Introduction
In the past few decades luminescent transition metal complexes based on polypyridine ligands, owing to their long-lived metal-to-ligand charge-transfer (MLCT) excited states, have already been used in various fields such as solar energy conversion [
1], information storage [
2], photocleavage of DNA [
3], and oxygen sensors [
4]. Although the photophysics and photochemistry of [Ru(bpy)
3]
2+ (bpy = 2,2’ bipiridine) have been the subject of extensive research [
1,
2,
3,
4], few other bidentate ligands, i.e. having two aza binding sites, have been prepared and the photophysical and/or photochemical properties of their complexes with transition metals studied [
5]. As a continuation of previous studies in this field [
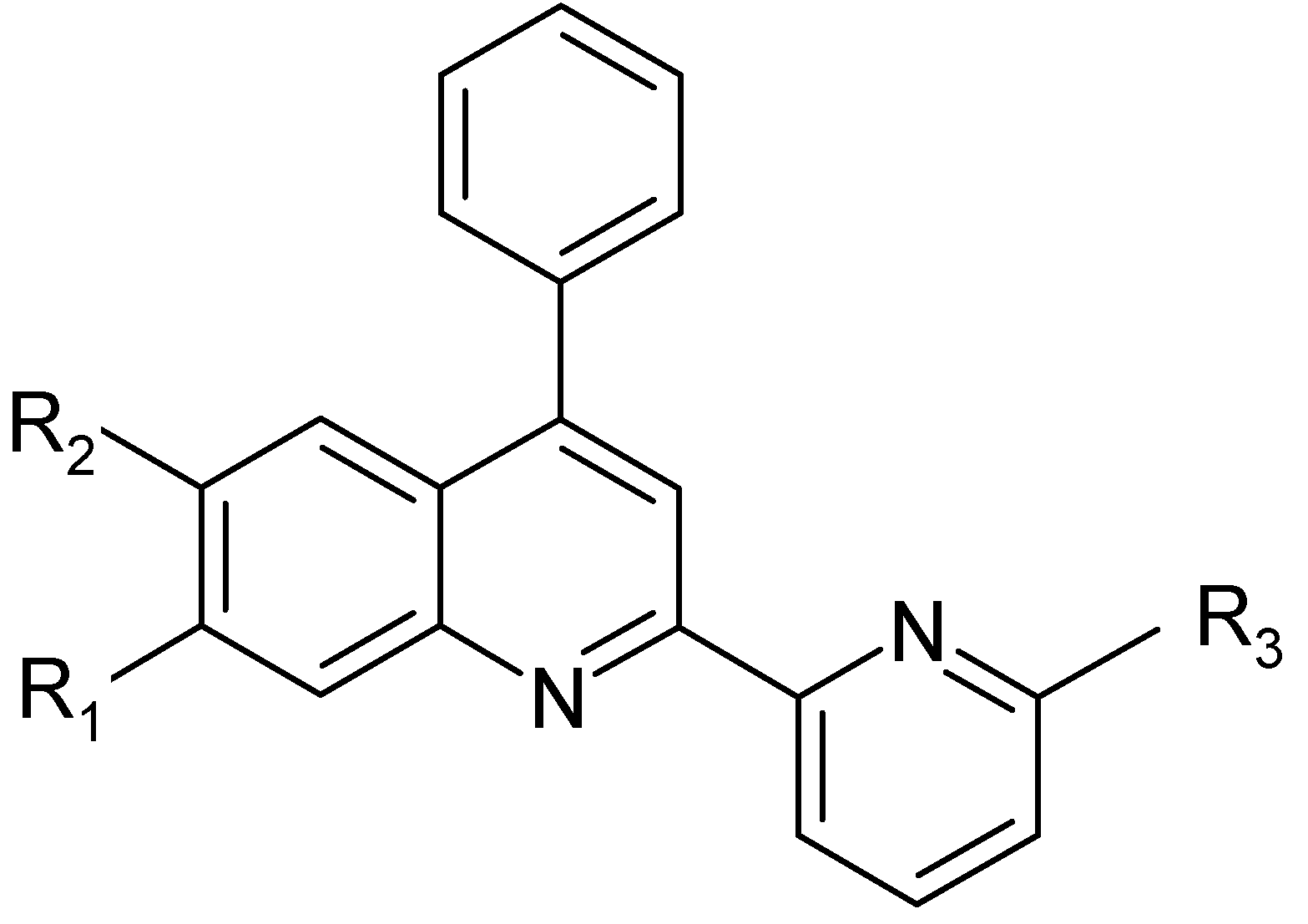
6], we now report the synthesis and characterization of the ligands shown in
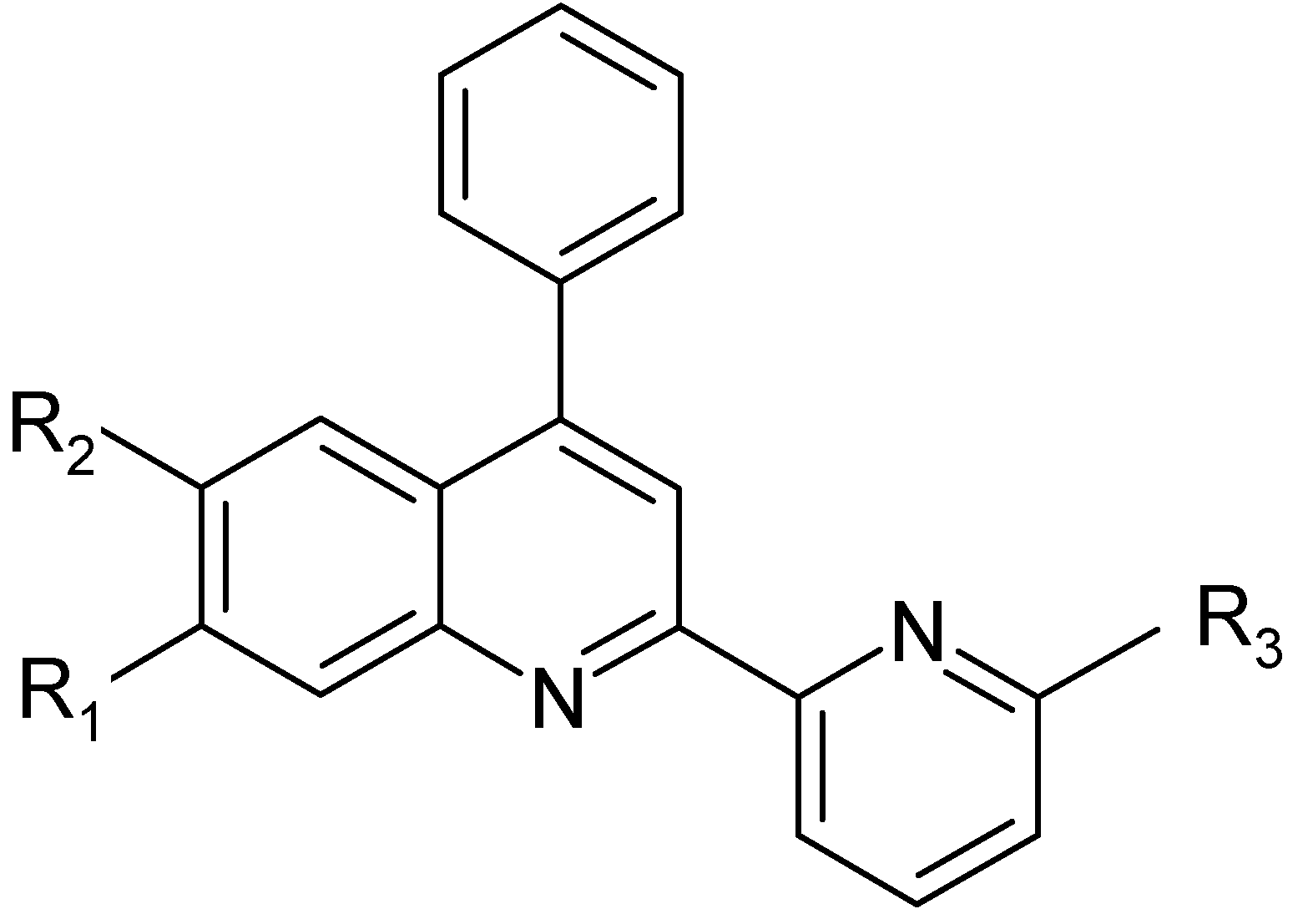
Scheme 1, with the aim of studing the photochemical properties of their complexes with transition metals such as ruthenium, osmium or iridium. Three of these asymmetric bidendate ligands (
L2 –
L4) are new. All the compounds were characterized by elemental analysis, EI or FAB mass,
1H and
13C NMR spectroscopies. Complete assignments of the
1H spectra of the various compounds were accomplished by using a combination of one- and two-dimensional NMR techniques.
Scheme 1.
| Ligands | R1 | R2 | R3 | Initials |
|---|
| L1 | H | H | H | ph-pq |
| L2 | CH3 | H | H | mph-pq |
| L3 | CH3 | H | CH3 | mph-mpq |
| L4 | H | Br | CH3 | brph-mpq |
Results and discussion
The literature describes numerous different ways to prepare substituted quinoline rings: i.e., by exploiting quinoline carboxamides [
7], acid-catalyzed condensation of
o-aminobenzophenones [
8] with ketones [
9], sequential vinylic substitution/annelation processes [
10], reactions of N-arylnitrilium salts with acetylenes [
11], cyclodehydration of
o-vinyl anilides [
12], intramolecular Wittig reactions [
13], and cyclization of oximes [
14]. Using
o-isocyanostyrenes only symmetric biquinoline may be prepared [
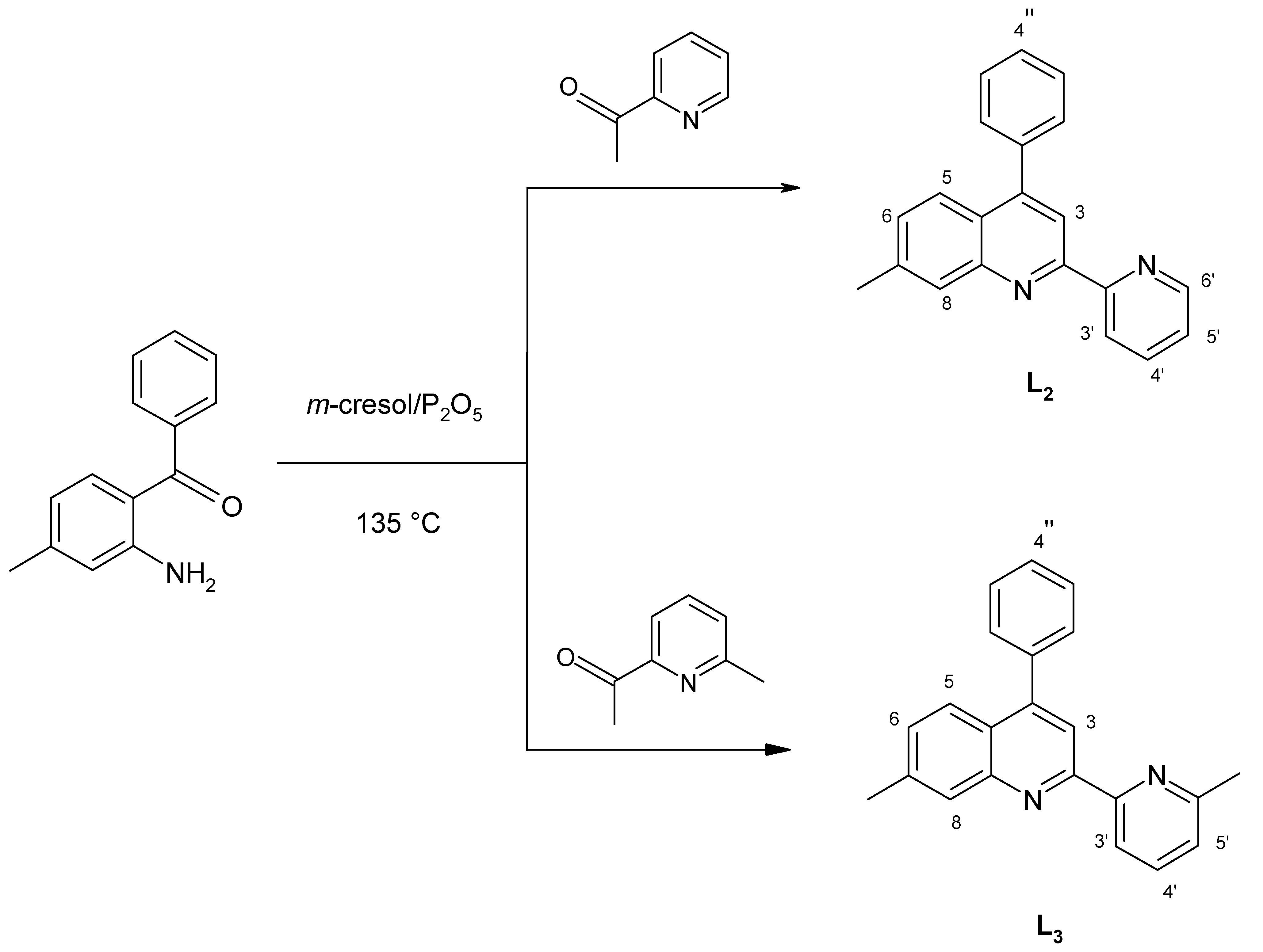
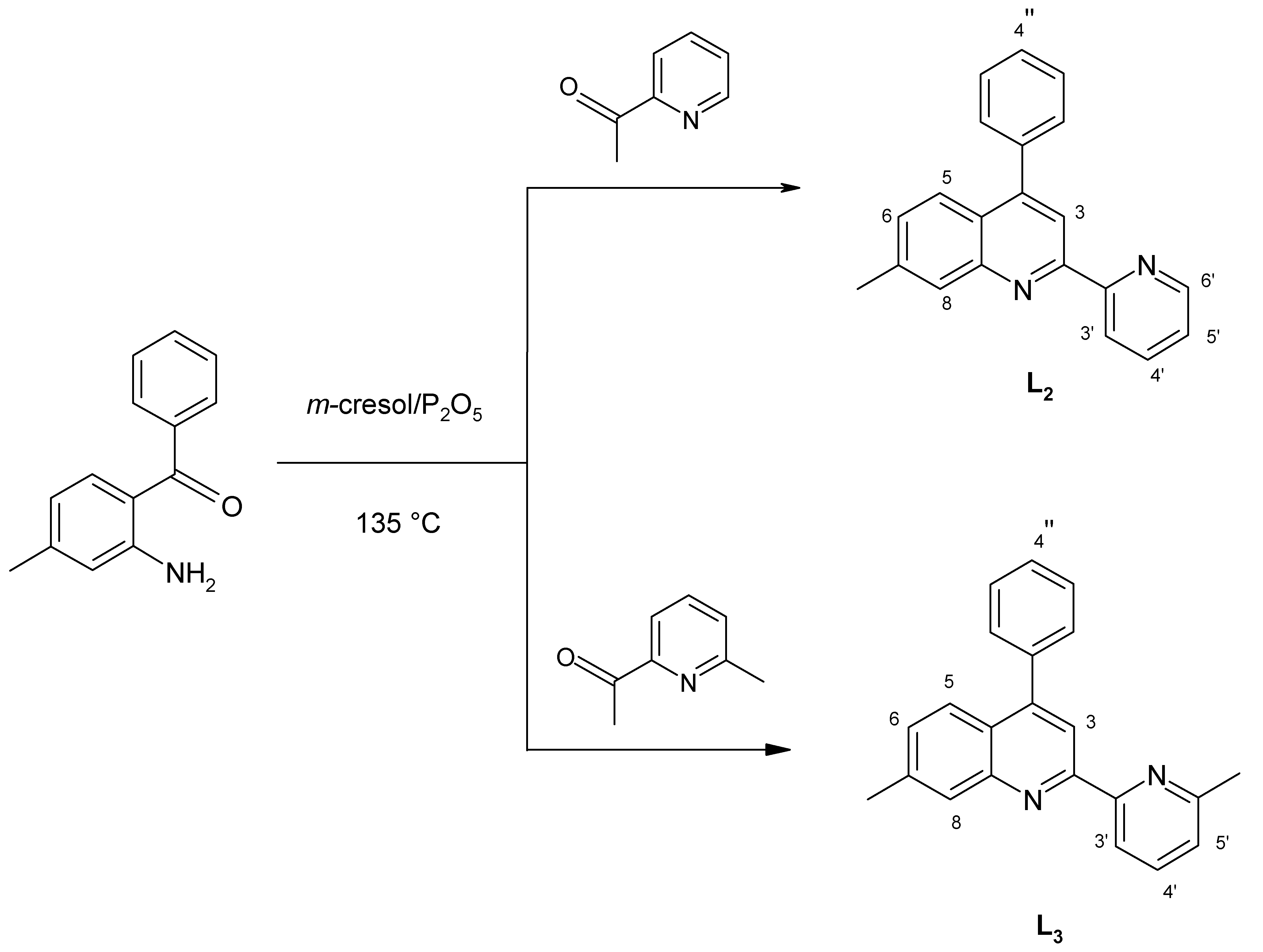
15]. Following the synthetic pathway previously used for the preparation of the unsubstituted ligand 4-phenyl-2-(2’-pyridyl)quinoline (
L1, ph-pq) [
16], namely the acid-catalyzed condensation of
o-amino-benzophenone with 2- acetylpyridine derivatives, as shown in
Scheme 2, we have now synthetized the ligands 4-phenyl-7-methyl-2-(2’-pyridyl)quinoline (
L2, mph-pq) and 4-phenyl-7-methyl-2-[2’-(6’-methyl)pyridyl]-quinoline (
L3, mph-mpq).
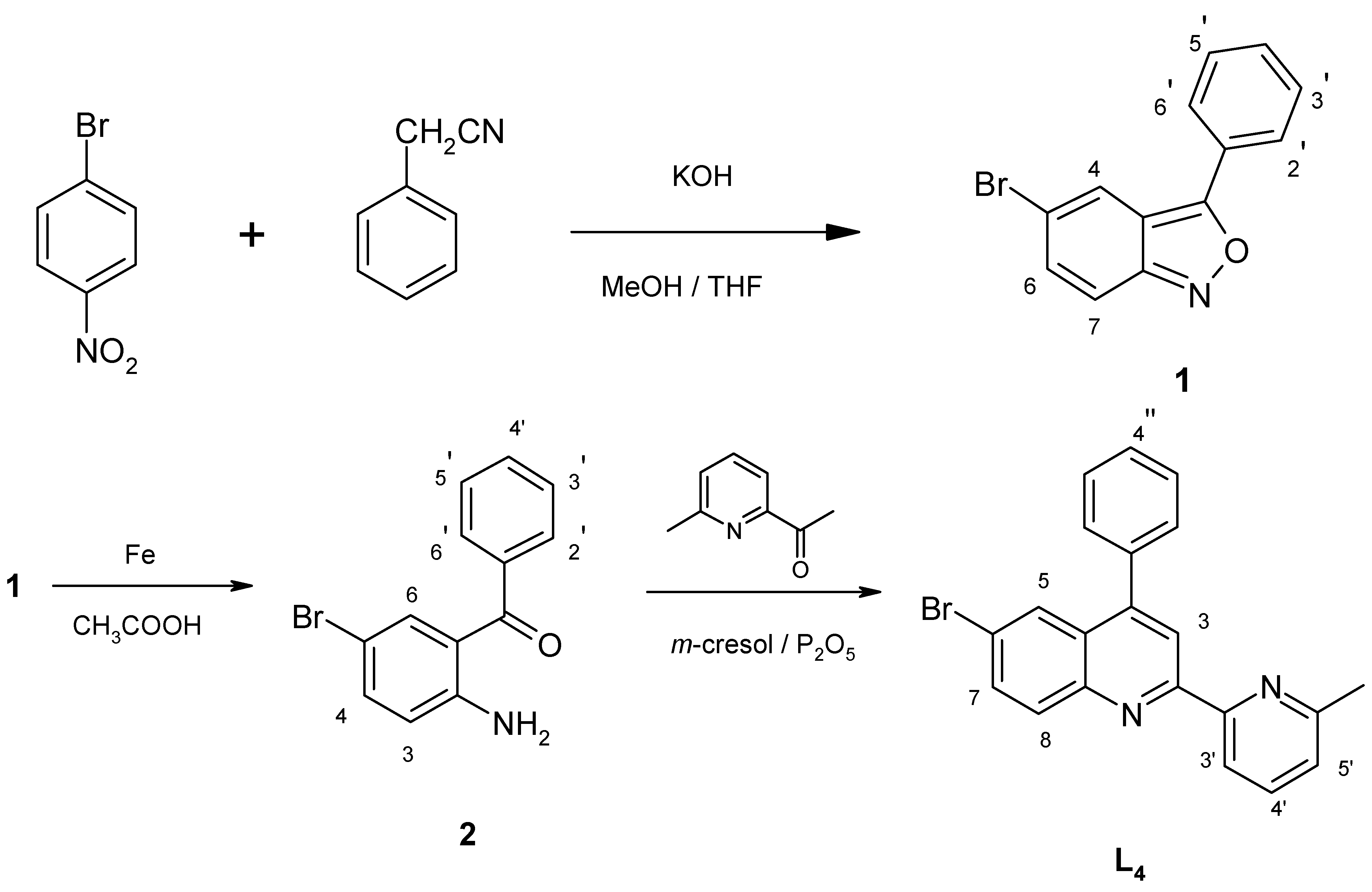
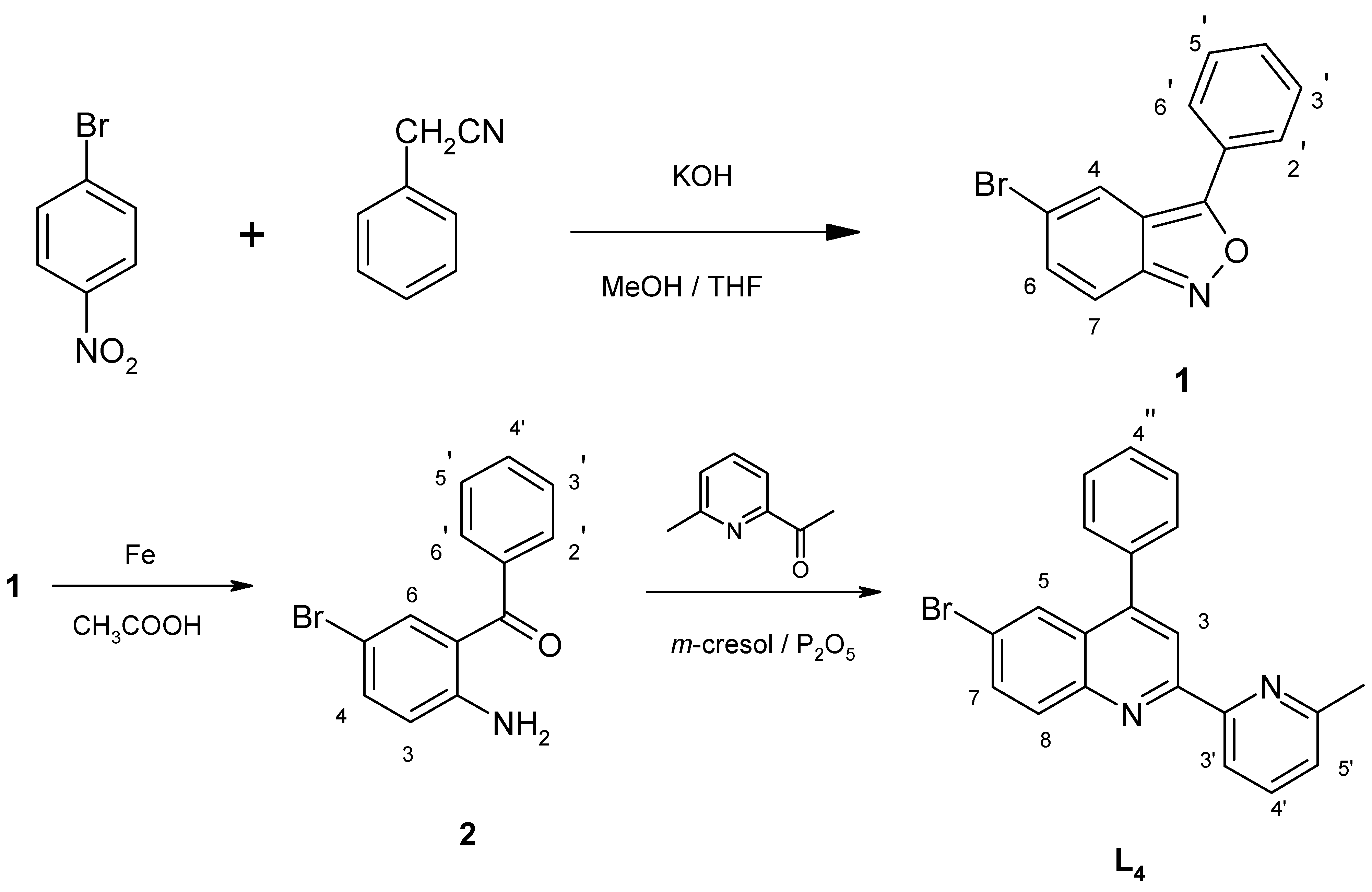
The ligand 4-phenyl-6-bromo-2-[2’-(6’-methyl)-pyridyl]quinoline (
L4, brph-mpq) was obtained in a three synthetic steps (
Scheme 3) starting from
p-nitrobromobenzene.
2-Amino-5-bromobenzophenone (
2) was obtained by condensation of
p-nitrobromobenzene with phenylacetonitrile in a basic methanol/tetrahydrofuran medium to give 3-phenyl-5-bromo-2,1-benzisoxazole (
1) (66%), which upon reductive cleavage (Fe/CH
3COOH) of the benzisoxazole ring was converted to the desired aminoketone
2 (70 %). A subsequent Friedlander reaction [
17] of the
o-aminobenzophenone
2 with 2-acetyl-6-methylpyridine, using a mixture of
m-cresol and phosphorous pentoxide gave ligand
L4 (71%).
Table I reports the results of a complete
1H-NMR analysis of ligands
L1–
L4. Proton chemical shifts and
J(H,H) values were measured at 500 MHz.
Table I.
1H NMR parameters of ligands L1 – L4
Table I.
1H NMR parameters of ligands L1 – L4
| Proton | L1 | L2 | L3 | L4 |
|---|
| 3 | 8.53 s | 8.47 s | 8.48 s | 8.57 s |
| 5 | 7.96 d | 7.85 d | 7.22 d | 8.06 d |
| J=8.0 | J=9.0 | J=7.5 | J=2.0 |
| 6 | 7.56-7.50 | 7.34 bd | 7.34 dd | - |
| m | J=6.0 | J=8.5, 1.5 |
| 7 | 7.75 dt | - | - | 7.79 dd |
| J=7.0, 1.5 | J=7.0, 2.0 |
| 8 | 8.26 d | 8.05 bs | 8.04 bs | 7.23 d |
| J=8.5 | J=7.5 |
| 3’ | 8.71 d | 8.69 d | 7.83 d | 8.45 d |
| J=7.5 | J=8.0 | J=8.5 | J=8.0 |
| 4’ | 7.90 dt | 7.88 dt | 7.77 t | 7.77 t |
| J=8.0, 2.0 | J=8.0, 1.5 | J=7.5 | J=8.0 |
| 5’ | 7.37 bt | 7.36 dt | 8.47 d | 8.10 d |
| J=6.5 | J=7.5, 1.5 | J=7.5 | J=8.5 |
| 6’ | 8.74 d | 8.73 dd | - | - |
| J=4.5 | J=4.5, 1.0 |
| Ph | 7.62-7.50 | 7.61-7.49 | 7.61-7.50 | 7.57-7.51 |
| m | m | m | m |
| Py-CH3 | - | - | 2.65 s | 2.64 s |
| q-CH3 | - | 2.60 s | 2.59 s | - |
Assignments were aided by the use of 2D homonuclear chemical shift correlated
1H-NMR (COSY) [
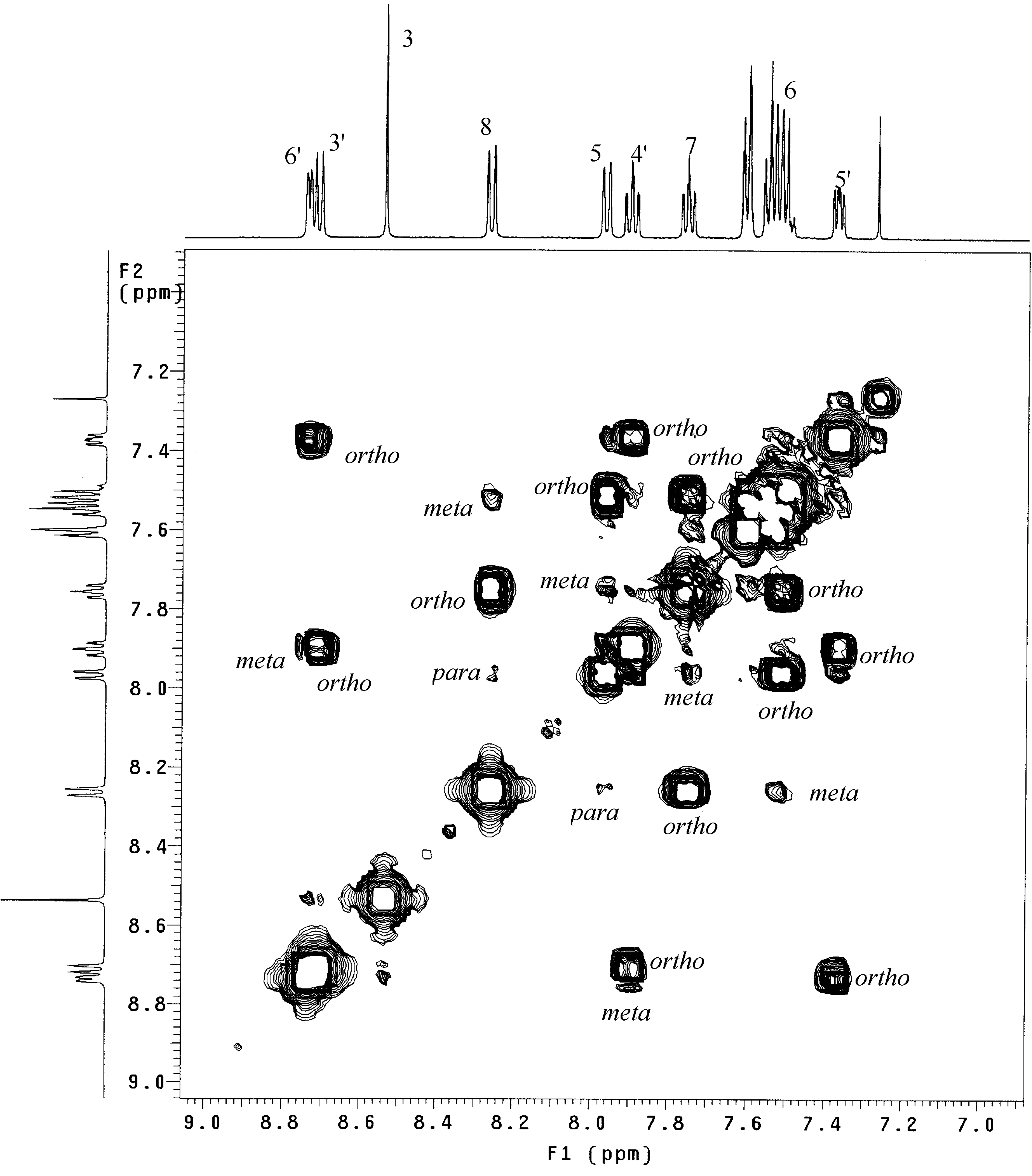
18]. As an example,
Figure 1 shows the COSY–45 experiment of
L1 and includes as the upper and left traces the related
1H-NMR spectrum, both run in deuterated chloroform (CDCl
3). The 1H singlet at 8.53 ppm was easily assigned by the integration ratio to the quinoline proton H
3. A four-spin system is identified, through the COSY spectrum, as connecting the
1H signals at 8.26, 7.96, 7.75, and 7.57-7.50 ppm. The doublet (
ortho coupling) at 8.26 ppm and the double triplet at 7.75 ppm have been assigned to H
8 and H
7, respectively, by comparison with the literature
1H data for
L1 in deuterated acetone [
16].
The resonances for H
5 and H
6 could be assigned to the signals at 7.96 and 7.57-7.50 ppm, respectively. The
1H double triplet at 7.90 ppm, diagnostic for a γ-pyridine [
19], and involved in another four spin system connecting the
1H signals at 8.74, 8.71, 7.90, and 7.37 ppm, was assigned to the pyridine proton H
4’. As a consequence of the
meta and
ortho couplings showed by H
4’, the doublets at 8.74, 8.71, and the broad triplet at 7.37 ppm, that in turn are correlated themselves, were easily assigned at H
6’, H
3’, and H
5’, respectively. It is worth noting that
ortho,
meta, and
para cross-peaks are observable in the COSY-45 spectrum and can be distinguished from the number and/or the intensity of the spots.
Figure 1.
500 MHz 1H/H1 COSY-45 spectrum of L1 in deuterated chloroform. The upper and left traces are 1D proton spectrum of L1.
Figure 1.
500 MHz 1H/H1 COSY-45 spectrum of L1 in deuterated chloroform. The upper and left traces are 1D proton spectrum of L1.
The highest downfield shift experienced by the H
3’ protons, due to deshielding by the non-bonding electrons of the nitrogen on the pyridine ring, is indicative of an
anti conformation for the ligands, in agreement with the conformation considered the most probable for bipyridine [
5]. According to literature data [
19], confirmed by our
1H-NMR analyses, these uncomplexed molecules show an
anti conformation (as depicted in
Scheme 1,
Scheme 2 and
Scheme 3) that changes to a
syn one when they act as ligands by using the nitrogen of the pyridine and quinoline rings as binding sites. According to the inductive and/or mesomeric effects of the substituents, their introduction onto the skeleton of the N-N bidendate ligand L
1 influence the upfield and/or downfield chemical shift of the nearest protons, and the reactivity of these molecules as well. The structures of ligands
L1-L4 was further confirmed by their
13C-NMR spectra (see
Table II), which displayed the expected patterns.
Table II.
13C NMR parameters of ligandsf L1 – L4
Table II.
13C NMR parameters of ligandsf L1 – L4
| Carbon | L1 | L2 | L3 | L4 |
|---|
| 2 | 156.40 | 156.54 | 155.97 | 156.35 |
| 3 | 119.24 | 118.48 | 118.62 | 118.85 |
| 4 | 149.23 | 149.02 | 148.88 | 148.24 |
| 5 | 125.82 | 125.47 | 125.45 | 128.55 |
| 6 | 128.30 | 129.03 | 128.90 | 120.80 |
| 7 | 129.39 | 139.62 | 139.55 | 132.79 |
| 8 | 130.21 | 129.20 | 129.16 | 131.87 |
| 9 | 148.51 | 148.74 | 148.70 | 147.08 |
| 10 | 126.78 | 124.78 | 124.76 | 123.80 |
| 2’ | 155.64 | 155.59 | 155.88 | 155.31 |
| 3’ | 121.87 | 121.78 | 118.82 | 120.11 |
| 4’ | 136.94 | 136.87 | 137.05 | 137.11 |
| 5’ | 124.03 | 123.92 | 123.48 | 123.79 |
| 6’ | 149.17 | 149.11 | 157.90 | 158.04 |
| 1” | 138.40 | 138.55 | 138.74 | 137.88 |
| 2”/6” | 128.44 | 128.41 | 128.43 | 128.69 |
| 3”5” | 129.67 | 129.63 | 129.64 | 129.55 |
| 4” | 126.78 | 128.21 | 128.17 | 127.93 |
| Py-CH3 | - | - | 24.64 | 24.61 |
| q-CH3 | - | 21.66 | 21.67 | - |
Experimental
General
The starting materials 2-acetylpyridine, 2-aminobenzophenone, 2-amino-4-methylbenzophenone,
p-nitrobromobenzene, and phenylacetonitrile were purchased from Aldrich. All other chemicals were reagent grade. 6-Methyl-2-acetylpyridine [
20] and the ligand 4-phenyl-2-(2’-pyridyl)pyridine (
L1) [
16], were prepared as described in the literature. All reactions were performed under an inert atmosphere of nitrogen except when otherwise stated and the solvents were dried and stored under nitrogen and over 4Å molecular sieves. Melting points are uncorrected. Elemental analyses were determined by a commercial laboratory.
1H- and
13C-NMR spectra were performed in deuterated chloroform (CDCl
3) with a Varian INOVA 500 instrument. Chemical shifts were calibrated relative to the solvent resonance considered at 7.26 ppm for residual CHCl
3 and at 77.0 ppm for CDCl
3. The analysis of the proton spectra was carried out according to the rules for the first-order splitting with the help of integral intensities. The
13C‑NMR spectra were measured with full decoupling from the protons, and the signals were assigned with the help of SCS. The quaternary carbon atoms and CH groups were differentiated by means of the APT pulse sequence. Positive ion FAB mass spectra were obtained on a Kratos MS 50 S double-focusing mass spectrometer equipped with a standard FAB source, using 3-nitrobenzyl alcohol as a matrix. The yields, melting points and elemental analyses of the ligands synthetized are presented in
Table III. The
1H- and
13C-NMR spectra with signal assignments are given in
Table I and
Table II, respectively.
3-phenyl-5-bromo-2,1-benzisoxazole (1): Phenylacetonitrile (1.75 g, 15 mmol) was slowly added to a vigorously stirred solution of potassium hydroxide (17.76 g, 310 mmol) in methanol (35 mL) at room temperature. After dissolution was complete, 36 mL of a methanol/tetrahydrofuran (2 : 1 v/v) solution containing p-nitrobromobenzene (3.0 g, 15 mmol) was added dropwise at 0 °C. The resulting dark mixture was stirred at 0 °C for 3 hours, at room temperature for 4 hours, refluxed overnight, and then poured into ice-water (300 mL), filtered, washed successively with cold water and methanol and recrystallized from methanol to afford compound 1 as yellow crystals; 2.22 g (66%); m.p. 112 °C; 1H-NMR (CDCl3) d: 8.05 (bs, 1H, benzisoxazole H4); 7.99 (d, 2H, J = 7.0 Hz, phenyl H2’/H6’), 7.58 (m, 3H, phenyl H4’ and H3’/H5’); 7.53 (dd, 1H, J = 10.0, 2.5 Hz, benzisoxazole H6); 7.38 (dd, 1H, J = 10.0, 1.5 Hz, benzisoxazole H7); MS, m/z 274 (MH+). Anal. Calcd. for C13H8BrNO: C, 56.95; H, 2.92; N, 5.11. Found: C, 57.19; H, 3.03; N, 4.86.
2 Amino-5-bromo-benzophenone (
2): Following the procedure of Simpson and Stephenson [
21], a solution, containing 0.44 g (1.6 mmol) of
1 in acetic acid (70 mL), was heated on a water-bath, and 1.0 g (18 mmol) of iron powder was added over 2.5 hours, during which time, 12 ml of water was also added. The mixture was filtered while hot and then 100 ml of water was added. The yellow precipitate was collected by filtration, washed with cold water until the water washings were clear and dried. The product was purified by column chromatography (silica; cyclohexane / ethyl acetate 9:1) followed by recrystallization from ethanol-water to afford
2 as a yellow powder; 0.31 g (70 %); m.p. 105 °C;
1H‑NMR (CDCl
3) d: 7.63 (d, 2H,
J = 8.5 Hz, phenyl H
2’/H
6’); 7.55 (m, 2H, benzene H
6 and phenyl H
4); 7.49 (d, 2H,
J = 8.5 Hz, phenyl H
3’/H
5’); 7.36 (dd, 1H,
J = 9.0, 2.0 Hz, benzene H
4); 6.65 (d, 1H,
J = 8.5 Hz, benzene H
3); 6.05 (bs, 2H, NH
2), MS,
m/z 276 (MH
+). Anal. Calcd. for C
13H
10BrNO: C, 56.54; H, 3.62; N, 5.07. Found: C, 56.28; H, 3.59; N, 4.95
The synthesis of L4 is given below as a general procedure for the synthesis of ligands.
4-phenyl-6-bromo-2-(2’-(6’-methyl)-pyridyl)quinoline (L4): A mixture of m-cresol (25 mL) and phosphorus pentoxide (0.81 g, 5.7 mmol) was stirred at 145 °C for 2.5 hours to afford a homogeneous solution. After cooling, 2-amino-5-bromobenzophenone (4.08 g, 15 mmol) and 2-acetyl-6-methyl-pyridine (2.03 g, 15 mmol) were added, followed by additional m-cresol (20 mL) to rinse the powder funnel. The reaction mixture was heated at 135 °C overnight. After cooling, the dark solution was poured into ethanol (200 mL) containing triethylamine (20mL). The resulting light grey precipitate was collected by filtration, continuosly extracted with a solution of ethanol/triethylamine for 24 hours, and recrystallized from n-hexane/methylene chloride to give L4 as an off white powder; 3.96 g (71%); m.p. = 212 °C.; MS, m/z 375 (MH+).
Table III.
Melting points, yield and elemental analyses of ligands L1 – L4
Table III.
Melting points, yield and elemental analyses of ligands L1 – L4
| Ligand | Recrystallization | M.p: | Yield | Formula / | Elemental Analysis
Calculated/Found (%) |
|---|
| Solvent(s) | (° C) | (%) | M. w. | C | H | N |
|---|
| L1 | EtOH | 152 | 70 | C20H14N2 | 85.05 | 5.00 | 9.90 |
| 296.35 | 85.00 | 5.05 | 10.00 |
| L2 | EtOH / CHCl3 | 138 | 62 | C21H16N2 | 85.10 | 5.44 | 9.45 |
| 296.35 | 85.02 | 5.63 | 9.32 |
| L3 | EtOH / H2O | 194 | 60 | C22H18N2 | 85.13 | 5.84 | 9.02 |
| 310.38 | 85.11 | 5.93 | 9.12 |
| L4 | n-C6H12 / CH2Cl2 | 212 | 71 | C21H15BrN2 | 67.21 | 4.03 | 7.46 |
| 375.26 | 67.33 | 4.34 | 7.33 |


{kind=link}
{kind=link}
{kind=link}
{kind=link}
