Synthesis of 3-Alkenyl-1-azaanthraquinones via Diels-Alder and Electron Transfer Reactions

{kind=link}
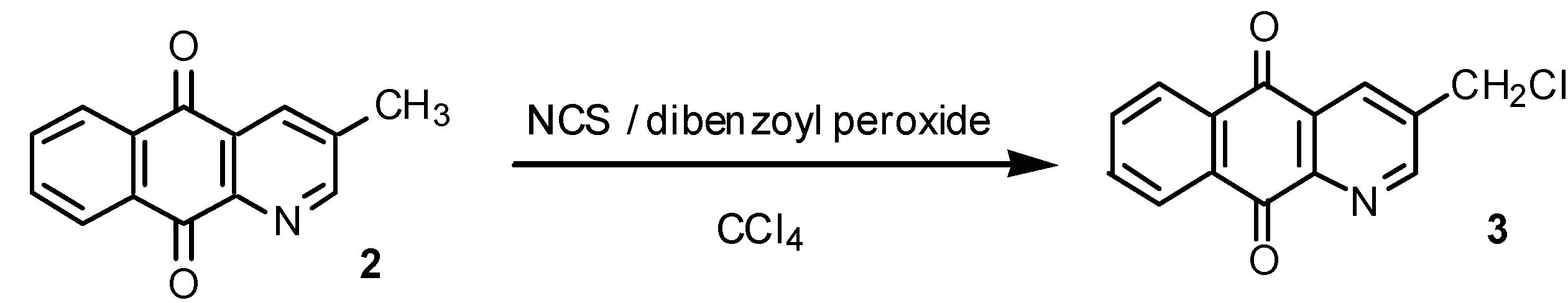{kind=link}
{kind=link}
Abstract
:Introduction
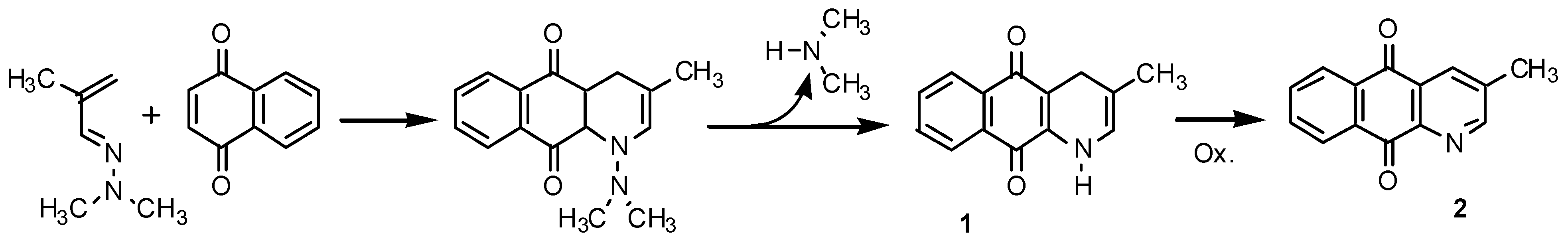
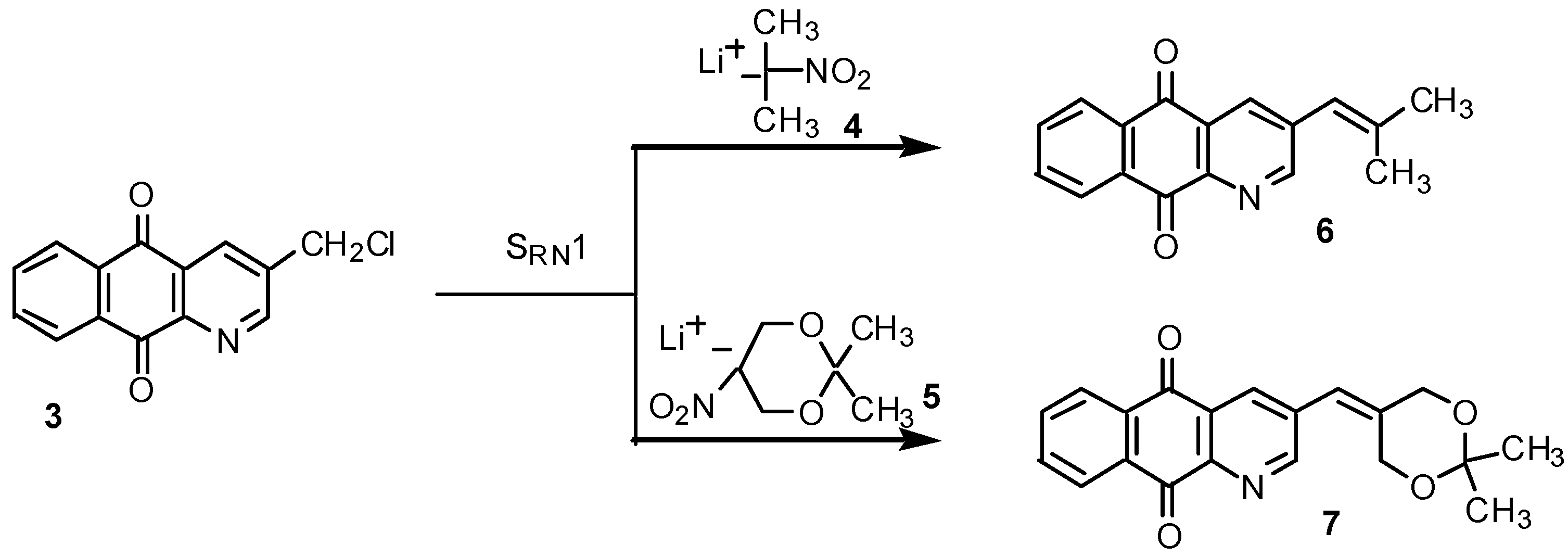
Results and Discussion

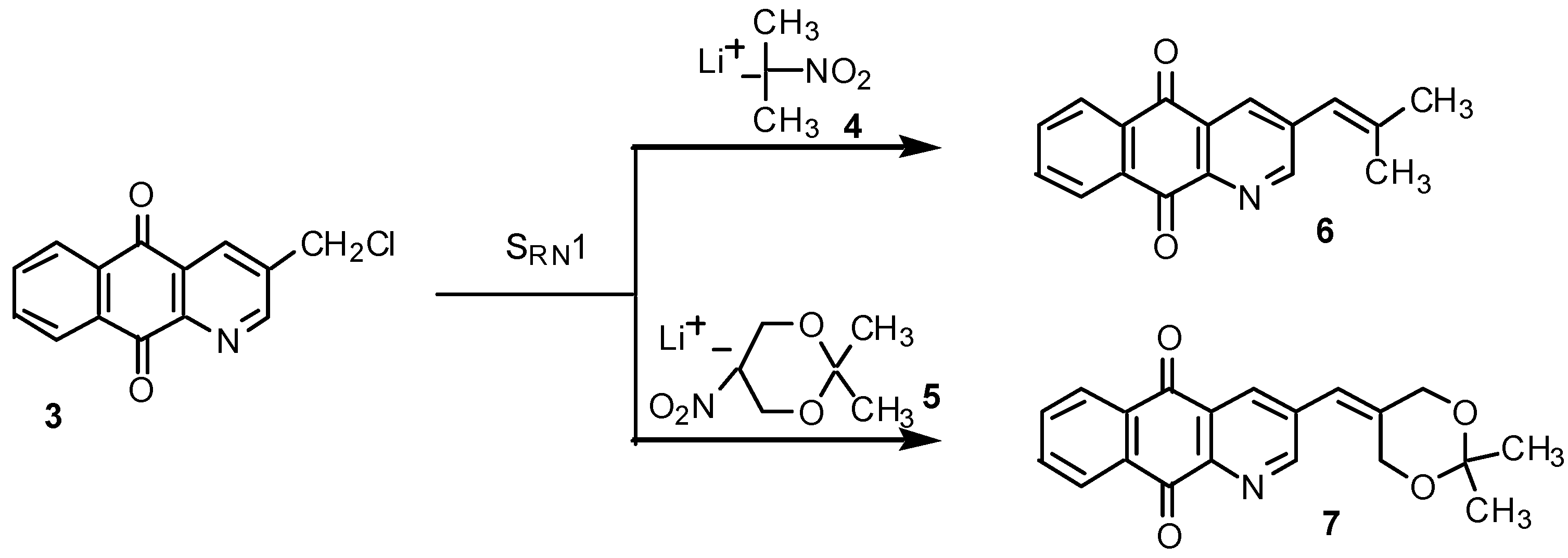
Conclusions
Experimental
General
Synthesis of 3-methyl-1-azaanthraquinone (2).
Free radical chlorination of 2: preparation of 3-chloromethyl-1-azaanthraquinone (3).
Preparation of nitronate lithium salts 4 and 5.
SRN1 reactions of chloride 3 with the lithium salts 4 and 5: preparation of 3-(2-methylpropenyl)-benzo[g]quinoline-5,10-dione (6) and 3-(2,2-dimethyl-[1,3]dioxan-5-ylidenemethyl)-benzo[g]-quinoline-5,10-dione (7).
Acknowledgments
References
- Arcamone, F. Cancer Res. 1985, 45, 5995–5999.
- Weiss, R. B.; Sarosy, G.; Clagett-Carr, K.; Russo, M.; Leyland-Jones, B. Cancer Chemother. Pharmacol. 1986, 18, 185–197.
- Pautet, F.; Nebois, P.; Bouaziz, Z.; Fillion, H. Heterocycles 2001, 54, 1095–1138.
- Serckx-Poncin, B.; Hesbain-Frisque, A.–M.; Ghosez, L. Tetrahedron Lett. 1982, 23, 3261–3264.
- Lee, H.; Hong, S. -S.; Kim, Y. -H. Bioorg. Med. Chem. Lett. 1996, 6, 933–936. [CrossRef]
- Potts, K. T.; Bhattachargee, D.; Walsh, E. B. J. Chem. Soc., Chem. Commun. 1984, 114–116.
- Newkome, G. R.; Kieffer, G. E.; Xia, Y. -J. Synthesis 1984, 8, 676–679. [CrossRef]
- Kornblum, N.; Pink, P. Tetrahedron 1963, 19, 17–22. [CrossRef]
- Kornblum, N.; Carlson, S. C.; Widmer, J.; Fifolt, M. J.; Newton, B. N.; Smith, R. G. J. Org. Chem. 1978, 43, 1394–1399. [CrossRef]
- Vanelle, P.; Madadi, N.; Roubaud, C.; Maldonado, J.; Crozet, M. P. Tetrahedron 1991, 47, 5173–5184.
- Samples Availability: Available from the authors.
© 2002 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Rathelot, P.; Rémusat, V.; Vanelle, P. Synthesis of 3-Alkenyl-1-azaanthraquinones via Diels-Alder and Electron Transfer Reactions. Molecules 2002, 7, 917-921. https://doi.org/10.3390/71200917
Rathelot P, Rémusat V, Vanelle P. Synthesis of 3-Alkenyl-1-azaanthraquinones via Diels-Alder and Electron Transfer Reactions. Molecules. 2002; 7(12):917-921. https://doi.org/10.3390/71200917
Chicago/Turabian StyleRathelot, Pascal, Vincent Rémusat, and Patrice Vanelle. 2002. "Synthesis of 3-Alkenyl-1-azaanthraquinones via Diels-Alder and Electron Transfer Reactions" Molecules 7, no. 12: 917-921. https://doi.org/10.3390/71200917
APA StyleRathelot, P., Rémusat, V., & Vanelle, P. (2002). Synthesis of 3-Alkenyl-1-azaanthraquinones via Diels-Alder and Electron Transfer Reactions. Molecules, 7(12), 917-921. https://doi.org/10.3390/71200917