | 225.000 |
| 80 | 80-62-6 | 2-Propenoic acid, 2-methyl-, methyl ester | 6.665 | 5.278 | 100.000 |
| 81 | 645-49-8 | (Z)-Stilbene | 22.354 | 27.371 | 307.000 |
| 82 | 103-84-4 | Acetamide, N-phenyl- | 17.128 | 27.646 | 304.000 |
| 83 | 106-49-0 | para-Toluidine | 11.770 | 24.079 | 200.000 |
| 84 | 90-90-4 | Methanone, (4-bromophenyl)phenyl- | 28.011 | 30.846 | 350.000 |
| 85 | 519-73-3 | Triphenylmethane | 29.768 | 34.483 | 359.000 |
| 86 | 832-69-9 | Phenanthrene, 1-methyl- | 25.093 | 35.107 | 359.000 |
| 87 | 60-29-7 | Ethoxyethane | 1.085 | 5.886 | 35.000 |
| 88 | 539-74-2 | Propanoic acid, 3-bromo-ethyl ester | 7.978 | 16.094 | 135.000 |
| 89 | 598-31-2 | 2-Propanone, 1-bromo- | 6.456 | 13.853 | 137.000 |
| 90 | 571-61-9 | Naphthalene, 1,5-dimethyl- | 18.065 | 25.165 | 265.000 |
| 91 | 1885-14-9 | Carbonochloridic acid, phenyl ester | 15.117 | 17.640 | 74.000 |
| 92 | 754-05-2 | Silane, ethenyltrimethyl- | 3.945 | 3.490 | 55.000 |
| 93 | 238-84-6 | 1,2-Benzofluorene | 30.666 | 42.448 | 407.000 |
| 94 | 99-08-1 | Benzene, 1-methyl-3-nitro- | 11.770 | 23.077 | 230.000 |
| 95 | 7209-38-3 | 1,4-bis(3-Aminopropyl)piperazine | 19.905 | 11.808 | 150.000 |
| 96 | 1558-33-4 | Silane, dichloro(chloromethyl)methyl- | 5.349 | 10.947 | 121.000 |
| 97 | 65-85-0 | Benzoic acid | 17.310 | 21.998 | 249.000 |
| 98 | 132-64-9 | Dibenzofuran | 21.822 | 26.034 | 154.000 |
| 99 | 213-46-7 | Picene (benzo[a]chrysene) | 39.917 | 57.587 | 525.000 |
| 100 | 191-07-1 | Coronene | 46.773 | 57.883 | 525.000 |
| 101 | 287-92-3 | Cyclopentane | 2.787 | 2.793 | 50.000 |
| 102 | 2782-91-4 | Thiourea, tetramethyl- | 15.021 | 29.789 | 245.000 |
| 103 | 109-07-9 | Piperazine, 2-methyl- | 5.612 | 11.565 | 155.000 |
| 104 | 7005-72-3 | Benzene, 1-chloro-4-phenoxy- | 21.923 | 31.152 | 284.000 |
| 105 | 532-27-4 | Ethanone, 2-chloro-1-phenyl- | 15.937 | 19.618 | 244.000 |
| 106 | 91-57-6 | Naphthalene, 2-methyl- | 17.298 | 22.531 | 241.000 |
| 107 | 109-01-3 | Piperazine, 1-methyl- | 5.461 | 12.701 | 138.000 |
| 108 | 591-35-5 | Phenol, 3,5-dichloro- | 17.553 | 23.380 | 233.000 |
| 109 | 454-89-7 | Benzaldehyde, 3-(trifluoromethyl)- | 4.909 | 10.983 | 83.000 |
| 110 | 99-04-7 | Benzoic acid, 3-methyl- | 18.077 | 24.559 | 263.000 |
| 111 | 120-72-9 | Indole | 14.205 | 20.915 | 253.000 |
| 112 | 109-86-4 | Ethanol, 2-methoxy- | 4.991 | 10.296 | 125.000 |
| 113 | 617-84-5 | N,N-Diethylformamide | 9.476 | 14.478 | 176.000 |
| 114 | 129-00-0 | Pyrene | 29.210 | 36.066 | 360.000 |
| 115 | 86-74-8 | Carbazole | 22.000 | 31.584 | 355.000 |
| 116 | 79-06-1 | Acrylamide | 6.990 | 8.215 | 125.000 |
| 117 | 589-18-4 | Benzene methanol, 4-methyl- | 14.733 | 21.268 | 217.000 |
| 118 | 123-07-9 | Phenol, 4-ethyl- | 14.733 | 23.994 | 218.000 |
| 119 | 75-78-5 | Silane, dichlorodimethyl- | 3.553 | 5.152 | 70.000 |
| 120 | 120-80-9 | 1,2-Benzenediol | 18.082 | 24.434 | 245.000 |
| 121 | 123-92-2 | 1-Butanol, 3-methyl-, acetate | 5.371 | 8.225 | 142.000 |
| 122 | 626-39-1 | Benzene, 1,3,5-tribromo- | 22.739 | 27.936 | 271.000 |
| 123 | 89-99-6 | Benzenemethanamine, 2-fluoro- | 9.428 | 10.347 | 73.000 |
| 124 | 366-18-7 | 2,2'-Dipyridine | 17.702 | 28.255 | 273.000 |
| 125 | 75-05-8 | Acetonitrile | 2.119 | 5.271 | 81.000 |
| 126 | 77-81-6 | Tabun | 7.771 | 24.781 | 246.000 |
| 127 | 7691-02-3 | CH2CHOS(CH3)(CH3)NS(CH3)(CH3)CHCH2 | 12.366 | 14.725 | 160.000 |
| 128 | 615-67-8 | 1,4-Benzenediol, 2-chloro- | 20.155 | 25.666 | 263.000 |
| 129 | 591-93-5 | 1,4-Pentadiene | 4.173 | -2.541 | 26.000 |
| 130 | 350-46-9 | Benzene, 1-fluoro-4-nitro- | 16.391 | 14.681 | 205.000 |
| 131 | 108-90-7 | Benzene, chloro- | 10.807 | 16.761 | 132.000 |
| 132 | 95-78-3 | Benzenamine, 2,5-dimethyl- | 12.537 | 21.017 | 218.000 |
| 133 | 557-11-9 | Urea, allyl- | 8.105 | 11.476 | 163.000 |
| 134 | 557-17-5 | Methyl propyl ether | 1.085 | 6.407 | 39.000 |
| 135 | 110-06-5 | di-tert-Butyldisulfide | 13.140 | 19.686 | 200.000 |
| 136 | 594-70-7 | Propane, 2-methyl-2-nitro- | 8.789 | 15.359 | 127.000 |
| 137 | 5582-62-7 | (Propargyloxy)trimethylsilane | 5.693 | 10.843 | 110.000 |
| 138 | 1072-43-1 | Thiirane, methyl- | 1.410 | 6.658 | 72.000 |
| 139 | 124-07-2 | Octanoic acid | 10.714 | 15.996 | 237.000 |
| 140 | 919-30-2 | 1-Propanamine, 3-(triethoxysilyl)- | 8.314 | 12.139 | 122.000 |
| 141 | 623-00-7 | 4-Bromobenzoic acid nitrile | 16.727 | 19.293 | 235.000 |
| 142 | 100-44-7 | Benzyl chloride | 12.036 | 16.857 | 177.000 |
| 143 | 109-55-7 | 1,3-Propanediamine, N,N-dimethyl- | 8.400 | 10.048 | 133.000 |
| 144 | 598-72-1 | 2-Bromopropanoic acid | 11.173 | 11.912 | 203.000 |
| 145 | 822-86-6 | Cyclohexane, 1,2-dichloro-(trans) | 6.936 | 13.335 | 193.000 |
| 146 | 67-71-0 | Dimethylsulfone | 4.844 | 22.383 | 238.000 |
| 147 | 56-33-7 | 1,1,3,3-Tetramethyl-1,3-diphenyldisiloxane | 20.964 | 14.576 | 155.000 |
| 148 | 112-57-2 | Tetraethylenepentamine | 18.198 | 37.473 | 340.000 |
| 149 | 4333-56-6 | Cyclopropyl bromide | 4.918 | 9.457 | 69.000 |
| 150 | 80-10-4 | Diphenyldichlorosilane | 20.633 | 31.348 | 305.000 |
| 151 | 96-23-1 | 2-Propanol, 1,3-dichloro- | 8.921 | 25.525 | 174.000 |
| 152 | 110-89-4 | Piperidine | 3.373 | 8.827 | 106.000 |
| 153 | 95-77-2 | Phenol, 3,4-dichloro- | 17.553 | 23.879 | 145.000 |
| 154 | 123-54-6 | Acetylacetone | 7.922 | 10.594 | 140.000 |
| 155 | 91-01-0 | Benzenemethanol, α-phenyl- | 23.734 | 31.844 | 297.000 |
| 156 | 115-19-5 | 3-Butyn-2-ol, 2-methyl- | 6.271 | 14.827 | 104.000 |
| 157 | 78-84-2 | Propanal, 2-methyl- | 3.082 | 6.660 | 63.000 |
| 158 | 104-54-1 | 2-Propen-1-ol, 3-phenyl- | 16.878 | 23.577 | 250.000 |
| 159 | 420-56-4 | Silane, fluorothiomethyl- | -1.061 | -2.005 | 57.000 |
| 160 | 98-02-2 | 2-Furanmethanethiol | 14.039 | 16.547 | 155.000 |
| 161 | 3970-62-5 | 3-Pentanol, 2,2-dimethyl- | 4.617 | 18.157 | 132.000 |
| 162 | 92-84-2 | Phenothiazine | 21.133 | 32.840 | 371.000 |
| 163 | 93-99-2 | Benzoic acid, phenyl ester | 23.751 | 27.643 | 298.000 |
| 164 | 109-67-1 | 1-Pentene | 2.703 | 3.244 | 30.000 |
| 165 | 451-40-1 | Ethanone, 1,2-diphenyl- | 23.901 | 24.618 | 320.000 |
| 166 | 625-30-9 | 2-Pentanamine | 2.563 | 7.876 | 91.000 |
| 167 | 2051-60-7 | 1,1'-Biphenyl, 2-chloro- | 21.515 | 27.031 | 274.000 |
| 168 | 2425-79-8 | Oxirane,2,2'[1,4-butanediylbis(oximethylene)]bis- | 7.299 | 13.704 | 155.000 |
| 169 | 623-73-4 | Ethyldiazoacetate | 8.784 | 18.857 | 140.000 |
| 170 | 103-11-7 | 2-Propenoic acid, 2-ethylhexyl ester | 9.070 | 15.387 | 215.000 |
| 171 | 107-05-1 | 1-Propene, 3-chloro- | 4.122 | 4.570 | 44.000 |
| 172 | 108-31-6 | 2,5-Furandione | 11.122 | 13.982 | 200.000 |
| 173 | 57-06-7 | Allylisothiocyanate | 6.281 | 4.360 | 150.000 |
| 174 | 77-75-8 | Meparfynol (1-pentyne-3-ol, 3-methyl) | 6.828 | 17.020 | 121.000 |
| 175 | 229-87-8 | Phenanthridine | 23.456 | 34.557 | 349.000 |
| 176 | 5510-99-6 | Phenol, 2,6-bis(1-methylpropyl)- | 16.829 | 33.099 | 255.000 |
| 177 | 3544-25-0 | 4-Aminophenylacetic acid nitrile | 14.885 | 25.592 | 312.000 |
| 178 | 501-65-5 | Diphenylethylene | 19.928 | 22.715 | 170.000 |
| 179 | 994-49-0 | Hexaethyldisiloxane | 2.852 | 17.928 | 129.000 |
| 180 | 189-64-0 | Dibenzo[a,h]pyrene | 44.801 | 59.141 | 596.000 |
| 181 | 127-19-5 | Acetamide, N,N-dimethyl- | 9.128 | 7.664 | 165.000 |
| 182 | 14548-46-0 | Phenyl, 4-pyridyl ketone | 22.473 | 25.002 | 315.000 |
| 183 | 1897-45-6 | Tetrachloroisophthalonitrile | 23.673 | 28.057 | 350.000 |
| 184 | 135-01-3 | Benzene, 1,2-diethyl- | 11.384 | 16.301 | 183.000 |
| 185 | 109-77-3 | Malononitrile | 5.234 | 5.889 | 220.000 |
| 186 | 1008-88-4 | Pyridine, 3-phenyl- | 18.572 | 22.606 | 269.000 |
| 187 | 3741-00-2 | Cyclopentane, pentyl- | 4.844 | 9.432 | 181.000 |
| 188 | 109-92-2 | Ethene, ethoxy- | 2.554 | 2.070 | 33.000 |
| 189 | 636-30-6 | Benzenamine, 2,4,5-trichloro- | 17.220 | 22.555 | 270.000 |
| 190 | 2916-68-9 | Trimethyl-2-hydroxyethylsilane | 6.001 | 5.437 | 90.000 |
| 191 | 126-73-8 | Tri-n-butylphosphate | 8.164 | 15.583 | 180.000 |
| 192 | 69-72-7 | Benzoic acid, 2-hydroxy- | 21.983 | 30.500 | 211.000 |
| 193 | 771-51-7 | 1H-indole-3-acetonitrile | 21.226 | 29.641 | 157.000 |
| 194 | 624-83-9 | Methane, isocyanato- | 2.312 | 13.655 | 37.000 |
| 195 | 191-24-2 | Benzo[ghi]perylene | 41.889 | 54.326 | 542.000 |
| 196 | 107-02-8 | 2-Propenal | 3.253 | 6.809 | 53.000 |
| 197 | 622-97-9 | Benzene, 1-ethenyl-4-methyl- | 12.296 | 12.533 | 175.000 |
| 198 | 762-49-2 | Ethane, 1-bromo-2-fluoro- | 0.212 | 10.710 | 71.000 |
| 199 | 5263-87-6 | Quinoline, 6-methoxy- | 16.835 | 25.734 | 193.000 |
| 200 | 108-01-0 | Ethanol, 2-(dimethylamino)- | 10.248 | 14.465 | 133.000 |
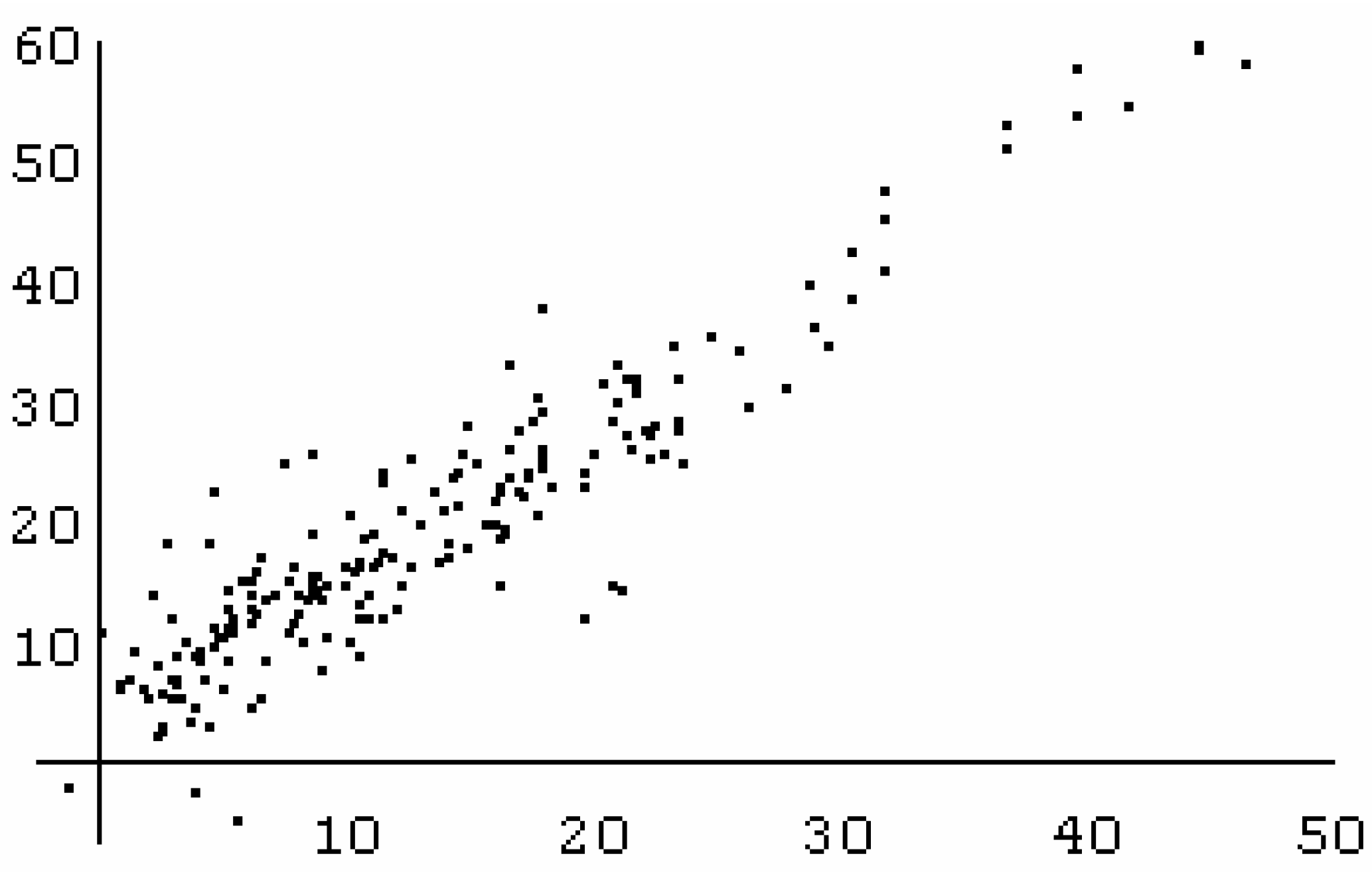
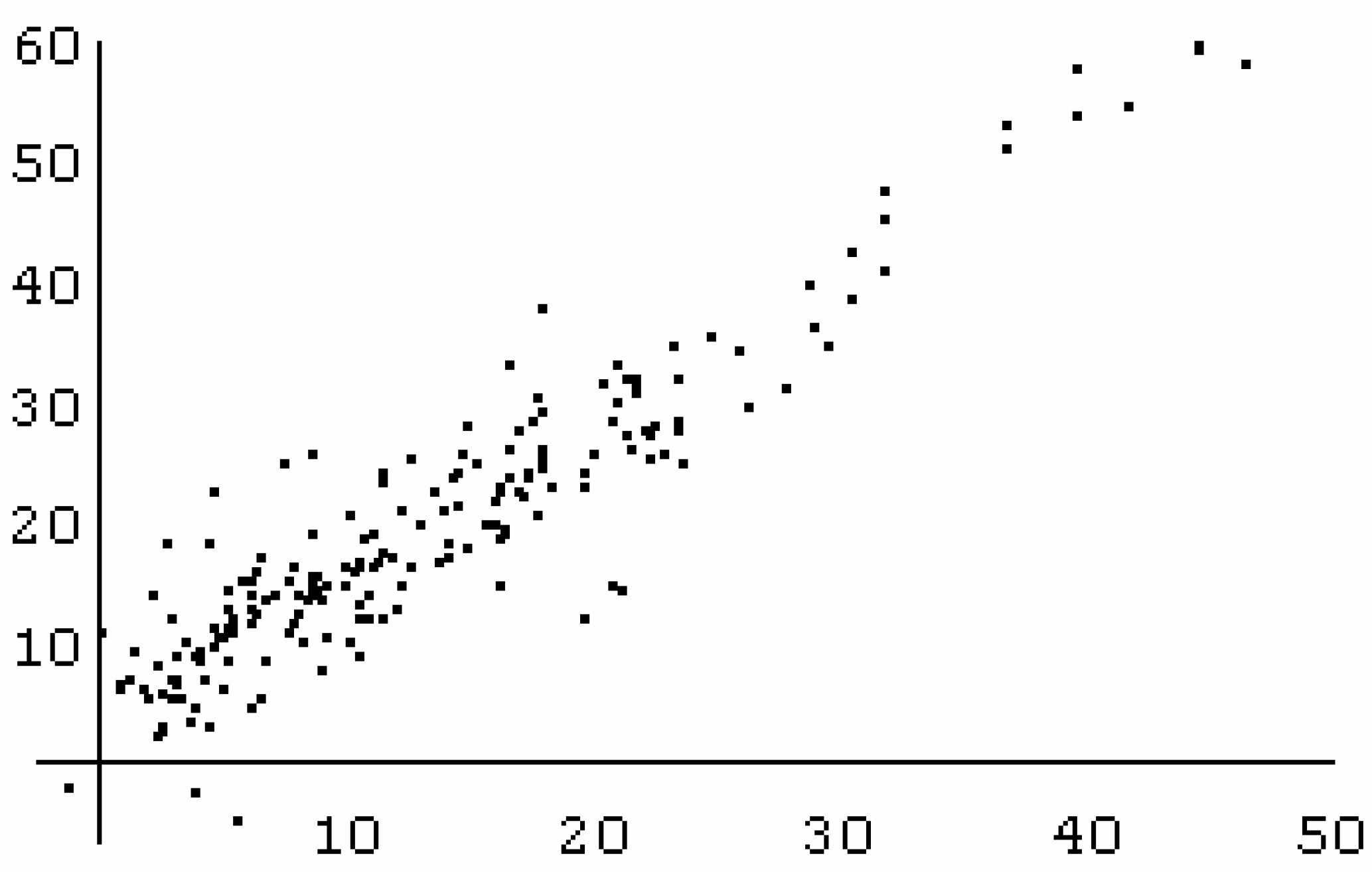
n = 200, r = 0.8910, S = 53.7, F = 763
n = 200, r = 0.892, S = 56.0, F = 783
n = 150, r = 0.8841, S = 55.1, F = 530 (training set)
n = 50, r = 0.9120, S = 49.3, F = 237 (test set)
n = 150, r = 0.9328, S = 42.5, F = 530 (training set)
n = 50, r = 0.8766, S = 57.6, F = 237 (test set)
n = 145, r = 0.9199, S = 46.8, F = 787 (training set)
n = 50, r = 0.9120, S = 46.6, F = 237 (test set)
n = 145, r = 0.9530, S = 36.1, F = 1414 (training set)
n = 50, r = 0.8765, S = 53.9, F = 159 (test set)
n = 158, r = 0.9586, S = 34.8, F = 1770 (complete set)
n = 126, r = 0.9633, S = 33.3, F = 1599 (training set)
n = 32, r = 0.9391, S = 39.1, F = 224 (test set)
n = 144, r = 0.9592, S = 33.9, F = 1633 (training set)
n = 37, r = 0.9564, S = 34.8, F = 376 (test set)
Conclusions
References
- King R., B. (Ed.) Chemical Applications of Topology and Graph Theory; Elsevier: Amsterdam, 1983.
- Diudea, M. V. (Ed.) QSPR/QSAR Studies by Molecular Descriptors; Nova Science Publishers, Inc.: Huntington, New York, 2001.
- Wiener, H. Structural Determination of Paraffin Boiling Points. J. Am. Chem. Soc. 1947, 56, 17–20. [Google Scholar]
- Hosoya, H. Topological Index. A Newly Proposed Quantity Characterizing the Topological Nature of Structural Isomers of Saturated Hydrocarbons. Bull. Chem. Soc. Jpn. 1971, 44, 2332–2339. [Google Scholar]
- Randic, M. On Characterization of Molecular Branching. J. Am. Chem. Soc. 1975, 97, 6609–6615. [Google Scholar]
- Trinajstic, N. Graph Theory; CRC Press: Boca Raton, FL, 1983. [Google Scholar]
- Harary, F. Graph Theory; Addison-Wesley: Reading, MA, 1969. [Google Scholar]
- Cramer, R. D. BC(DEF) Parameters. 2. An Empirical structure-Based Scheme for the Prediction of Some Physical Properties. J. Am. Chem. Soc. 1979, 102, 1849–1859. [Google Scholar]
- Monnery, W. D.; Svreck, W. Y.; Mehrota, A. K. Voscicity: A Critical Review of Practical Predictive and Correlative Methods. Can. J. Chem. Eng. 1995, 73, 3–40. [Google Scholar]
- Stein, S. E.; Brown, R. L. Estimation of Normal Boiling Points from Group Contributions. J. Chem. Inf. Comput. Sci. 1994, 34, 581–587. [Google Scholar]
- Pouchly, J.; Quin, A.; Munk, P. Excess Volume of Mixing and Equation of State Theory. J. Solution Chem. 1993, 22, 399–418. [Google Scholar]
- Elbro, H. S.; Fredenslund, A.; Rasmussen, P. Group Contribution Meted for the Prediction of Liquid Densities as a Function of Temperatures for Solvents, Oligomers and Polymers. Ind. Eng. Chem. Res. 1991, 30, 2576–2593. [Google Scholar]
- Fisher, C. H. Boiling Point Gives Critical Temperatures. Chem. Eng. 1989, 96, 157–158. [Google Scholar]
- Satyanarayana, K.; Kakati, M. C. Note: Correlation of Flash Points. FIRE Mater. 1991, 15, 97–100. [Google Scholar]
- Rechsteiner, C. E. Handbook of Chemical Property Estimation Methods; Lyman, W. J., Reehl, W. F., Rosenblatt, D. H., Eds.; McGraw-Hill: New York, 1982; Chapter 12. [Google Scholar]
- Katritzky, A.R.; Mu, L.; Lobanov, V. S.; Karelson, M. Correlation of Boiling Points with Molecular Structure. 1. A Training Set of 298 Diverse Organics and a Test Set of 9 Simple Inorganics. J. Phys. Chem. 1996, 100, 10400–10407. [Google Scholar]
- Horvath, A.L. Molecular Design: Chemical Structure Generation from the Properties of Pure Organic Compounds; Elsevier: Amsterdam, 1992. [Google Scholar]
- Wessel, M. D.; Jurs, P. C. Prediction of Normal Boiling Points for a Diverse Set of Industrially Important Organic Compounds from Molecular Structure. J. Chem. Inf. Comput. Sci. 1995, 35, 841–850. [Google Scholar]
- Lee, T. D.; Weers, J. G. QSPR and GCA Models for Predicting the Normal Boiling Points of Fluorocarbons. J. Phys. Chem. 1995, 99, 6739–6747. [Google Scholar]
- Komasa, A. Prediction of Boiling Points of Ketones Using a Quantitative Structure-Property Relationships Treatment. Polish J. Chem. 2003, 77, 1491–1499. [Google Scholar]
- Kompany-Zareh, M. A QSPR Study of Boiling Point of Saturated Alcohols Using Genetic Algorithm. Acta Chim. Slov. 2003, 50, 259–273. [Google Scholar]
- Öberg, T. Boiling Points of Halogenated Aliphatic Compounds: A Quantitative Structure-Property Relationship for Prediction and Validation. J. Chem. Inf. Comput. Sci. 2004, 44, 187–192. [Google Scholar]
- Randic, M.; Basak, S. C. Variable Molecular Descriptors, in Some Aspects of Mathematical Chemistry; Sinha, D. K., Basak, S. C., Mohanty, R. K., Busamallick, I. N., Eds.; Visva-Bharati University Press: Santiniketan (India), 1999. [Google Scholar]
- Randic, M. Novel Graph Theoretical Approach to Heteroatoms in QSAR. Chemom. Intel. Labl. Syst. 1991, 10, 213–223. [Google Scholar]
- Toropova, A.P.; Toropov, A. A.; Ishankhodzhaeva, M. M.; Parpiev, N. A. QSPR Modeling of Stability Constants of Coordination Compounds by Optimization Weights of Local Graph Invariants. Russ. J. Inorg. Chem. 2000, 45, 1057–1059. [Google Scholar]
- Toropov, A. A.; Voropaeva, N. L.; Ruban, I. N.; Rashidova, S. Sh. Quantitative Structure-Property Relationships for Binary Polymer-Solvent Systems: Correlation Weighting of the Local Invariants of Molecular Graphs. Polymer Science Ser. A 1999, 41, 975–985. [Google Scholar]
- Toropov, A.; Toropova, A.; Ismailov, T.; Bonchev, D. 3D Weighting of Molecular Descriptors for QSPR/QSAR by the Method of Ideal Symmetry (MIS). 1. Application to Boiling Points of Alkanes. J. Mol. Struct. THEOCHEM 1998, 424, 237–247. [Google Scholar]
- Krenkel, G.; Castro, E. A.; Toropov, A. A. Improved Molecular Descriptors Based on the Optimization of Correlation Weights of Local Graphs. Int. J. Molec. Sci. 2001, 2, 57–65. [Google Scholar]
- Toropov, A. A.; Toropova, A. A. Prediction of Heteroatomic Amine Mutagenicity by Means of Correlation Weighting of Atomic Orbital Graphs of Local Invariants. J. Mol. Struct. THEOCHEM 2001, 538, 287–293. [Google Scholar]
- Toropov, A. A.; Toropova, A. P. Modeling the Lipophilicity by Means of Correlation Weighting of Local Graph Invariants. J. Mol. Struct. THEOCHEM 2001, 538, 197–199. [Google Scholar]
- Mercader, A.; Castro, E. A.; Toropov, A. A. QSPR Modeling the Enthalpy of Formation from Elements by Means of Correlation Weighting of Local Invariants of Atomic Orbital Molecular Graphs. Chem. Phys. Lett. 2000, 330, 612–623. [Google Scholar]
- Toropov, A. A. A. P. Toropova, QSAR Modeling of Toxicity on Optimization of Correlation Weights of Morgan Extended Connectivity. J. Mol. Struct. THEOCHEM 2002, 578, 129–134. [Google Scholar]
- Toropov, A. A.; Toropova, A. P. QSPR Modeling of Alkanes Properties Based on Graph of Atomic Orbitals. J. Mol. Struct. THEOCHEM 2003, 637, 1–10. [Google Scholar]
- Toropov, A. A.; Nesterov, I. V.; Nabiev, O. M. QSPR Modeling of Cycloalkanes Properties by Correlation Weighting of Extended Graph Valence Shells. J. Mol. Struct. THEOCHEM 2003, 637, 37–42. [Google Scholar]
- Basak, S. C.; Grunwald, G. D. Predicting mutagenicity of chemicals using topological and quantum chemical parameters: A similarity based study. Chemosphere 1995, 31, 2529. [Google Scholar]
- Toropov, A. A.; Toropova, A. P. QSPR modeling of the formation constants for complexes using Atomic Orbital Graphs. Russ. J. Coord. Chem. 2000, 26, 398. [Google Scholar]
- Toropov, A. A. A. P. Toropova, Optimization of correlation weights of the local graph invariants: use of the enthalpies of formation of complexes compounds for the QSPR modeling. Russ. J. Coord. Chem. 1998, 24, 81. [Google Scholar]
- Pérez-González, M.; González Díaz, H.; Molina Ruiz, R.; Cabrera, M. A.; Ramos de Armas, R. TOPS-MODE Based QSARs Derived from Heterogeneous Series of Compounds. Applications to the Design of New Herbicides. J. Chem. Inf. Comput. Sci. 2003, 43, 1192–1199. [Google Scholar]
- Kowalski, R. B.; Wold, S. Pattern Recognition in Chemistry. In Handbook of Statistics; Krishnaiah, P. R., Kanal, L. N., Eds.; North Holland Publishing Company: Amsterdam, 1982; pp. 673–697. [Google Scholar]
- McFarland, J. W.; Gans, D. J. Cluster Significance Analysis. In Methods and Principles in Medicinal Chemistry; Manhnhold, R., Krgsgaard, L., Timmerman, H., Eds.; VCH: Weinheim, 1995; Vol. 2 , (Chemometric Methods in Molecular Design, van Waterbeemd, H. ed.); pp. 295–307. [Google Scholar]
© 2004 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
González, M.P.; Toropov, A.A.; Duchowicz, P.R.; Castro, E.A. QSPR Calculation of Normal Boiling Points of Organic Molecules Based on the Use of Correlation Weighting of Atomic Orbitals with Extended Connectivity of Zero- and First-Order Graphs of Atomic Orbitals. Molecules 2004, 9, 1019-1033. https://doi.org/10.3390/91201019
González MP, Toropov AA, Duchowicz PR, Castro EA. QSPR Calculation of Normal Boiling Points of Organic Molecules Based on the Use of Correlation Weighting of Atomic Orbitals with Extended Connectivity of Zero- and First-Order Graphs of Atomic Orbitals. Molecules. 2004; 9(12):1019-1033. https://doi.org/10.3390/91201019
Chicago/Turabian StyleGonzález, Maykel Pérez, Andrey A. Toropov, Pablo R. Duchowicz, and Eduardo A. Castro. 2004. "QSPR Calculation of Normal Boiling Points of Organic Molecules Based on the Use of Correlation Weighting of Atomic Orbitals with Extended Connectivity of Zero- and First-Order Graphs of Atomic Orbitals" Molecules 9, no. 12: 1019-1033. https://doi.org/10.3390/91201019
APA StyleGonzález, M. P., Toropov, A. A., Duchowicz, P. R., & Castro, E. A. (2004). QSPR Calculation of Normal Boiling Points of Organic Molecules Based on the Use of Correlation Weighting of Atomic Orbitals with Extended Connectivity of Zero- and First-Order Graphs of Atomic Orbitals. Molecules, 9(12), 1019-1033. https://doi.org/10.3390/91201019