Abstract
The reactions of acenaphthenequinone and its derivatives with different nucleophiles, organic and inorganic reagents are reviewed. This survey also covers their oxidation and reduction reactions, in addition to many known reactions such as Friedel Crafts, Diels-Alder, bromination and thiolation.
Contents
- Introduction
- Reactions of Acenaphthenequinones
- 2.1
- Action of Alkali and Oxidation
- 2.2
- Reduction
- 2.3
- Protonation
- 2.4
- Reaction with active methylene compounds
- 2.5
- Reaction with aldehydes and ketones
- 2.6
- Reaction with Wittig reagents
- 2.7
- Reaction with magnesium and lithium reagents
- 2.8
- Friedel Craft reaction
- 2.9
- Diels-Alder reactions
- 2.10
- Reaction with phenolic compounds
- 2.11
- Bromination reactions
- 2.12
- Thiation reactions
- 2.13
- Alkylation reactions
- 2.14
- Ketal derivatives
- 2.15
- Reaction with Phosphite and Phospholanes
- 2.16
- Reaction with carbenes
- 2.17
- Decarbonylation
- 2.18
- Reaction with nitrogen nucleophiles
1. Introduction
The broad spectrum of applications of acenaphthenequinone and its derivatives as biologically active compounds, dyes, etc has prompted us to review their chemistry and uses. The syntheses of acenaphthenequinone (1) and its derivatives, which are based mainly on the use of starting materials having the carbon skeleton of 1 and their reactivity towards nitrogen nucleophiles have been discussed in the first part of this series [1]. In this part, the reactions of acenaphthenequinones are reviewed.
2. Reactions of Acenaphthenequinones
2.1 Ring opening and enlargement
Ring cleavage of acenaphthenequinone (1) with an aqueous potassium hydroxide solution in dimethylsulphoxide at room temperature led to formation of 1, 8-naphthaldehydic acid (2) which exists in equilibrium with the corresponding cyclic structure [2,3,4]. The reaction of 1 in aqueous alkali gave 2,3-dimethylbenzoic acid (3) [5]. The alkaline permanganate oxidation of 1 gave 2,6-dicarboxy-phenylglyoxylic acid [6]. On the other hand, oxidation of 1 with molecular oxygen in propionic acid containing a homogeneous catalyst such as cobalt (II) acetate or manganese (II) acetate gave 1,8-naphthalic anhydride (6) [7]. Addition of potassium bromide to this reaction mixture increased the rate of reaction. A similar transformation was effected by oxygen in the presence of copper (I) chloride and pyridine [8]. On the other hand, the oxidative cleavage of 1 by the oxygen adduct of cobaltocene gave cobaltocinium carboxylate (4) that upon reaction with hydrogen chloride in ether, acid halides or dialkyl sulphates gave naphthalene-1,8-dicarboxylic acid (5), or its anhydride or ester, respectively [9]. When ozonolyses of vinyl ethers were conducted in presence of 1, it afforded 6 in addition to unreacted 1 [10]. This was attributed to the transfer of an oxygen atom from the carbonyl oxides, generated from the viny1 ethers, to 1 to give a Baeyer-Villiger type product. Heating of 1 with sodamide and treating with water gave naphthalene and oxalamide [11].
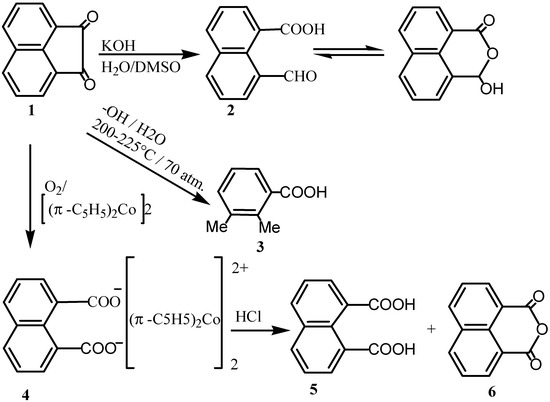
Scheme 1.
Photolysis of acenaphthenequinone in methylene chloride saturated with oxygen [12,13] gave 1,8-naphthalic anhydride (6). When an olefin such as cyclohexene was included in the reaction, it was converted to a mixture of oxidized products consisting, mainly the allylic hydroperoxide (7), epoxide (8), and adipaldehyde (9) in addition to 6. The quantum efficiency for quinone oxidation was independent of quinone and olefin concentrations. A mechanism was suggested in which an initial reaction between excited quinone and oxygen resulted in covalent bond formation whose subsequent rearrangement accounted for the formation of the products.
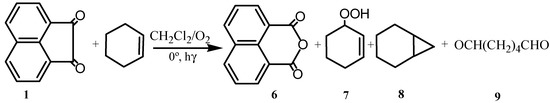
Scheme 2.
The acenaphthenequinone was cleaved electrochemically in presence of oxygen to give, after methylation, the corresponding ester of 1,8-naphthalene dicarboxylic acid [14]. Schmidt rearrangement of 1 with sodium azide gave naphthalic anhydride [15]. Reaction of 1 with the diazoalkanes 10 or 11 yielded 3-substituted-2-perinaphthen-2-o1-1-one 12 or 13, respectively (Scheme 3)[16]. The products could be extracted from the reaction mixture with dilute aqueous alkali in order to prevent undesirable side reactions [17] which led to difficulties in isolating the products.
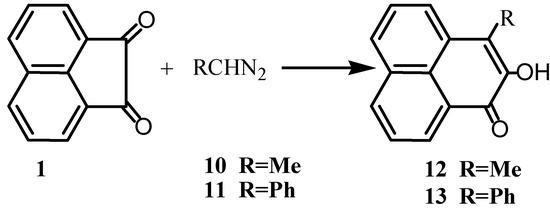
Scheme 3.
The cyanohydrin 14 undergoes facile base-catalyzed carbon-to-oxygen acy1 rearrangement to peri ring-expanded naphthalides 15 [18]. The proposed mechanism (Scheme 4) involved base-catalyzed formation of an intermediate α-oxanol followed by bridgehead carbon-carbon bond cleavage to an aromatic carbanion isoelectronic with the 14 π-electron phenaleny1 carbanion. The reaction could also be extended to other analogues of 14 where the CN is replaced by other substituents.
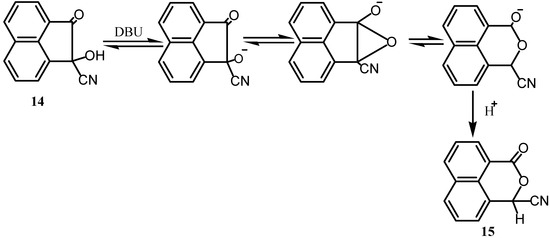
Scheme 4.
2.2 Reduction
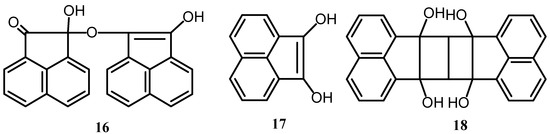
Acenaphthenequinone is easily reduced as a consequence of the involvement of its carbony1 groups in the conjugated system [19]. Treatment of 1 with iron in acetic acid, until a water soluble colorless compound is formed, yields easily soluble alkali salts, which are of a violet-blue color, in the presence of an excess of caustic alkali. Condensing the reduced products with 3-hydroxy-1-thionaphthene or indoxy1 derivatives gave vat-dyeing materials [20]. Reduction products of acenaphthenequinone were obtained by confining the reduction to the formation of compound 16 which is poorly soluble in water and yields with alkalies deep blue salts which are also poorly soluble in water (Scheme 5). On the other hand reduction of 1 could be carried until the formation of 17, which is soluble in water. It forms with excess of alkalies readily soluble violet-blue salts [21,22]. When acenaphthenequinone absorbed five moles of hydrogen, in the presence of platinum in aqueous ammonium hydroxide or dilute alkali [23], it yielded exclusively the bimolecular substance 18. Its catalytic hydrogenation in presence of nickel salts was also studied [24].
Scheme 5.
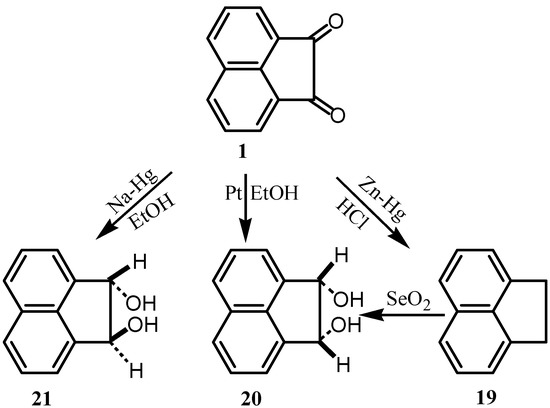
Clemmensen reduction of 1 with amalgamated zinc in hydrochloric acid gave acenaphthene 19 (Scheme 6) [25]. When the reduction was carried out with amalgamated sodium in ethanol, in an atmosphere of nitrogen, it gave 38% of the transglycol 21 [26]. The product did not give a condensation product with acetone and it did not decolorize bromine in warm chloroform. Catalytic reduction of 1 in presence of platinum in ethanol gave a mixture of cis-and trans acenaphthylene glycols (20 and 21). Reduction with LiA1H4 gave also the trans diol accompanied by the cis diol whose derivatives were prepared [27]. The cis-diol could be prepared by selenium dioxide oxidation of acenaphthene [25].
Scheme 6.
Treatment of 1 with alkali metals e.g. sodium and potassium in tetrahydrofuran gave three reduced forms [28], which behave like a monovalent, a divalent, and a trivalent base, respectively. It was found that tris(tripheny1phosphine)chlororhodium is an effective catalyst for the homogeneous reductive hydrosilylation of quinones [29] which offer an easy procedure for protecting the highly reactive quinonic moiety. Thus, reductive silylation of 1 with Et3SiH over tris(triphenylphosphine)-chlororhodium as a catalyst gave the bis(silyl) ethers of the hydroquinone which could be oxidatively desilylated with PhI(OAc) 2 [30]. The respective 1,2-bis(trimethy1siloxy)ethene analogue was prepared from reaction of 1 with hexamethyldisilane in presence of Pd or Pt catalyst [31].
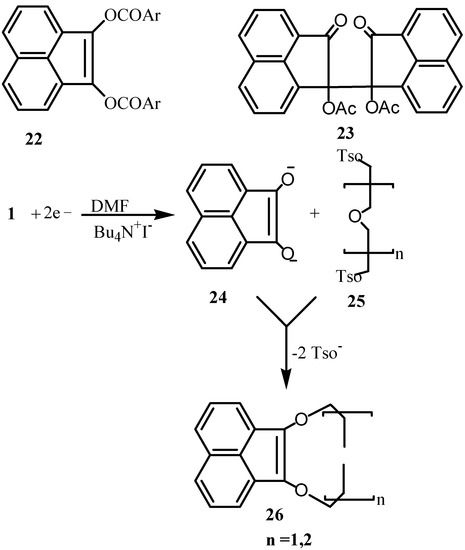
Electrochemical reductions of 1 at a mercury cathode were carried out under a constant potential, in presence of nonelectroactive aroy1 chlorides to give the 1,2-diaroyloxyacenaphthylene derivatives 22, in good yields (Scheme 7) [31,33]. Their formation corresponds to the transfer of an overall two-electron process. However, when acetic anhydride was used, a one-electron transfer process had taken place to give meso-bis (1-acetoxy-2-oxoacenaphthen-1-y1) (23). The structure of the last compound was determined by x-ray crystallography [32]. The effect of metal ions and solvents on the polarographic reduction of 1 was studied [34]. The dependence of limiting currents and half-wave potentials were determined [35]. The mechanism and kinetics of the polarographic reduction of 1 in DMF and in the presence of phenol as proton donor was found to involve 4-electrons in successive 1-electron steps [36]. Reductive methylation of 1 had taken place electrochemically in presence of methylhalides via coupling of the radical anion of 1 with the methyl radical [37]. Electrochemical reduction in DMF-Bu4NI gave a binucleophile 24, which underwent cyclization with 25 to give heterocyclic macrocycles 26 [38].
Scheme 7.
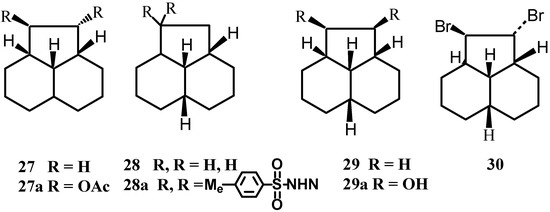
The three stereoisomers 27-29 of the six possible dodecahydroacenaphthylene were prepared (Scheme 8)[39]. The configurations were confirmed inter alia by X-ray analysis of the precursors.
Scheme 8.
2.3 Protonation
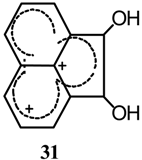
Protonation of acenaphthenequinone (1) gave a diprotonated species [40], the structure of which was determined as 31 by 1H- and 13C-NMR. The relative photochemical reactivity of acenaphthenequinone as an α-diketone was investigated in hydrogen donating solvents [41].
2.4 Reaction with active methylene compounds
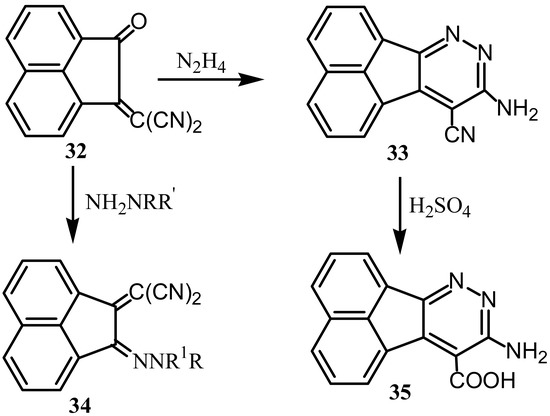
Reaction of 1 with malononitrile gave 1-(dicyanomethylene) acenaphthen-2-one (32) whose reaction with hydrazine gave 33 that hydrolyzed with sulfuric acid to give 35 (Scheme 9) [42]. Reaction of 32 with substituted hydrazines gave deeply colored hydrazones 34, which are classified as azacyanine types polymethine dyes [43]. Reaction of 1 with malononitrile in presence of bases yielded a blue compound namely 6β-hydroxy-8-imino-7, 8-dihydro-6β-H-cyclopenta[a] acenaphthylene-7,7,9-tricarbonitrile [44].
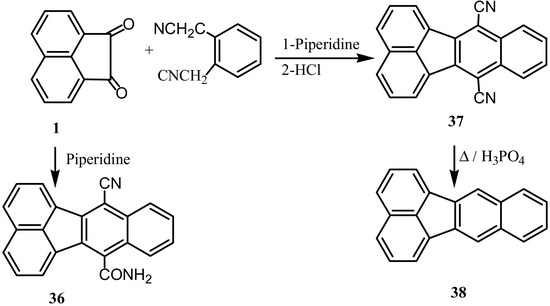
Reaction of 1 with o-phenylene diacetonitrile in presence of piperidine at room temperature [45,46] gave the dinaphthylenenitrile amide (36) and not the expected dinitrile 37 (Scheme 10). The product could not be hydrolyzed to the dicarboxylic acid; hydrolysis ceases at the diamide stage [47]. However, the cyclocondensation of o-phenylene diacetonitrile with 1 was reported in a more recent publication to give 37 that followed by decyanation to give benzo[k]fluoranthene (38) [48].
Scheme 9.
Scheme 10.
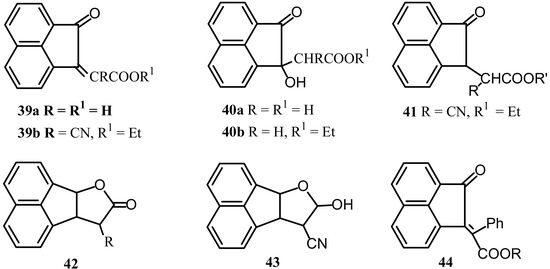
Condensation of ethyl cyanoacetate and malonic acid with 1 gave the acid 39a and the ester 39b, respectively (Scheme 11) [49]. Esterification of 40a gave the corresponding ester 40b. Dehydration of 40a gave 39a.
Hydrogenation of 39b in presence of Adams’ catalyst gave 41. Reduction of 41 gave either the lactone 42 (R=CN) or the hemiacetal 43 depending upon the conditions employed. The lactone (42, R=H) was prepared by reaction of hemiacetal 43 with hot alkali followed by acidification. Claisen-Stobbe condensation of 1 with phenylacetic esters afforded the benzylidene derivatives 44 [50].
Scheme 11.
Reaction of 1 with diethylacetone dicarboxylate gave the substituted cyclopentadienone (45) (Scheme 12) [51]. Reduction of 45 by zinc and acetic acid followed by hydrolysis and decarboxylation afforded the ketones 46, 47 and 48.
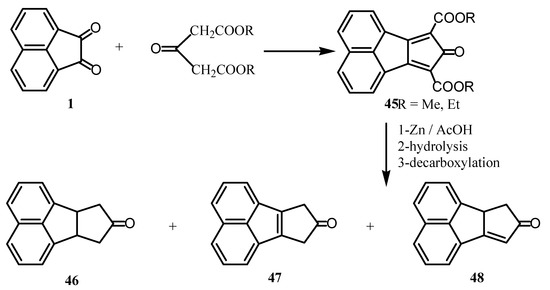
Scheme 12.
The ketone 46 is a valuable a precursor for the synthesis of peri-diketone 52 via the conversions to 49-52 (Scheme 13).
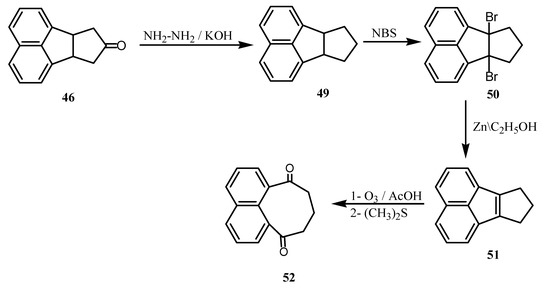
Scheme 13.
Condensation of acenaphthenequinone (1) with dimethyl pentenedioate (dimethyl glucatonate) [52] gave the epimeric diesters 53 and 54 (Scheme 14). The diester 53 was oxidized by lead tetraacetate to 55 whose reduction was effected by magnesium in methanol in order to reduce the conjugated 8,9‑double bond while preserving the keto-ester groups, whereby compounds 56, 57, and 58 were obtained.
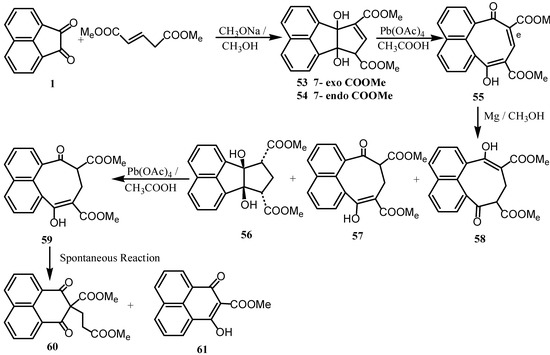
Scheme 14.
Reaction of 56 with lead tetraacetate gave 59, which could be a precursor for the peri-diketone 52, however, 59 was spontaneously transformed to 60 and 61 and consequently this approach for 52 was precluded. Acenaphthenequinone (1) was condensed with S(CH2CO2Et) 2 or p-NO2BnSCH2CO2Et in the presence of base to give tetrahydroacenaphthothiophenes 62a and 62b (Scheme 15). Dehydration of 62a in sulfuric acid or acetic anhydride gave acenaphtho[1,2-c]thiophene (64a) [53]. Heating of 64a with copper and quinoline gave 65 [54]. On the other hand, when 1 was treated with O(CH2CO2Et)2, it gave 63, which upon reaction with acetic anhydride gave 66 [55].
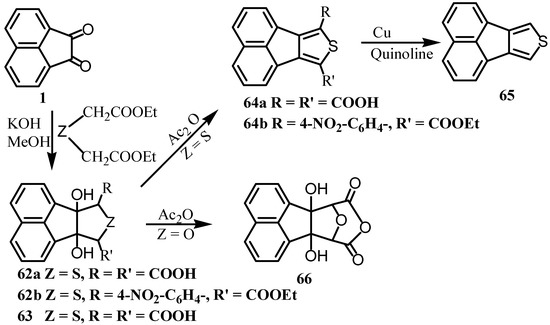
Scheme 15.
Condensation of 1 with 1,3-indandione (Scheme 16) gave 2,2-bis(1,3-indandion-2-y1) acenaphthene-1-one (67) [56,57,58]. The reaction of 1 and 2,2-dihydroxy-1,3-phenylenedione gave 1,8-naphthalic anhydride in high yield [59]. Condensation of 1 with the appropriate 2-propanones gave 68 [60,61].
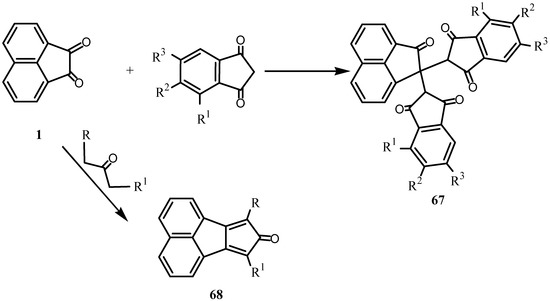
Scheme 16.
Reaction of acenaphthenequinone and 6-chloro-3-hydroxy-thionaphthene gave 2-(6-chloro-thionaphthene)acenaphthylene indigo [6-chloro-2-(2-oxo-1-acenphthylidene)-3(2H) thionaphthenone] which was used as a dye [62]. Similarly, condensation of substituted acenaphthenequinone with 3-hydroxythionaphthenes or indoxyl in the presence of a catalytic amount of hydrochloric acid gave 69 or its isomer which was not specifically identified (Scheme 17) [63,64,65,66]. Reaction of 1 or its halogen derivatives with thiohydantion [67], pseudothiohydantion [68] or rhodanine gave products of the type 70 or 71 [69].
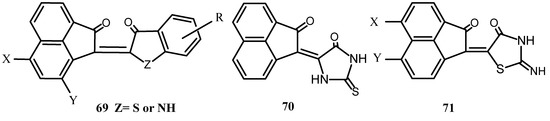
Scheme 17.
Reaction of 1 with 3,4–dehydro-DL-proline gave a product in which the pyrroline ring is converted into an N-substituted pyrrole [70]. Cyclocondensation of 2,3-dimethylquinoxaline-1,4-dioxide with 1 gave phenazine dioxide 72 (Scheme 18) [71]. When dialkyl-N-aminoazinium salts 73 or the quinazoline derivative 74 have been condensed with 1, they gave the new heterocycles 75 and 76, respectively, with a quaternary N in the bridgehead position (Scheme 19) [72,73].
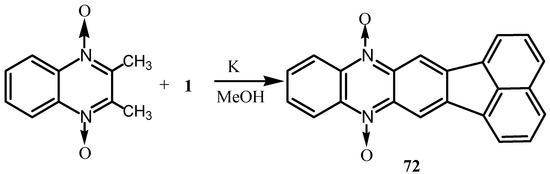
Scheme 18.
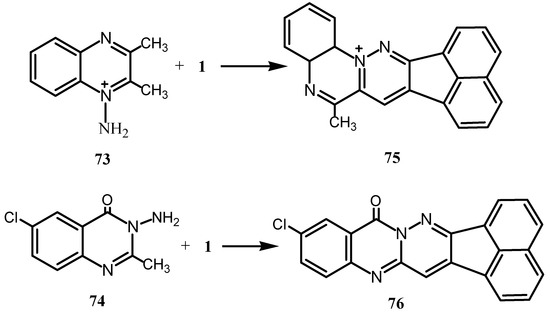
Scheme 19.
When 1 was treated with the furoxan derivative 77, an addition product was formed whose heating gave the diisocyanate 78 (Scheme 20) [74].
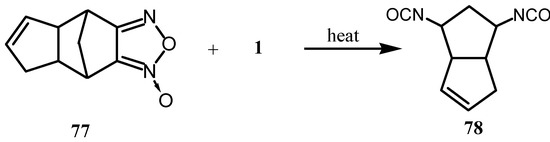
Scheme 20.
The Westphal condensation was used for the synthesis of different types of heterocycles from 1. Thus, condensation of 2-methylpyridinium, quinolinium or isoquinolinium salts with 1 in presence of a sodium acetate yielded the quinolizium salts 79 [75,76]. When 1-ethoxycarbonylmethyl-2, 6-dimethylpyridinium salt was heated with acenaphthenequinone in the presence of di-n-butylamine, deep purple precipitate of [2,3,3]cyclizin -1-one derivatives 80 were formed whose formation was rationalized as presented in Scheme 21. All cyclazinone derivatives 80 were isolated as hydrobromides 81 [77].
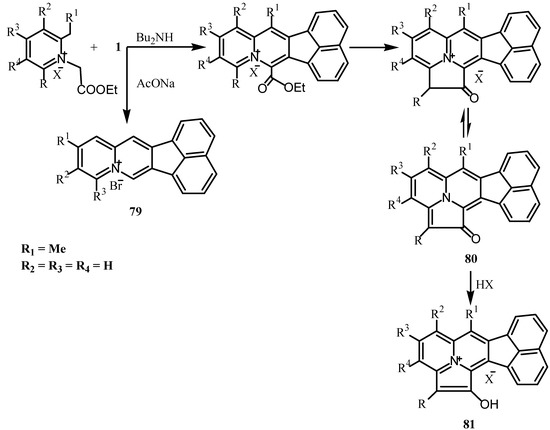
Scheme 21.
The [2,3,3] cyclazin-6-one was prepared by condensation of acetoxymethylpyridinium bromide and 1 in presence of sodium acetate to yield the 4-ethoxycarbonylquinolizinium-1-olate (82) which upon reaction with hydrobromic acid produced 83 (Scheme 22). Treatment of the latter with sodium carbonate followed by DMAD gave [2,3,3] cyclazin-6-one derivative 84.
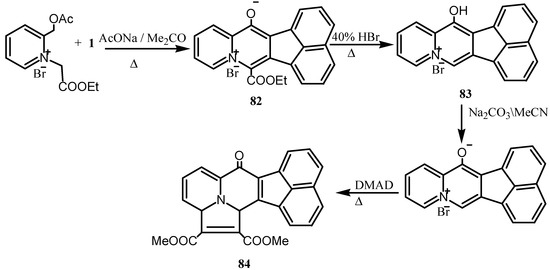
Scheme 22.
The Westphal condensation was also used for the synthesis of π-donor-π-acceptor heterocycles such as pyridopyrrolopyrazinium 86 by the condensation of 1 with pyrrolopyrazinium compounds 85 (Scheme 23) [78]. Similarly, the 2-methylthiazolium salts (87) were used as 1,4-dinucleophiles for the synthesis of thiazolo [3,2-a] pyridinium salts 88 [79].
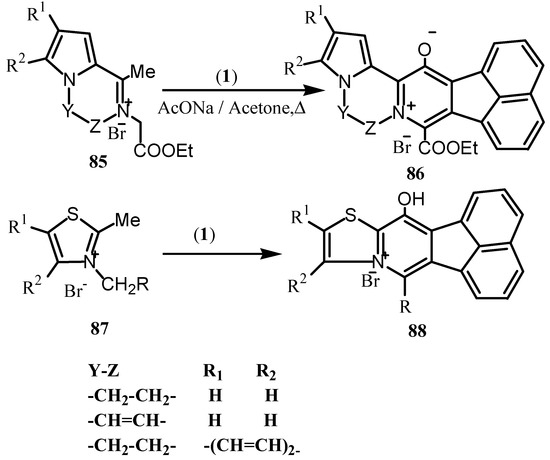
Scheme 23.
Condensation of 1 with 2-alkyl-1-aminopyridinium, quinolinium or 1-alkyl-2- aminoisoquinolinium salts gave in presence of base, pyrido[1,2-b] pyridazinium salts 89 in good yield [80]. Similarly, the pyridazinopyrrolopyrazinium derivatives 91 were prepared from 90 (Scheme 24) [80].
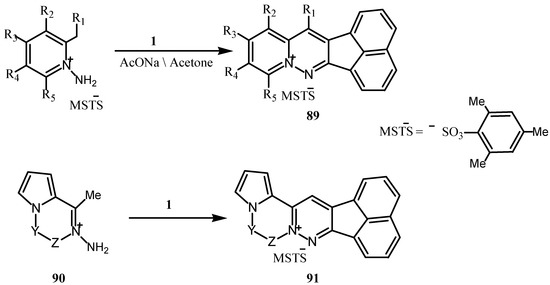
Scheme 24.
Reaction of 1 with nitromethane in alkali followed by acidification gave an adduct C13H9NO4 which was formed by a 1,2-addition on one of the carbonyl groups [81].
2.5. Reaction with aldehydes and ketones
It has been shown that 1 could be condensed with aldehydes in a general manner to afford 92 that gave a violet-red color with concentrated sulfuric acid (Scheme 25) [82]. Reaction of acenaphthenequinone with acetone in presence of potassium hydroxide gave mono-acetoneacenaphthenequinone [83]. Reaction of 1 with p-chlorobenzaldehyde, p-acetamidobenzaldehyde, o-nitrobenzaldehyde in presence of ammonia gave oxazoles 99 at 0 ºC and imidazoles 100 at higher temperatures. On the other hand, o-chloro-benzaldehyde, o-hydroxybenzaldehyde and m-hydroxybenzaldehyde under similar condition gave a mixture of oxazoles and imidazoles, which cannot be separated [83], and at higher temperatures, only imidazoles were obtained. p-nitrobenzaldehyde, p-hydroxybenzaldehyde and p-methoxybenzaldehyde gave only imidazoles. Vanillin and p-bromosalicyladehyde react very slightly at 0ºC, but at higher temperature imidazoles 98 were formed. Heating the corresponding oxazole with ammonia in a sealed tube caused a partial conversion into the imidazole. When the reaction of 1 with o-hydroxybenzaldehyde was exposed to light for one month, it gave the monosalicylyl derivative of acenaphthenequinol [83].
When a suspension of 1 in isoamyl alcohol or anhydrous ethanol was treated with benzaldehyde in presence of ammonia, a variety of products 93-97 were obtained depending upon the condition of the reaction [84]. The structure of 97 was though to be either 97a or 97b. Its hydrolysis with dilute hydrochloric acid gave 1, naphthylimide and an unidentified compound C12H8N2O.Substituted benzaldehydes were also used. Reaction of 1 with various aldehydes in boiling ammonium hydroxide and in dry ammonia gave ary1 acenaphthimidazoles [83]. When dry ammonia was passed through a hot solution of 1 and p-acetamidobenzaldehyde in ammonium hydroxide, 4-acetyl-amino-2-phenyl-acenaphthoxazole (99), and the iminazole (100, R=NHAc) were obtained [85]. Treatment of 1 with ammonium acetate in presence of p-nitrobenzaldehyde gave (101,R=NO2) [86]. It may be supposed that oxazoles are first formed which by subsequent replacement of the ring oxygen atom by NH forms the iminazoles.
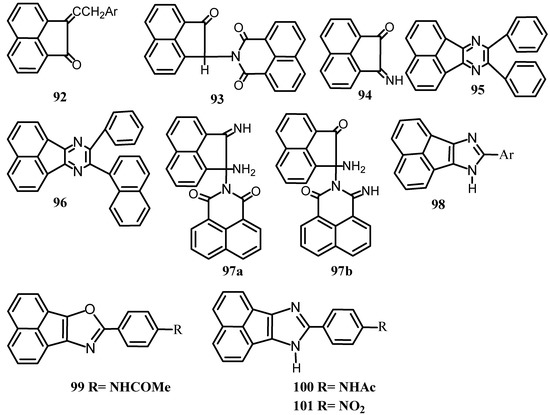
Scheme 25.
2.6. Reaction with Wittig reagents
Reaction of 1 with several Wittig reagents has been studied [87]. Thus, its reaction with equimolar amount of benzylidenetriphenylphosphorane at room temperature gave the corresponding benzylidene-acenaphthenones in fairly good yields. When the reaction of 1 was done with two molar equivalents of benzylidenetriphenylphosphorane under severe conditions it afforded also the benzylidene-acenaphthenone and no dibenzylidene derivative could be obtained. When 1 was reacted with 3‑(methoxyphenethyl) triphenylphosphonium bromide followed by cyclization-dehydration of the intermediate 102 gave 10-methoxybenzo[j]fluoranthene (103) exclusively (Scheme 26) [88].
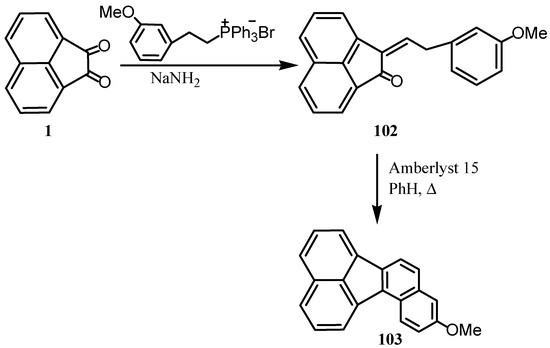
Scheme 26.
The adducts 104 were isolated from the reaction of 1 with triethyl phosphonoacetate (Scheme 27) [89]. The reaction of 1 with a resonance-stabilized phosphorane [90], such as acetonylidene-, phenacylidene-and p-chlorophenacylidene-phosphoranes afforded the expected α,β-unsaturated ketones 105. The ethoxycarbonyl-methylene acenaphthenone was obtained from the reaction of 1 with diethyl ethoxycarbonyl methyl phosphonate. Methylene-phosphorane was reacted with 1 to give methyleneacenaphthenone in poor yield. On the contrary, the reaction with ethylidenephosphorane gave 2,2’-methylenebisacenaphthenone, which was also formed by the reaction of acenaphthenone with glyoxal, in good yield.
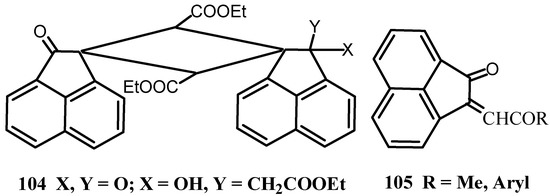
Scheme 27.
When equimolar amounts of the bisphosphonium salt 106 and 1 were treated with aqueous 5 M-lithium hydroxide (Scheme 28), 8,10-dimethylfluorantheno[8,9-c]thiophene (110) and 2-(2,4,5-trimethyl-3-thienyl-idene)acenaphthylen-2-one (111) were obtained [91]. Compound 111 was isolated in only one configuration and its formation may arise either via the o-quinomethaneylide intermediate 109 or the monoylide 108. Intramolecular Witting reaction of 109 gave 110.
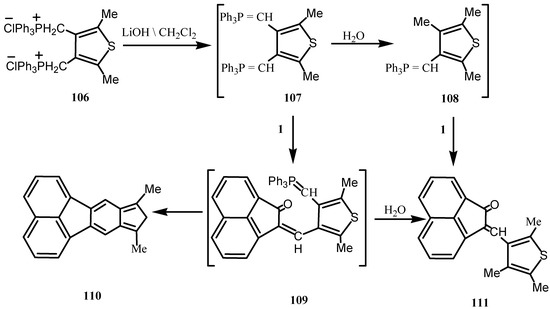
Scheme 28.
Wittig reaction of bis(triphenylphosphonium) dibromides with 1 under phase-transfer conditions gave 112 (Scheme 29) [92].
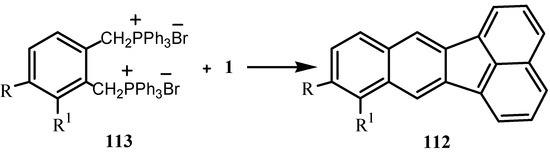
Scheme 29.
Reaction of 113 with hydroxylamine gave the oxime 114 and the pyrrole derivative 115, whereas its reaction with hydrazines afforded the pyridazinones 116 (Scheme 30) [93]. Reaction of the monoxime of 1 with Ph3P=CHCO2Me afforded stereoisomeric products of 114, whereas the reaction with ylide Ph3P=CHCOMe gave the pyridine derivatives 118. Hydrogenation of 114 and thermal cyclization gave the polycyclic compounds 117 [94].
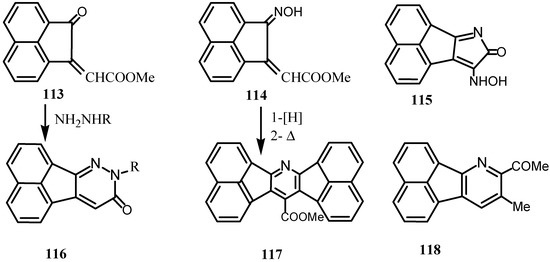
Scheme 30.
Spiro acenaphthyleneisoxazoles 119 were prepared by the regioselective 1,3-dipolar cycloaddition reaction of 113 with nitrile oxides (Scheme 31) [95]. The reaction of 1 with nitrile oxide gave acenaphthylenedioxazoles 120, which underwent a Wittig reaction with Ph3P=CHCO2Et to give 121. The cycloaddition reaction of 120 with PhCNO gave a mixture of the dispiro compounds 122 and 123. Catalytic hydrogenation of 121-123 over Pd/C cleaved the dioxazole rings, whereas the isoxazole ring of 119 was cleaved by reduction over Raney nickel or by treatment with sodium ethoxide.
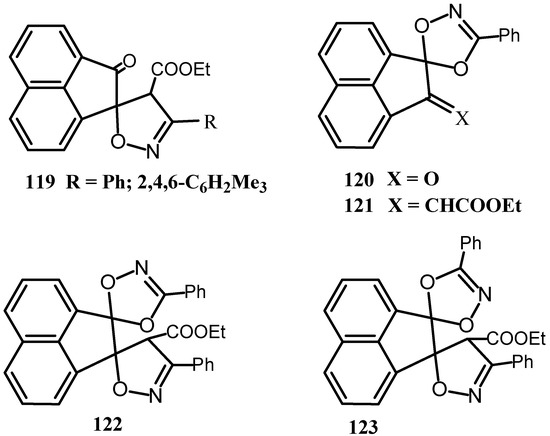
Scheme 31.
2.7. Reaction with magnesium and lithium reagents
Various reports on the reaction of Grignard reagents with 1 have been published [96,97,98,99,100,101,102]. Thus, reaction of 1 with EtMgBr gave 1,2-diethylacenaphthoglycol 124 [96] whose dehydration with acid gave 1,2-diethylideneacenaphthene 125 (Scheme 32). The latter could be oxidized back to 1 with sodium dichromate in acetic acid.
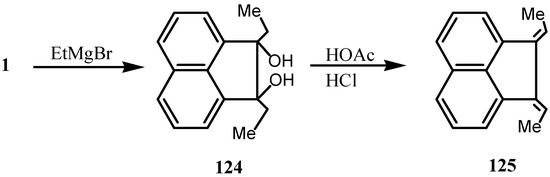
Scheme 32.
The dehydration behavior of 124 is unlike that of the diphenyl derivative, which gave a pinacoline under this treatment [96]. Reaction of 1 with arylmagnesium bromides gave 7,8-diarylnaphthenediols (126) (Scheme 33) [97,98] in which the simple aryl group migrates exclusively to give the 7,7-diaryl-acenaphthenones (127) [99]. The latter could be cleaved into the naphthoic acid derivatives 128 by alkali. Some derivatives of 126 gave low yields of 127 due to the formation of 131. The latter were prepared from 126 via 130. Ring opening of 126 was achieved by chromic acid to give the aroyl naphthalene 129 [99,100,101,102,103].
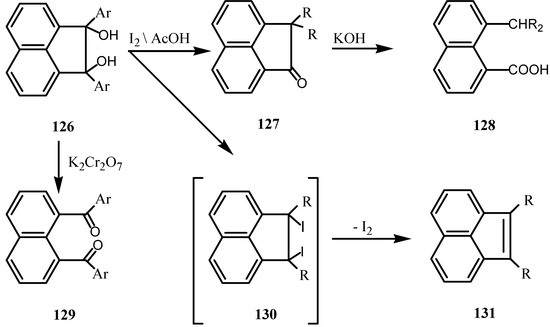
Scheme 33.
Reaction of acenaphthenequinone with several organolithium and organomagnesium reagents gave 132 (Scheme 34) [104]. This and its derived pinacol rearrangement product failed to cyclize under a variety of the acidic conditions, perhaps due to the poor stereoelectronic alignment for cyclization. On the other hand, when a less rigid analogue such as l-lithio-3,4-dihydronaphthalene was used the tricyclo[4.3.0.0]nonane 133 was formed by di-oxy-Cope rearrangement followed by the unprecedented criss-cross 2π+2π cycloaddition of the two enolate ions formed [104]. The structure was confirmed by X-ray crystallography.
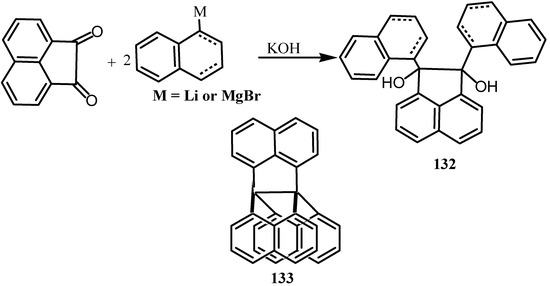
Scheme 34.
The addition of mesityl or triisopropyl magnesium bromide to 1 led (via a single electron transfer) to the formation of the corresponding semiquinone [105] to give 134 that upon reduction with lithium aluminum hydride gave 135 (Scheme 35). Reaction of the latter with phenyl lithium gave 136 that upon Birch reduction gave the diaryl derivative 137 [106]. Reaction of vinyl chloride with 134 gave the corresponding vinyl ethers [107].
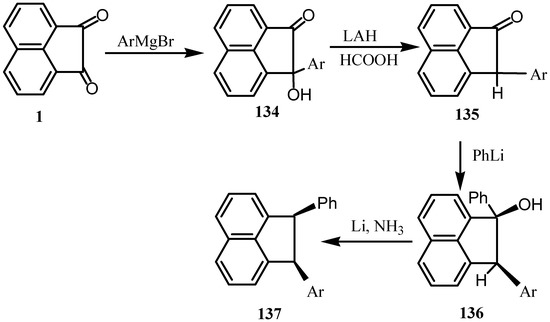
Scheme 35.
When 1,8-diiodonaphthalene and 5, 6-dibromoacenaphthene were lithiatied and treated with acenaphthenequinone ,and then the diol cycloaddition products were treated with hydrofluoric acid, acenaphth[1,2-a]acenaphthylenes, 138-140 were obtained (Scheme 36) [108].
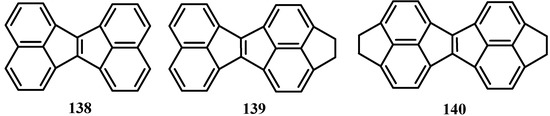
Scheme 36.
2.8. Friedel Crafts reaction
Condensation of acenaphthenequinone with benzene in presence of aluminum chloride gave 1,1-diphenyl-2-acenaphthenone (141) (Scheme 37) [109]. Boiling alcoholic potassium hydroxide converts 141 into 8-diphenylmethyl-1-naphthoic acid (142). Its distillation with barium hydroxide gave diphenyl naphthylmethane (143) whereas its treatment with chromium trioxide in acetic aid gave the lactone derivative 144.
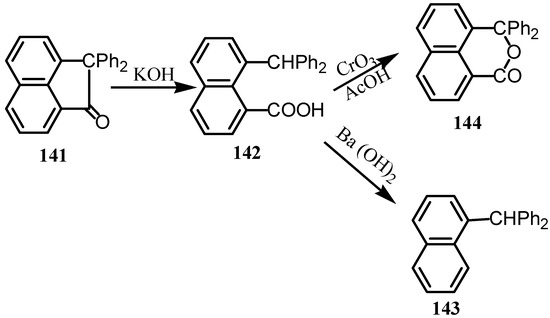
Scheme 37.
2.9. Diels-Alder reaction
Photochemical Diel’s-Alder reaction of acenaphthenequinone with olefin led to the formation of 145 and 146 (Scheme 38) [110].
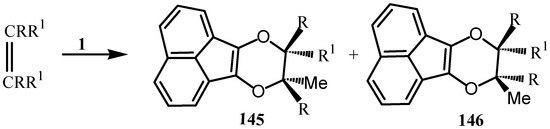
Scheme 38.
2.10. Reaction with phenolic compounds
The reaction of phenol with 1 was successful in presence of concentrated sulfuric acid [111,112]. Since the para-position is the most reactive in phenol, the product may probably be given the structure 1,1-bis [4’-hydroxyphenyl]-2-acenaphthenone (147) (Scheme 39). Reaction of 1 with resorcinol in presence of zinc chloride gave 148 [109], and the reaction with hydroquinone gave 149 [111]. To explain the formation of such anhydro derivatives it must be assumed that the oxygen of the quinone reacts with a hydrogen atom in the o-position to one of the hydroxyl groups in the dihydroxy compound [110]. The compound from catechol formed no anhydride and as the hydrogen atom in the p-position to one of the hydroxy groups in the catechol is the most reactive, the product must be 1,1-bis[3’,4’-dihydroxyphenyl]-2-acenaphthenone. Condensation of acenaphthenequinone with asymmetric o‑xylenol, or gave 150. Thymol yields dithymol acenaphthenone. Nitrophenols reacted more sluggishly with 1 and a large excess of the phenol and the condensing agent was required [114]. This was explained on the basis of theoretical electrostatic considerations. Reaction with o-nitrophenol gave 1,1-bis(3-nitro-4-hydroxy-phenyl)-2-acenaphthenone, and with p-nitrophenol gave anhydro-1,1-bis(5-nitro-2-hydroxyphenyl)-2-acenaphthenone.
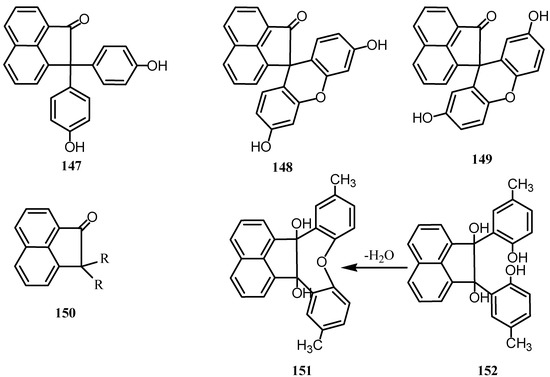
Scheme 39.
It was found that condensation of acenaphthenequinone with cresols and naphthols does not always give, like mono-and dihydric phenols, compounds of the type 150. Thus, reaction of 1 with p-cresol, gave 151 and 152; the relative amounts of which depend on the amount of sulfuric acid used as condensing agent [115].
2.11. Halogenation reactions
Treatment of 1 with N-bromosuccinimide in polar solvents gave 5-bromoacenaphthenequinone [116]. Reaction of 1 with bromine gave the 3-bromo derivative [114]. No further bromination could be effected with bromine alone but in presence of iron filings, acenaphthenequinone gave the 2,3,5-tribromo derivative. Prolonged bromination of 1 in presence of iron gave the 2,3,4,5-tetrabromo derivative. The position of the bromine atoms has not been definitely determined [117]. On the other hand, treatment of 1 with bromine in chlorobenzene as a solvent gave 1,8-naphthalic anhydride whereas the reaction did not take place in nitrobenzene [118].
Haloacenaphthenes or haloacenaphthylenes were successively brominated, dehydrobrominated, chlorinated and hydrolyzed with sulfuric acid to give haloacenaphthenequinones [119]. Reaction of 1 with PCl5 gave dichloroacenaphthenone whose reduction with powdered Fe in glacial acetic acid gave acenaphthenone (Scheme 40) [120,121]. Haloacenaphthenequinones 153 were prepared from the corresponding tetrachlorides 154 by successive reaction with sulfuric acid and sodium sulfite and hydrolysis (Scheme 40). The tetrachloro derivatives 154 were prepared by chlorination [122] of dibromo derivatives 155, which were prepared by bromination of 156.
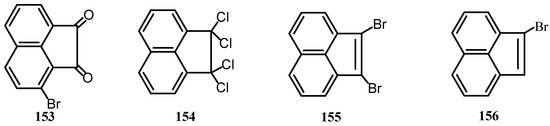
Scheme 40.
3-Bromo- and 3-iodoacenaphthenequinone were obtained from the 2–bromo- and 2-iodo-acenaphthenes, respectively by oxidation with sodium dichromate [63]. Reaction of 3-iodo-acenaphthenequinone and sodium dichromate in acetic acid gave 2-iodo-1,8-naphthalic anhydride.
2.12. Thiation reactions
Acenaphthenequinone underwent mono thiation [123] on treatment with dithiadiphosphetane disulfide 157.
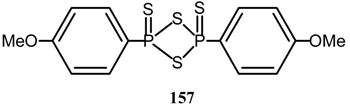
2.13. Alkylation reactions
When acenaphthenequinone was treated with sodium in dry tetrahydrofuran, followed by 1,4-dichlorobutane], the product was 1,4-dioxacine derivative 158 (Scheme 41) [124]. Mercuration of acenaphthene-quinone under various conditions was failed [125]. Irradiation of a solution of 1 in acetonitrile in presence of allylic stannanes afforded homoallylic alcohols in good yields. When unsymmetrical allylstannens were used, the allylic groups were introduced predominantly at the α-positions. Complete regioselective introduction could be achieved by irradiation in presence of sodium hydroxide or cobalt chloride [126]. Reaction of 1 with trialkylallyltin gave the corresponding allylhydroquinone 159 [127] which was catalyzed by the Lewis acid [127].
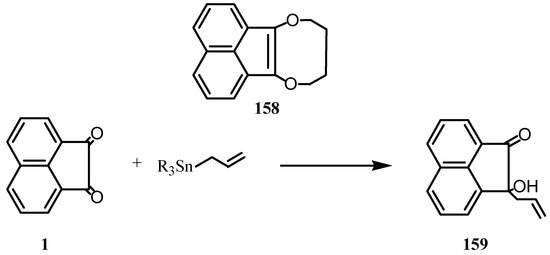
Scheme 41.
The photoaddition of 1 to cycloheptatriene gave various cycloadducts, (2+2)π-, and (2+6)π- cycloadducts together with ene product [128]. Irradiation of 1 in benzene in the presence of 2,3-dimethyl-2-butene led to a facile formation of a single photoproduct (Scheme 42) [129].
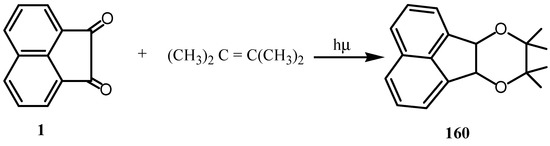
Scheme 42.
The photoaddition reaction of 1 with  -silyl n-electron donors via triplet single electron transfer desilylation and triplet hydrogens abstraction pathways was explored [129]. Thus, photoaddition of Et2NCH2SiMe3 to 1 produced 2-hydroxy-2-[(diethylamino) methyl] acenaphthylen-1-one (161), whereas the photoaddition of n-PrSCH2SiMe3 to 1 generates two photoproducts 162 and 163 along with a photoreduction dimer of 1 (Scheme 43).
-silyl n-electron donors via triplet single electron transfer desilylation and triplet hydrogens abstraction pathways was explored [129]. Thus, photoaddition of Et2NCH2SiMe3 to 1 produced 2-hydroxy-2-[(diethylamino) methyl] acenaphthylen-1-one (161), whereas the photoaddition of n-PrSCH2SiMe3 to 1 generates two photoproducts 162 and 163 along with a photoreduction dimer of 1 (Scheme 43).
-silyl n-electron donors via triplet single electron transfer desilylation and triplet hydrogens abstraction pathways was explored [129]. Thus, photoaddition of Et2NCH2SiMe3 to 1 produced 2-hydroxy-2-[(diethylamino) methyl] acenaphthylen-1-one (161), whereas the photoaddition of n-PrSCH2SiMe3 to 1 generates two photoproducts 162 and 163 along with a photoreduction dimer of 1 (Scheme 43).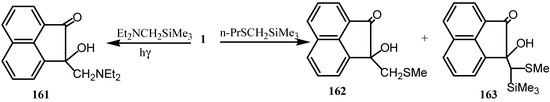
Scheme 43.
2.14. Ketal derivatives
The products of the reaction of 1 and ethylene glycol in benzene were identified by using mass spectroscopy [130,131]. The least polar compound was 164 whereas the products of the highest and intermediate polarity were 165 and 166, respectively. Reaction of simple mercaptans with 1 gave monomercaptols (Scheme 44) [132].
The antiphlogistic compound acenphth[1,2-b]oxazole-8-propionic acid (167) was prepared from 1 via its monoethylene ketal and 1-hydroxy-2-acenaphthenone and subsequent esterification with succinic anhydride and reaction with ammonium acetate [133].
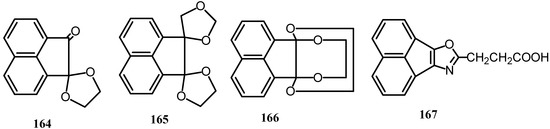
Scheme 44.
Condensation of 1 with N-(hydroxymethyl) trichloroacetamide gave 1,2-dioxo-4-trichloroacetyl-aminomethyl acenaphthene that was oxidized by dilute nitric acid in a sealed tube to give a C6H2(CO2H)4 ( prehnitic acid) [2].
2.15. Reaction with phosphites and phospholanes
Reaction of 1 with dialkyl phosphites yielded the phosphonates 168 [134] whose heating gave the starting quinone. Treatment of 168 with hydrogen peroxide-sodium hydroxide gave naphthalene-1, 8-dicarboxylic acid (Scheme 45).
The reaction of P(OMe)3 with 1 under air afforded P(O)(OMe)3 and a 1:2 adduct 169 which was rearranged into the δ-lactone 170 by addition of water, while 169 was only obtained quantitatively under nitrogen atmosphere [135,136]. E.S.R and U.V spectra, decolorization of 1,1-diphenyl-2-picryl-hydrazyl, and initiation of styrene polymerization suggest the transient formation of radical ions. A mechanism, which involves one-electron transfer from phosphite to 1 followed by autoxidation, was proposed for the reaction under air [135]. The kinetic of the reaction to form a 1:1 adduct that cyclized was studied in anhydrous dioxan [137]. Treatment of 1 with sodium in tetrahydrofuran followed by Cl2P (X) OR gave a fused di-oxophospholes 171 [138].
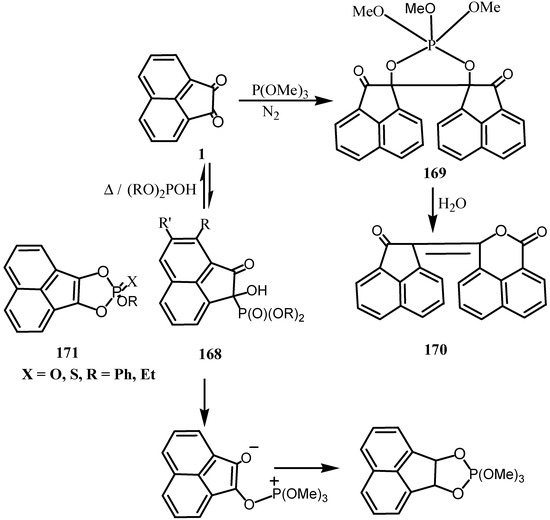
Scheme 45.
The reaction of 1 with 2-N-pyrrolidino-1,3-dimethyl-1,3,2-diazaphospholane (172) was quite vigorous in methylene chloride solution even at -70°C (Scheme 46) [139]. When the solution was allowed to reach 20°C, a deep brown mixture was produced from which the only isolable product was 2-N-pyrrolidino-2-oxo-1,3-dimethyl-1,3,2-diazaphospholane (176). A similar behavior was noted when 1 was treated with 2-dimethylamino-1,3-dimethyl-1,3,2-diazaphospholane (173). The only isolable product was 2-dimethylamino-2-oxo-1,3-dimethyl-1,3,2-diazaphospholane (177). No intermediate could be detected in these reactions, but it was assumed by analogy with previous reactions that the phosphorus of the cyclic aminophosphines attacked the oxygen of 1 to give a 1:1 dipolar adduct 174 or 175. The dipolar adduct apparently was too unstable to form the cyclic phospholene analogous to those of benzil. Instead, the 1:1dipolar adduct lost phosphoramidate to yield a carbenoid fragment 178 which underwent further transformations. Reaction of the diazides of 1 with triphenylphosphine gave the phophazine (179) [140].
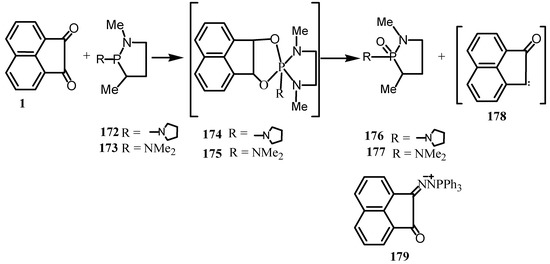
Scheme 46.
2.16. Reaction with carbenes
Addition of 1,3-diphenylimidazolidin-2-yildene to 1 gave 180 (Scheme 47) [141].
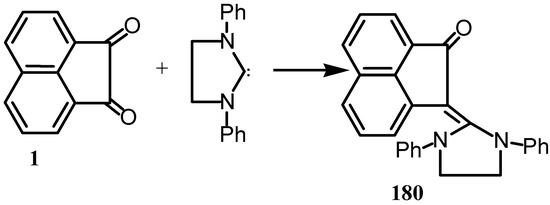
Scheme 47.
2.17. Decarbonylation
Stepwise elimination of carbonyl groups occurs when a vapor of 1 was passed through glow discharge plasmas to give 1,8-dehydronaphthalene, part of which dimerizes to give perylene [142].
2.18. Reaction with nitrogen nucleophiles
The reactions of 1 with nitrogen nucleophiles namely ammonia, amines, urea, hydroxylamines, aminoacids, o-diamines and hydrazines are included in the previous review [1]. These reactions produce heterocyclic compounds or products that are precursors for the synthesis of heterocyclic compounds. Various types of heterocyclic compounds could be prepared via the use of such nitrogen nucleophiles.
References
- El Ashry, E. S.; Abdel Hamid, H.; Shoukry, M. Ind. J. Heterocycl. Chem. 1998, 7, 313.
- De Diesbach, H.; Lachat, P.; Poggi, M.; Baladi , B.; Friderich, R.; Walker, H. Helv. Chim. Acta 1940, 23, 1232.
- Bader, H.; Chiang, Y. H. U.S. Pat. 1974. 3,812,115; [Chem. Abstr. 1974, 81, 25456p].
- Bader, H.; Chiang, Y. H. Synthesis 1976, 4, 249.
- Proskuryakov, V. A.; Chistyakov, A. .N.; Soboleva, T. P. U.S.S.R. Pat. 1971.
- Randall, R. B.; Benger, M.; Groocock, C.M. Proc. Roy. Soc. 1938, A165, 432, [Chem. Abstr. 1938, 32, 5374].
- Tkacheva, G. d.; Suvorov, B. V. Izv. Akad. Nauk Kaz. SSR, Ser. Khim. 1987, 3, 63, [Chem. Abstr., 1988, 108, 94341k]..
- Tsuji, J.; Kobayashi, Y. Jpn. Pat. 1971, 71, 18–969, [Chem. Abstr., 1972, 76, 34020r].
- Kojima, H.; Takahashi, S.; Hagihara, N. Tetrahedron Lett. 1973, 22, 1991.
- Tabuchi,, T.; Nojima, M. J. Org. Chem. 1991, 56, 6591.
- Kasiwagi, I. Bull. Chem. Soc. Jpn. 1926, 1, 66.
- J-Younge, Koo; Schuster, G. B. J. Org. Chem. 1979, 44, 847.
- Schuster, G. B.; Ja-Young, K. Gov. Rep. Announce Index (U.S.) 1978, 78, 116, [Chem. Abstr. 1979, 90, 18601w].
- Boujlel, K.; Simonet, J. Tetrahedron Lett. 1979, 12, 1063.
- Edwards, W. G. H; Petrow, V. J. Chem. Soc. 1948, 1713.
- Eistert, B.; Schoenberg, A. Ber. 1962, 95, 2416.
- Eistert, B.; Selzer, V. Ber. 1963, 96, 314.
- Miller, A. R. J. Org. Chem. 1979, 44, 1931.
- Schwabe, I. K.; Berg, H. Z. ElektroChem. 1952, 56, 952, [Chem. Abstr. 1954, 48, 2005].
- Elbel, K.; Biebrich. U.S. Pat. 1910. 965,170; [Chem. Abstr. 1910, 4, 2738].
- Kalle and co. Ger. Pat. 1909, 224, 979, [Chem. Abstr. 1911, 5, 213].
- Kalle and co. Brit.Pat. 1909, 21, 579, [Chem. Abstr. 1911, 5, 1336].
- Skita, A. Ber. 1927, 60, 2522.
- Braun, J.V.; Bayer, O. Ber. 1926, 59B, 920.
- Goldestien, H.; Glauser, W. Helv. Chim. Acta 1934, 17, 788.
- Jack, K. M.; Rule, H. G. J. Chem. Soc. 1938, 188.Ghigi, E. Gazz. Chim. Ital. 1938, 68, 184, [Chem. Abstr., 1938, 32, 7910].Graebe, C.; Jequier, E. Ann. 1896, 290, 202.
- Trevoy, L.W.; Brown, W. G. J. Am. Chem. Soc. 1949, 71, 1675.
- Panaiotov, I. M.; Rashkov, I. B. Dokl. Bolg. Akad. Nauk 1968, 21, 885, [Chem. Abstr. 1969, 70, 28277q].
- Bakola-Christianopoulou, M. N. J. Organomet. Chem. 1986, 308, C24.
- Bakola-Christianopoulou, M. N. J. Mol. Catal. 1991, 65, 307, [Chem. Abstr. 1991, 115, 28843u].
- Yamashita, H.; Reddy, N. P.; Tanka, M. Chem. Lett. 1993, 2, 315.
- Guirado, A.; Barba, F. ; Hursthouse, M. B.; Arcas, A. J. Org. Chem. 1989, 54, 3205.
- Guirado, A.; Barba, F.; Tevar, A. Synth. Commun. 1984, 14, 333, [Chem. Abstr. 1984, 101, 190629m].
- Kalinwaski, M. K.; Tenderende-Guminska, B. J. Electroanal. Chem. Interfacial Electrochem. 1974, 55, 277, [Chem. Abstr. 1975, 82, 36634s].
- Rozhnova, T. K.; Serazetdinova, V. A.; Sembaev, D. Kh.; Suvorov, B. V. Zh. Anal. Khim. 1975, 30, 2462, [Chem. Abstr. 1976, 85, 71789e].
- Ghe, A. M.; Valcher, S.; Del Monte, M. G. Ann. Chim. (Rome) 1970, 60, 729, [Chem. Abstr. 1971, 75, 44255q].
- Boujlel, K.; Simonet, J. Tetrahedron Lett. 1979, 17, 1497.
- Simonet, J.; Lund, H. Bull. Soc. Chim. Fr. 1975, 2547, [Chem. Abstr. 1976, 85, 77232q].
- Boldt, P.; Arensmann, E.; Blenkle, M.; Kersten, H.; Tendler, H.; Trog, R. S.; Jones, P. G.; Doring, D. Chem. Ber. 1992, 125, 1147.
- Bruck, D.; Minsky, A.; Dagan, A.; Rabinovitz, M. Tetrahedron Lett. 1981, 22, 3545.
- Murayama, K.; Ono, K.; Osugi, J. Bull. Chem. Soc. Jpn. 1972, 45, 847.
- Junek, H.; Hamboeck, H.; Hornischer, B. Monatsh. Chem. 1967, 98, 315.
- Hans, J.; Albin, H.; Andre, B. M. Monatsh. Chem. 1975, 106, 715.
- Junek, H.; Hornischer, B.; Sterk, H. Monatsh. Chem. 1968, 99, 2121.
- Moureu, H.; Chovin, P.; Rivoal, G. Bull. Soc. Chim. France 1946, 106, [Chem. Abstr. 1946, 40, 6070].
- Moureu, H.; Chovin, P.; Rivoal, G. Compt. Rend. 1946, 223, 951, [Chem. Abstr. 1947, 41, 2032].
- Moureu, H.; Chovin, P.; Rivoal, G. Bull. Soc. Chim. France 1948, 99.
- Vickery, E. H.; Eisenbraun, E. J. Org. Prep. Proced. Int. 1979, 11, 259, [Chem. Abstr. 1980, 92, 94121q.
- Bedford, M. J.; Crombie, D. A. Proc. R. Soc. Edinburgh, Sect. A 1974, 71A, 279, [Chem. Abstr. 1975, 82, 97871p.
- Patwardhan, B. H.; Bagavant, G. Indian. J. Chem. 1973, 11, 1333.
- Tucker, S. H. J. Chem. Soc. 1958, 1462.
- Jackson, D. A.; Lacy, Ph. H.; Smith, D. C. C. J. Chem. Soc., Perkin Trans.1 1989, 215.
- Koshelev, V. I.; Palkidin, V. L. Zh. Org. Khim. 1973, 9, 597, [Chem. Abstr. 1973, 79, 5199n].
- Birch, A.; Crombie, D. A. Chem. Ind.(London) 1971, 6, 177.
- Rangnekar, D. W.; Mavalankar, S. V. J. Heterocycl. Chem. 1991, 28, 1455.
- Vanags, G.; Geita, L. Zhur. Obshchei Khim. 1956, 26, 511, [Chem. Abstr. 1956, 50, 13852a].
- Geita, L.; Vanags, G. Latvijas PSR Zinatnu Akad. Vestis 1958, 127, [Chem. Abstr. 1959, 53, 11371d].
- Vanags, G.; Geita, L. J. Gen. Chem. U.S.S.R. 1956, 26, 539, [Chem. Abstr. 1957, 51, 2686g].
- Ezoe, K.; Kurosawa, K. Bull. Chem. Soc. Jpn. 1977, 50, 443.
- Banerjee, P. K.; Bhattacharya, A. J. J. Indian J. Chem., Sect. B 1977, 15B, 953.
- Samanta, S. R.; Mukherjee, A. K.; Battacharyya, A. J. Curr. Sci. 1988, 57, 926.
- Guha, S. K.; Sinha, A. K. J. Indian Chem. Soc. 1952, 29, 415.
- Karishin, A. P.; Krivoshapko, N. G.; Osobik, D. I. Khim. Geterotsikl. S oedin 1968, 1, 61, [Chem. Abstr. 1969, 70, 12633s].
- Karishin, A. P.; Fedorenko, T. P. Ukrain, Khim. Zhur. 1953, 19, 631, [Chem. Abstr. 1955, 49, 1214e].
- Karishin, A. P. Ukrain. Khim. Zhur. 1952, 18, 504, [Chem. Abstr. 1955, 49, 1682i].
- Karishin, A. P.; Kustol, D. M. Ukrain. Khim Zhur. 1956, 22, 229, [Chem. Abstr. 1957, 51, 365c].
- Karishin, A. P., Timchenko; Dzhurka, G.F.; Samusenko, Yu. V.; Baklan, T. F.; Lysenko, G. M. Khim. Geterotskil. Soedin, Akad. Nauk Latv. SSR 1965, 5, 704, [Chem. Abstr. 1966, 64, 9708g].
- Karishin, A. P.; Samusenko, Yu. V. Zh. Organ. Khim. 1965, 1, 1003, [Chem. Abstr. 1965, 63, 11534f].
- Karishin, A. P.; Solomakha, L. A. Zh. Organ. Khim. 1965, 1, 2062, [Chem. Abstr. 1966, 64, 8166b].
- Hudson, C. B.; Robertson, A. V. Aust. J. [Chem. 1967, 20, 1511, [Chem. Abstr. 1967, 67, 117251g].
- Issidorides, C. H.; Atfah, M. A.; Sabounji, J. J.; Sidani, A. R.; Haddadin, M. J. Tetrahedron 1978, 34, 217.
- Matia, M. P.; Garci-Navio, J. L.; Vaquero, J. J.; Alvarez-Builla, J. Liebigs Ann. Chem. 1992, 7, 777.
- Erwin, D.; Frank, P. Eur. Pat. 1989, 339, 556, [Chem. Abstr. 1990, 112, 181391c].
- Crosby, J.; Milner, J. A. Ger. Pat. 2 1977, 714, 668, [Chem. Abstr. 1978, 88, 23601c].
- Alvarez-Builla, J.; Gonzalez Trigo, G.; Ezquerra, J.; Fombella, M. E. J. Hetrocycl. Chem. 1985, 22, 681.
- Ezquerra, J.; Builla, J. A. J. Heterocycl. Chem. 1986, 23, 1151.
- Pastor, J.; Paz Matia, M.; Garcia Nario, J. L.; Vaquero, J. J.; Alvarez-Builla, J. Heterocycles 1989, 29, 2369.
- Matia, M. P.; Ezquerra, J.; Sanchez-Ferrnado, F.; Garcia Navio, J. L.; Vaquero, J. J.; Alvarez-Builla, J. Tetrahedron 1991, 47, 7329.
- Galera, C.; Vaquero, J. J.; Garcia Navio, J. L.; Alvarez-Builla, J. J. Heterocycl. Chem. 1986, 23, 1889.
- Matia, M. P.; Garcia Navio, J. L.; Vaquero, J. J.; Alvarez-Builla, J. J. Heterocycl. Chem. 1990, 27, 661.
- Nightingale, D. V.; Erickson, F. B.; Shackelford, J. M. J. Org. Chem. 1952, 17, 1005.
- De Fazi, R.; Monforte, F. Atti accad. Lincei 1929, 10, 653, [Chem. Abstr. 1930, 24, 24423].
- Sircar, A. C.; Sen, S. C. J. Indian Chem. Soc. 1931, 8, 605.
- Tsuge, O.; Gunjima, T. Asahi Garasu Kogyo Gijustsu Shorei-Kai Kenkyu Hokou 1966, 12, 209, [Chem. Abstr. 1968, 68, 78248k].
- Sircar, A. C.; Sen, S. C. J. Indian Chem.. Soc. 1936, 13, 482.
- White, D. N. J. Org. Chem. 1970, 35, 2452.
- Otohiko, T.; Masashi, T.; Ichiro, S. Bull. Chem. Soc. Jpn. 1969, 42, 181.
- Rice, J. E; Shih, H. C.; Hussain, N.; La Voie, E. J. J. Org. Chem. 1987, 52, 849.
- Boulos, L. S.; Abd El-Rahman, N. M. Phosphorous, Sulfur- Silicon Relat. Elem. 1992, 68, 241.
- Helena, S.; Acad, C. R. Sci, Ser. C 1971, 273, 1194, [Chem. Abstr. 1972, 76, 59135v].
- Nicolaides, D. N.; Litinas, K. E.; Argyropoulos, N. G. J. Chem. Soc., Perkin Trans. 1 1986, 415.
- Rice, J. E.; Czech, A.; Hussain, N.; LaVoie,, E. J. J. Org. Chem. 1988, 53, 1775.
- Lefkaditis, D. A.; Nicolaides, D. N.; Papageorgiou, G. K.; Stephanidou-Stephanatou, J. J. Heterocycl. Chem. 1990, 27, 227.
- Papageeorgiou, G.; Nicolaides, D.; Stephanidou-Stephanatou, J. Liebigs Ann. Chem. 1989, 4, 397, [Chem. Abstr. 1989, 110, 212325z].
- Lefkaditis, D. A.; Argyropoulos, N. G.; Nicolaides, D.N. Liebigs Ann. Chem. 1986, 11, 1863, [Chem. Abstr. 1987, 106, 4947u].
- Maxim, N. Bull. Soc. Chim. 1928, 43, 769, [Chem. Abstr. 1928, 22, 4121].
- Acree, S. F. Am. Chem. J. 1905, 33, 186.
- Beschke, E.; Kitay, M. Ann. 1909, 369, 200.
- Bachmann, W. E.; Chu, E. J. J. Am. Chem. Soc. 1936, 58, 1118.
- Bartlett, P. D.; Brown, R. F. J. Am. Chem. Soc. 1940, 63, 2927.
- Brown, R. F. J. Am. Chem. Soc. 1954, 76, 1279.
- Moriconi, E. J.; O’Conner, W. F.; Kuhn, L. P.; Keneally, E. A.; Wallenberger, F. T. J. Am. Chem. Soc. 1959, 81, 6472.
- Jingshun, J. Huaxue Xuebao 1987, 45, 1211, [Chem. Abstr. 1988, 109, 92967r].
- Alder, R. W.; Colclough, D.; Grams, F.; Orpen, A. G. Tetrahedron 1990, 46, 7933.
- Miller, A. R.; Curtin, D. Y. J. Am. Chem. Soc. 1976, 98, 1860.
- Levchenko, A. I.; Morzo, R. A.; Zatolokin, E. I.; Suprun, V. Z.; Beloglazova, V. V.; Gaidukova, R. G.; Stal’nova, L.K. Kim. Atsetilena, Tr. Vses. Konf., 3rd 1986, 311, [Chem. Abstr. 1973, 79, 53803b].
- Mitchell, R. H.; Fyles, T.; Ralph, L. M. Can. J. Chem. 1977, 55, 1480, [Chem. Abstr. 1987, 88, 37475b].
- Blomberg, C.; Grootveld, H. H.; Gerner, T. H.; Bickelhaupt, F. J. Organometal. Chem. 1970, 24, 549.
- Zsuffa, M. Ber. 1910, 43, 2915, [Chem. Abstr. 1911, 5, 495].
- Tai-Shan, F.; Wang Ping, M.; Tsing Hsing, C.; Shih-Chen, S. J. J. Chin. Soc. 1985, 32, 457, [Chem. Abstr. 1987, 106, 196342n].
- Matei, I. Ber. 1929, 62B, 2095.
- Salazkin, S. N.; Korshak, V. V.; Vinogradova, S. V.; Beridze, L. A.; Pankratov, V. A. Deposited Doc., VINITI 1976, 2833, [Chem. Abstr. 1978, 89, 129169].
- Matei, I.; Bogdan, E. Ber. 1938, 71B, 2292.
- Matei, I.; Ccea, E. Ber. 1944, 77B, 714.
- Matei, I.; Bogdan, E. Ber. 1934, 67B, 1834.
- Dewhurst, F.; Shah, P. K. J. J. Chem. Soc., C 1970, 12, 1737.Graebe, C.; Guinsburg, M. Ann. 1903, 327, 85.
- Mayer, F.; Schonfelder, H. Ber. 1922, 55B, 2972.
- Rule, H. G.; Thompson, S. B. J. Chem. Soc. 1937, 1761.
- Petrenko, G. P.; Terent’eva, G. N.; Usachenko, V. G. U.S.S.R. Pat. 1973, 406, 827, [Chem. Abstr. 1974, 80, 82499h].Dziewonski, K.; Zarkrzewska-Barnaowska, M. Bull. Intern. Acad. Polon. Sci. 1927, 1-2A, 65.
- Ghigi, E. Gazz . Chem. Ital. 1938, 68, 184.
- Graebe; Jequier. Ann. 1896, 290, 202, [Chem. Abstr. 1938, 32, 7910].
- Petrenko, G. P.; Terent’eva, G. N.; Usachenko, V. G. Zh. Org. Khim. 1973, 9, 2313, [Chem. Abstr. 1974, 80,47690s].
- El Kateb, A. A.; Hennaway, I.T.; R.Shabana, Osman, F. H. Phosphorus Sulfur 1984, 20, 329, [Chem. Abstr. 1985, 103, 53784k].
- Singh, M. S.; Mehrotra, K. N. Indian J. Chem., Sect.B 1984, 23B, 1289, [Chem. Abstr. 1985, 102, 203950g].
- Ogata, Y.; Tsuchida, M. J. Org. Chem. 1955, 20, 1631.
- Akio, T.; Yutaka, N.; Koichi, Y.; Hidetoshi, I. Chem. Lett. 1990, 4639, [Chem. Abstr. 1990, 113, 77826h].
- Yoshinori, N. J. Am. Chem. Soc. 1980, 102, 3774.
- Brown, R. E.; Legg, K. D.; Wolf, M. W.; Singer, L. A.; Parks, J. H. Anal. Chem. 1974, 46, 1690.
- Yoon, U. C.; Kim, Y. C.; Choi, J. J.; Kim, D. U.; P. S., Mariano; Cho, I. S.; Jeon, Y. T. J. Org. Chem. 1992, 57, 1422.
- Cohen, A. I.; Harper, I. T.; Levine, S. D. J. Chem. Soc. D 1970, 23, 1610.
- Cohen, A. I.; Harper, I. T.; Puar, M. S.; Levine, S. D. J. Org. Chem. 1972, 37, 3147.
- Schönberg, A.; Schutz, O.; Arend, G.; Peter, J. Ber. 1927, 60B, 2344.
- Seymour David, L. Ger. Pat.2 1971, 102, 843, [Chem. Abstr. 1971, 75, 140829a].
- Sidky, M. M.; Osman, F. H. J. Prakt. Chem. 1973, 315, 881.
- Ogata, Y.; Yamashita, M. Bull. Chem. Soc. Jpn. 1973, 46, 2208.
- Ramirez, F.; Ramanathan, N. J. Org. Chem. 1961, 26, 3041.
- Ogata, Y.; Yamashita, M. J. Chem. Soc., Perkin Trans 2 1972, 493.
- Singh, M. S.; Mishra, G.; Mehrotra, K. N. Phosphorus Sulfur Silicon Relat. Elem. 1991, 63, 177.
- Ramirez, F.; Patwardhan, A. V.; Kugler, H. J.; Smith, C. P. Tetrahedron 1968, 24, 2275.
- Ried, W. Hg. Appeal Z. Naturforschung 1960, 15b, 684, [Chem. Abstr. 1961, 55, 21062g].
- Burmistrov, S. I.; Kondrat’eva, S. E. Nov. Khim. Karbenov, Mater. Vses. Soveshch. Khim. Karbenov Ikh. Analogv, 1ts 1972 (pub.1973). 240, [Chem. Abstr. 1975, 82, 43265a].
- Andras, S.; Harald, S.; Mundiyath, V. Justus Liebigs Ann. [Chem. 1977, 5, 747, [Chem. Abstr. 1977, 87, 134800r].
- Sample Availability: Not applicable.
© 2002 by MDPI (http://www.mdpi.org). Reproduction is permitted for non commercial purposes.