Use of Cyclic Allylic Bromides in the Zinc–Mediated Aqueous Barbier–Grignard Reaction

Abstract
:Introduction
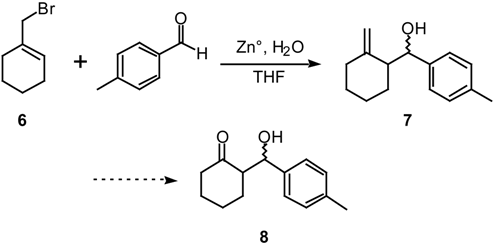
Results and Discussion
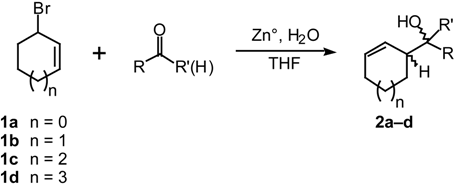

| Cyclic Bromide | Substrate | % Yield b | Ratio of diastereomers erythro/threoc |
| 1b | benzaldehyde | 87 | 83:17 |

Conclusions
Experimental
General
3–Bromo–1–methylcyclohexene (3) [11].
1-(Bromomethyl)cyclohexene (6) [12].
General Procedure for the Reaction of Cyclic Allylic Bromides with Aldehydes and Ketones.
Acknowledgments
References and Notes
- (a) Yamamoto, Y.; Asao, N. Chem. Rev. 1993, 93, 2207–2293. (b) Kobayashi, S.; Nishio, K. J. Org. Chem. 1994, 59, 6620–6628, and references therein.
- (a) Li, C. J.; Chan, T. H. Organic Reactions in Aqueous Media; John Wiley & Sons: New York, 1997; pp. 65–114. [Google Scholar] (b) Li, C. J. Tetrahedron 1996, 52, 5643–5668.
- Loh, T. P.; Zhou, J. R. Tetrahedron Lett. 1999, 40, 9115–9118.
- (a) Zhou, J. Y.; Chen, Z. G.; Wu, S. H. Syn. Comm. 1994, 24, 2661–2664. (b) Isaac, M. B.; Chan, T. H. Tetrahedron Lett. 1995, 36, 8957–8960. (c) Rubsam, F.; Seck, S.; Giannis, A. Tetrahedron 1997, 53, 2823–2834. See also Killinger, T. A.; Boughton, N. A.; Runge, T. A.; Wolinsky, J. J. Organometallic Chem. 1997, 124, 131–134.
- (a) Young, D.; Kitching, W. Aust. J. Chem. 1985, 38, 1767–1777. (b) Yadav, V. K. Syn. Comm. 1990, 20, 1023–1032. (c) Tamaru, Y.; Tanaka, A.; Yasui, K.; Goto, S.; Tanaka, S. Angew. Chem. Int. Ed. Engl. 1995, 34, 787–789.
- March, J. Advanced Organic Chemistry, 4th ed.; John Wiley & Sons: New York, 1992; pp. 449–451. [Google Scholar]
- (a) Griesbeck, A. G.; Stadtmuller, S. J. Am. Chem. Soc. 1991, 113, 6923–6928. (b) Takai, K.; Toratsu, C. J. Org. Chem. 1998, 63, 6450–6451.
- Maeda, H.; Shono, K.; Ohmori, H. Chem. Pharm. Bull. 1994, 42, 1808–1812.
- For a related reaction exhibiting similar regioselectivity, see reference 4b.
- Monson, R. S. Advanced Organic Synthesis; Academic Press: New York, 1971; pp. 48–49. [Google Scholar]
- Bottini, A. T.; Corson, F. P.; Fitzgerald, R.; Frost, K. A. Tetrahedron 1972, 28, 4883–4904.
- Pages, L.; Llebaria, A.; Camps, F.; Molins, E. J. Am. Chem. Soc. 1992, 114, 10449–10461.
- Sample Availability: Available from the authors.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Breton, G.W.; Shugart, J.H.; Hughey, C.A.; Conrad, B.P.; Perala, S.M. Use of Cyclic Allylic Bromides in the Zinc–Mediated Aqueous Barbier–Grignard Reaction. Molecules 2001, 6, 655-662. https://doi.org/10.3390/60800655
Breton GW, Shugart JH, Hughey CA, Conrad BP, Perala SM. Use of Cyclic Allylic Bromides in the Zinc–Mediated Aqueous Barbier–Grignard Reaction. Molecules. 2001; 6(8):655-662. https://doi.org/10.3390/60800655
Chicago/Turabian StyleBreton, Gary W., John H. Shugart, Christine A. Hughey, Brian P. Conrad, and Suzanne M. Perala. 2001. "Use of Cyclic Allylic Bromides in the Zinc–Mediated Aqueous Barbier–Grignard Reaction" Molecules 6, no. 8: 655-662. https://doi.org/10.3390/60800655
APA StyleBreton, G. W., Shugart, J. H., Hughey, C. A., Conrad, B. P., & Perala, S. M. (2001). Use of Cyclic Allylic Bromides in the Zinc–Mediated Aqueous Barbier–Grignard Reaction. Molecules, 6(8), 655-662. https://doi.org/10.3390/60800655