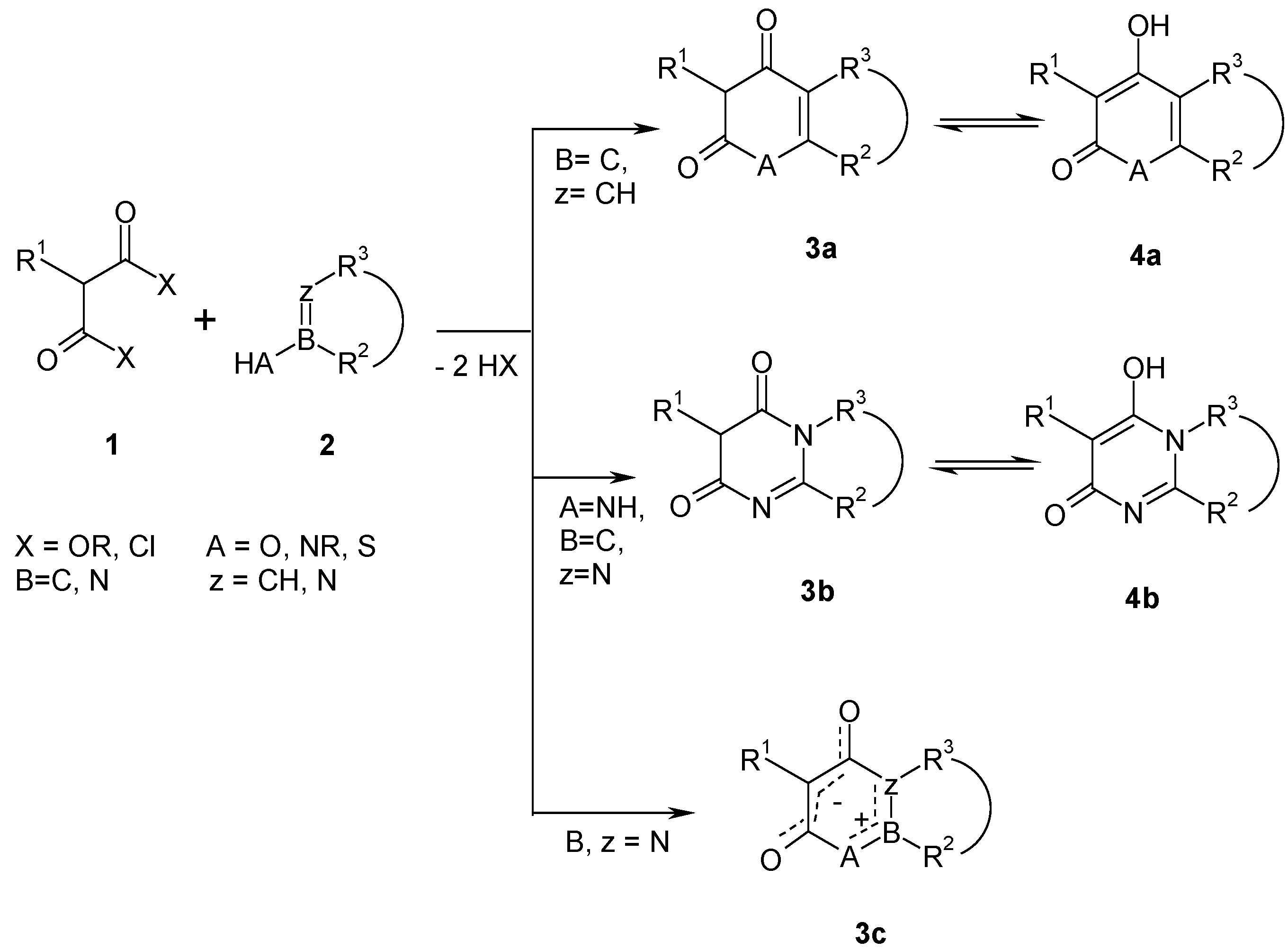
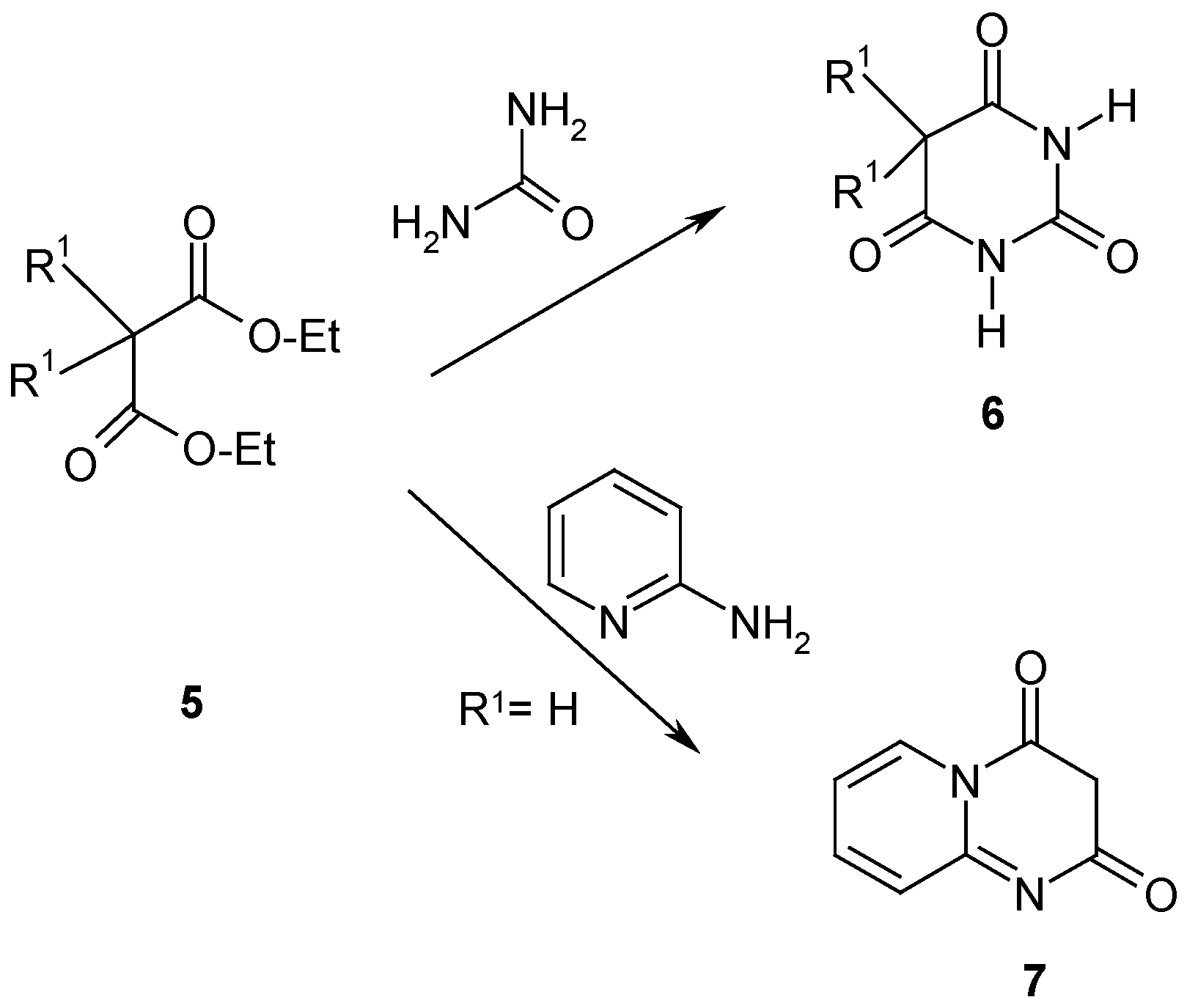
Results and Discussion
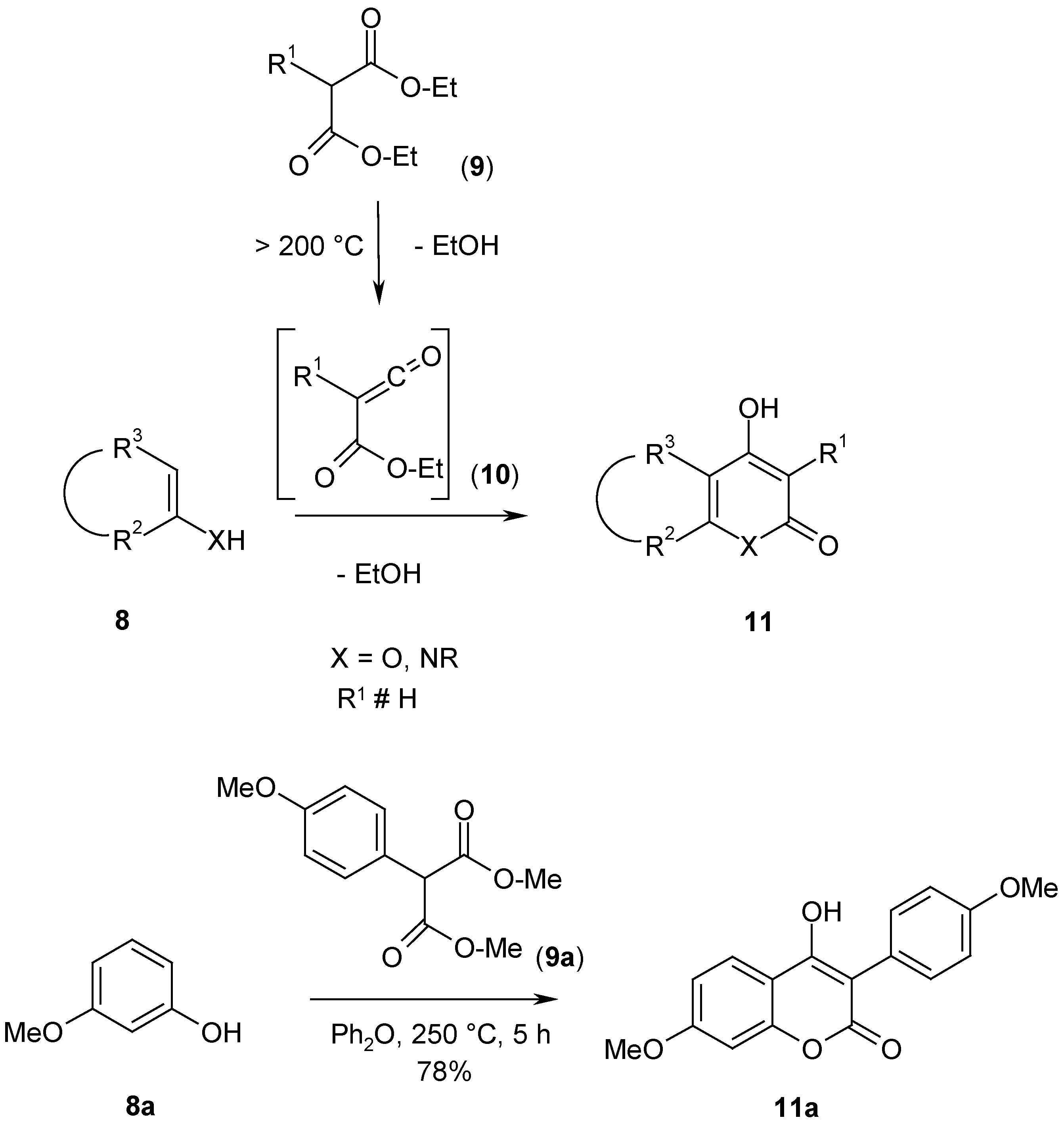
Pyrones and benzoanalogous coumarins (
11, X = O) have been obtained from enols or phenols (
8, X = O) and 2-unsubstituted or 2-monosubstituted diethyl malonates (
9), e.g. ref. [
3,
4]. Pyridones and benzoanalogous quinolones (
11, X = NR) have been obtained from azomethines or anilines (
8, X = NR) and malonates (
9), e.g. ref. [
4,
5]. As an example procedure the synthesis of 4-hydroxy-7-methoxy-3-(4-methoxyphenyl)-coumarin (
11a, R
2-R
3 = -CH=C(OMe)-CH=CH-, X = O, R
1 = 4-MeO-C
6H
4) from diethyl 2-(4-methoxyphenyl)malonate (
9a, R
1 = 4-MeO-C
6H
4) and 2-methoxyphenol (
8a, R
2-R
3 = -CH=C(OMe)-CH=CH-, X = O) is described in the experimental part (
Scheme 3).
Reported procedures for compounds of type
11 involve multistep procedures (see e.g. ref. [
6], which describes the reaction of
8a with bis(2,4,6-trichlorophenyl) 2-(4-methoxyphenyl)malonate
18, R
1 = 4-MeO-C
6H
4). We were able to show that especially 2-arylmalonates
9 (R
1 = Ph) provide at reaction temperatures above 250°C good synthons for simple and quick cyclization reactions in good to moderate yields [
5] (probably
via arylketene ester intermediates
10, R
1 = Ph [
7]). The only disadvantage of this reaction sequence is its high reaction temperature, which prevents its use for sensitive substrates and substituents. The ketene mechanism is also supported by observations obtained during the reaction: at temperatures below 200°C the first mole of alcohol is observed to be liberated when the open chain ester or amide is formed, then it needs a temperature of more than 250°C to liberate the second mole of alcohol and to form the ketene intermediate
10. Thermal investigations of the thermolysis reaction of the phenol
8a and the malonate
9a by differential scanning calorimetry (DSC) show, that at about 160°C a first exothermic reaction starts immediately after the endothermic boiling point of the phenol
8a (in accordance with findings during the formation of malonamides), then a weak exothermic reaction begins at about 240°C followed by a strong exothermic reaction (onset 260°C). These data are in accordance with the observations obtained from the synthetic experiments as described above.
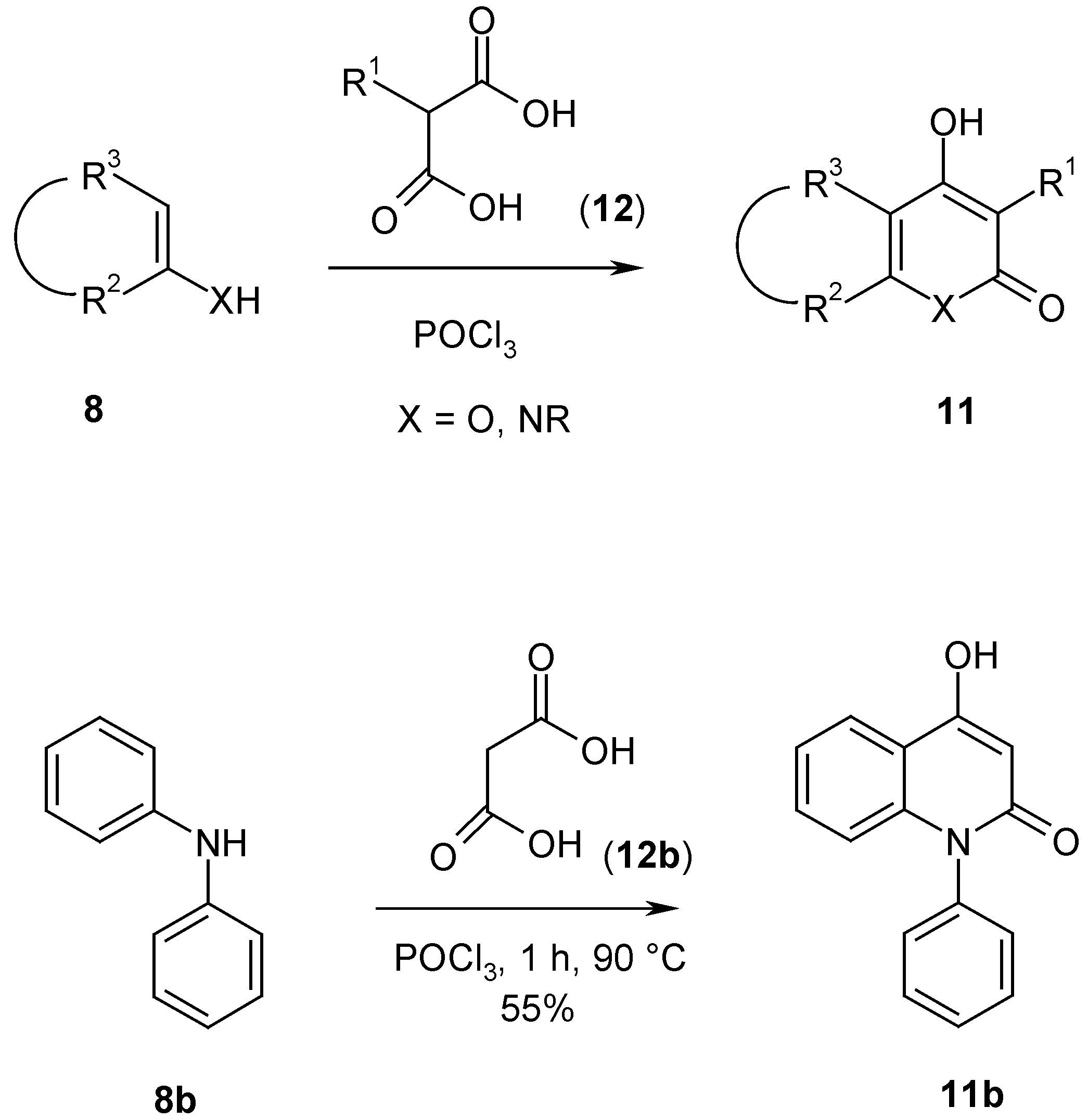
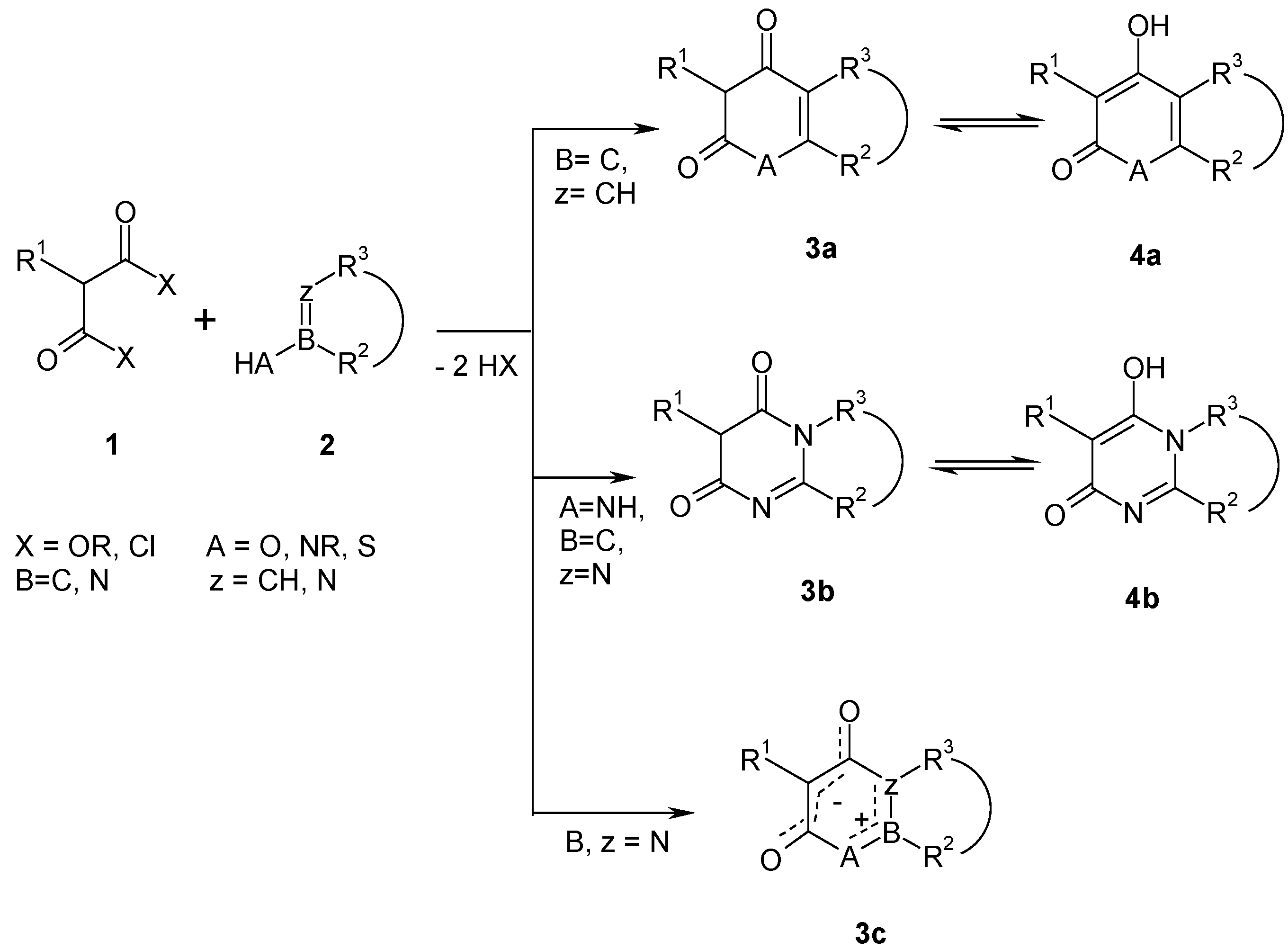
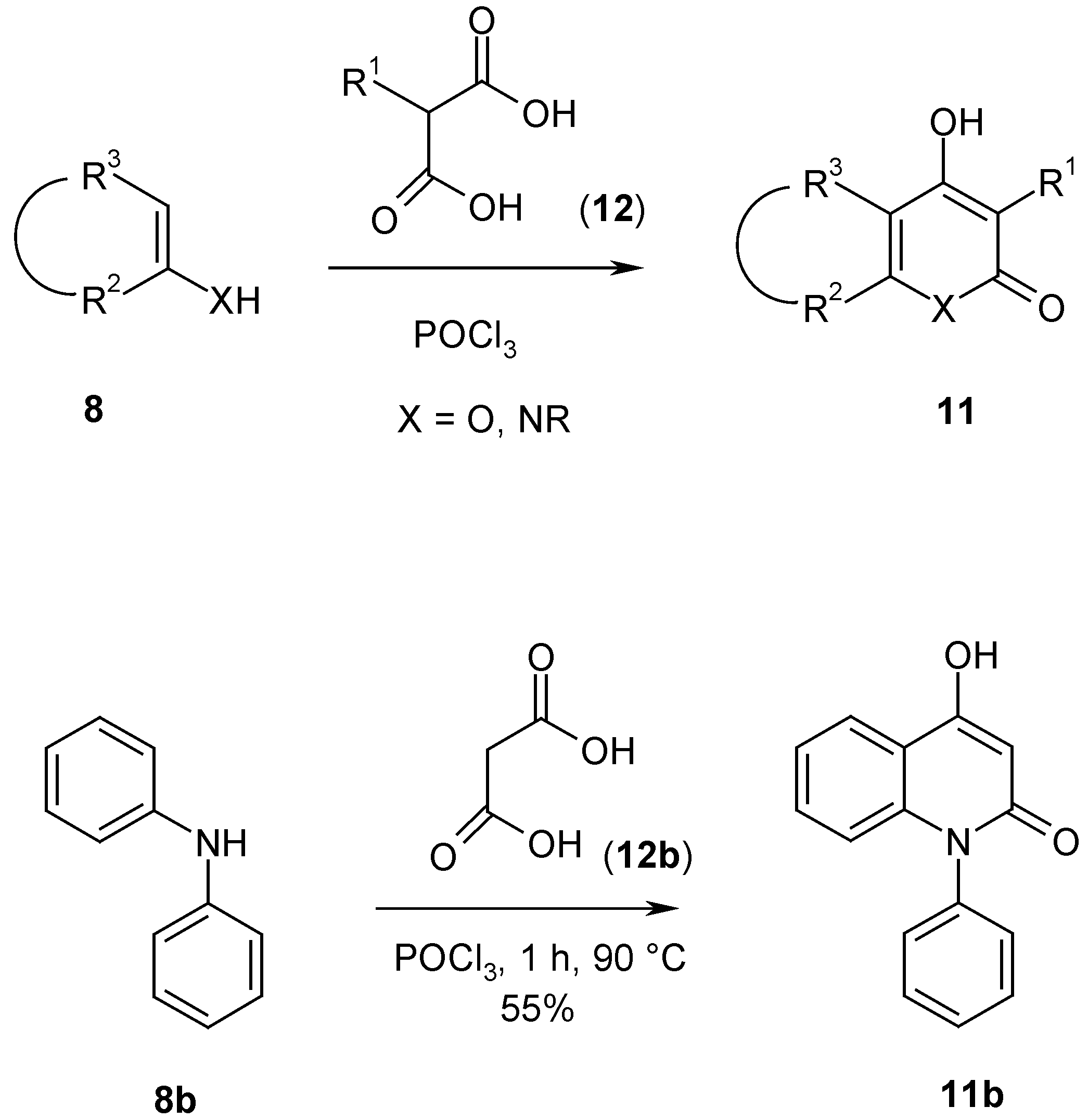
Malonic acid derivatives
12 are not reactive enough to react with dinucleophiles
8. However, when they are converted
in situ with phosphoryl chloride [
8] to the corresponding acid chlorides, they give pyrones and pyridones
11 with dinucleophiles
8. As an example procedure the one step synthesis of 4-hydroxy-1-phenyl-2-quinolone (
11b) from malonic acid (
12b, R
1 = H), phosphoryl chloride and diphenylamine (
8b, R
2-R
3 = -CH=CH-CH=CH=, X = N-Ph) is described. The reported synthesis of
12b involves a 3-step procedure from malonates
9 (R
1 = H) [
3]. Although the yields are rather low (30-50%), the quick and short pathway overcomes this disadvantage (
Scheme 4).
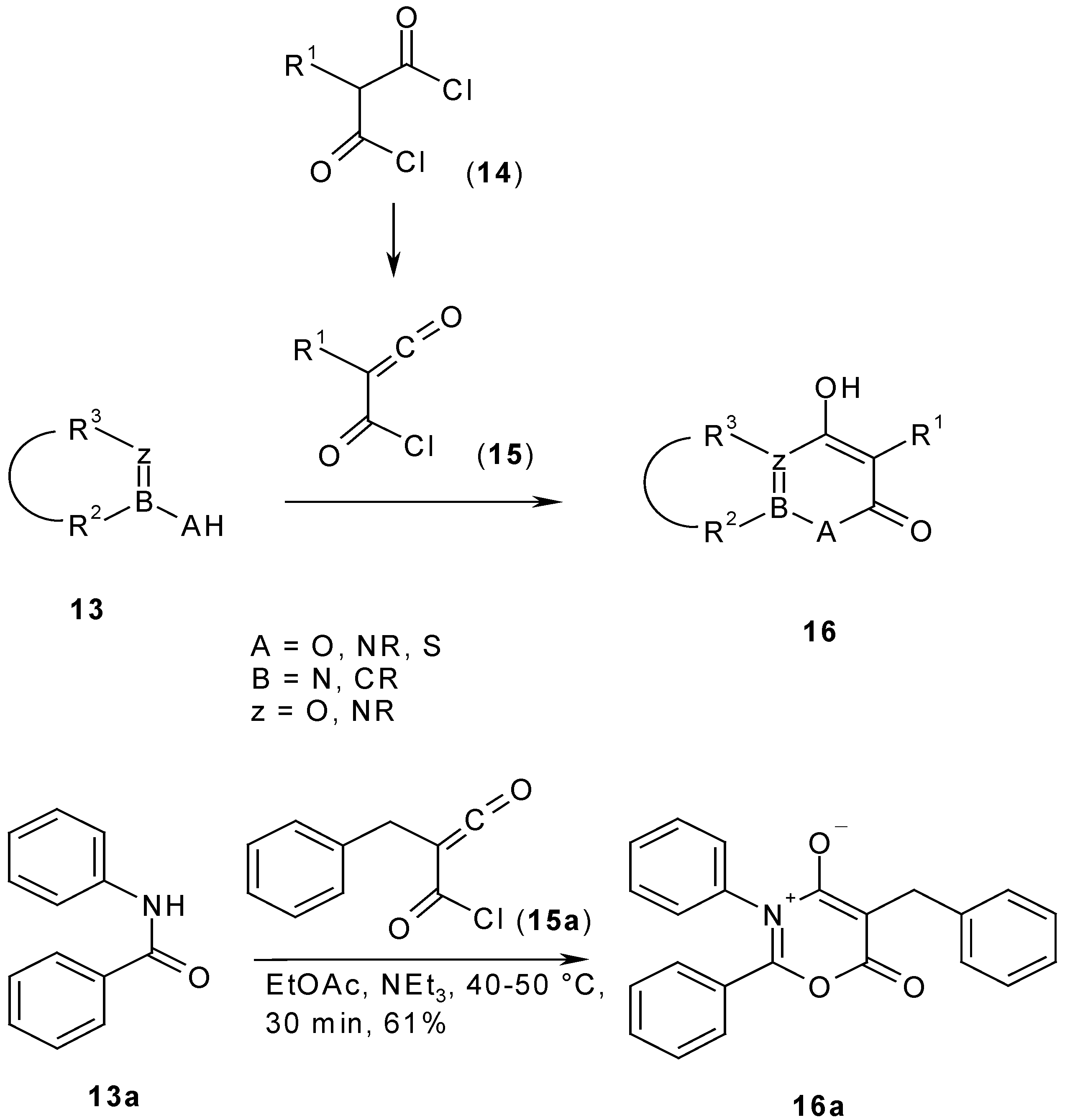
Less reactive or sensitive substrates of type
2 require more reactive malonyl derivatives. Malonyl dichlorides
14 already react at lower temperatures in aprotic solvents with dinucleophiles
13 to give heterocycles
16 [
9]. Above 80°C, malonyl dichlorides lose hydrogen chloride to form chlorocarbonyl ketenes
15, which are more reactive than malonyl dichlorides
14 and react in the presence of bases such as triethylamine with many substrates at room temperature [
10]. As an example procedure, the improved synthesis of 5-benzyl-2,3-diphenyl-6
H-6-oxo-1,3-oxazin-3-ium-4-olate (
16a, A = O, B = C, z = N, R
1 = CH
2Ph, R
2 = R
3 = Ph), having a mesoionic betaine structure, is described (
Scheme 5). Attempts to obtain
16a with other less reactive malonates gave only rearrangement products because of the high temperature which was necessary for the cyclocondensation [
11].
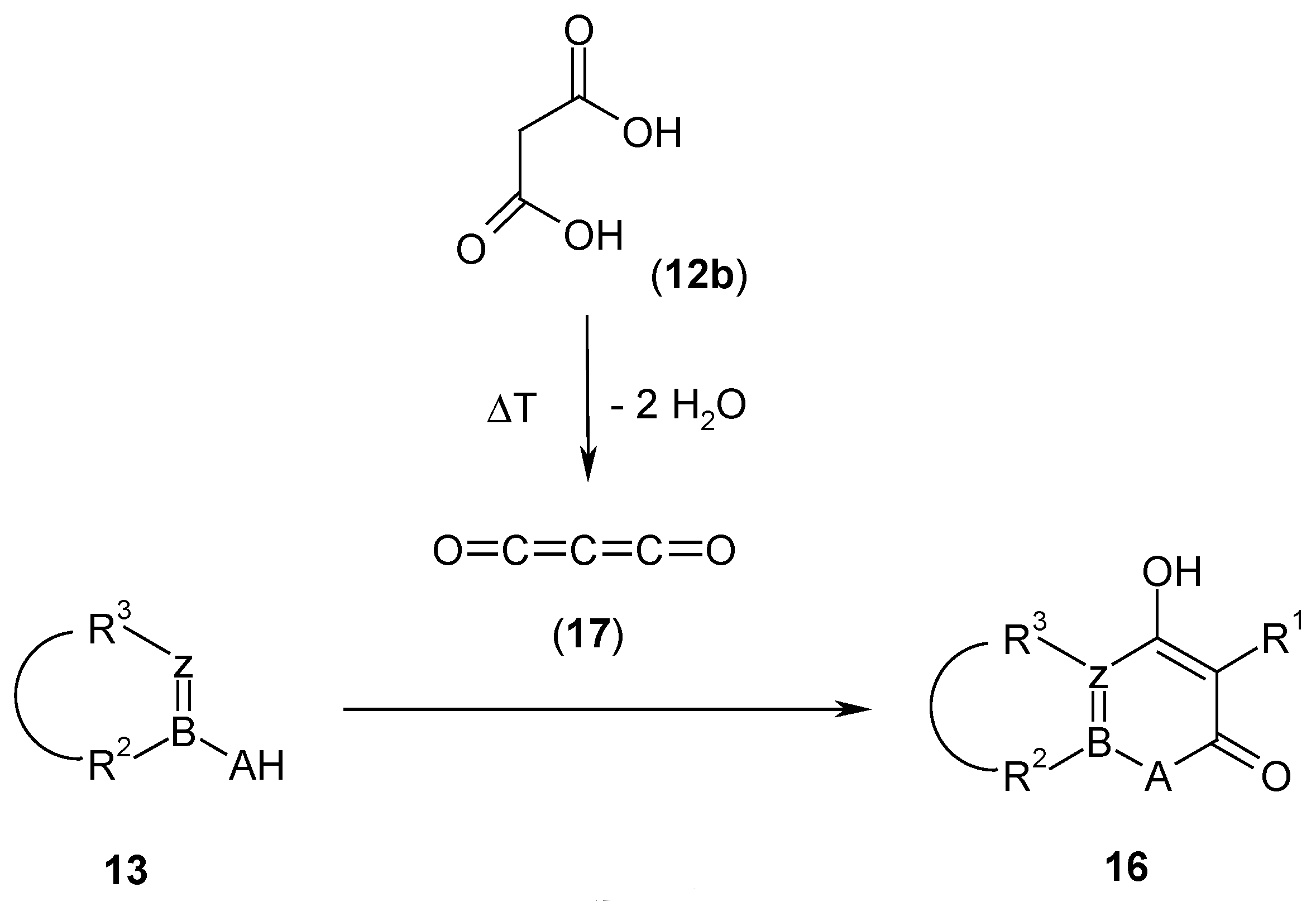
The most electrophilic malonic acid derivative has been found in carbon suboxide
17 [
12], which reacts with dinucleophiles
13 at low temperatures (-78°C) to give five- or six-membered malonyl heterocycles
16 (
Scheme 6). Its preparation is performed either by pyrolysis of diacetyl tartaric anhydride, or from malonic acid
12b and phosphorus pentoxide, or from bis(trimethylsilyl) malonate
19b and phosphorus pentoxide [
12].
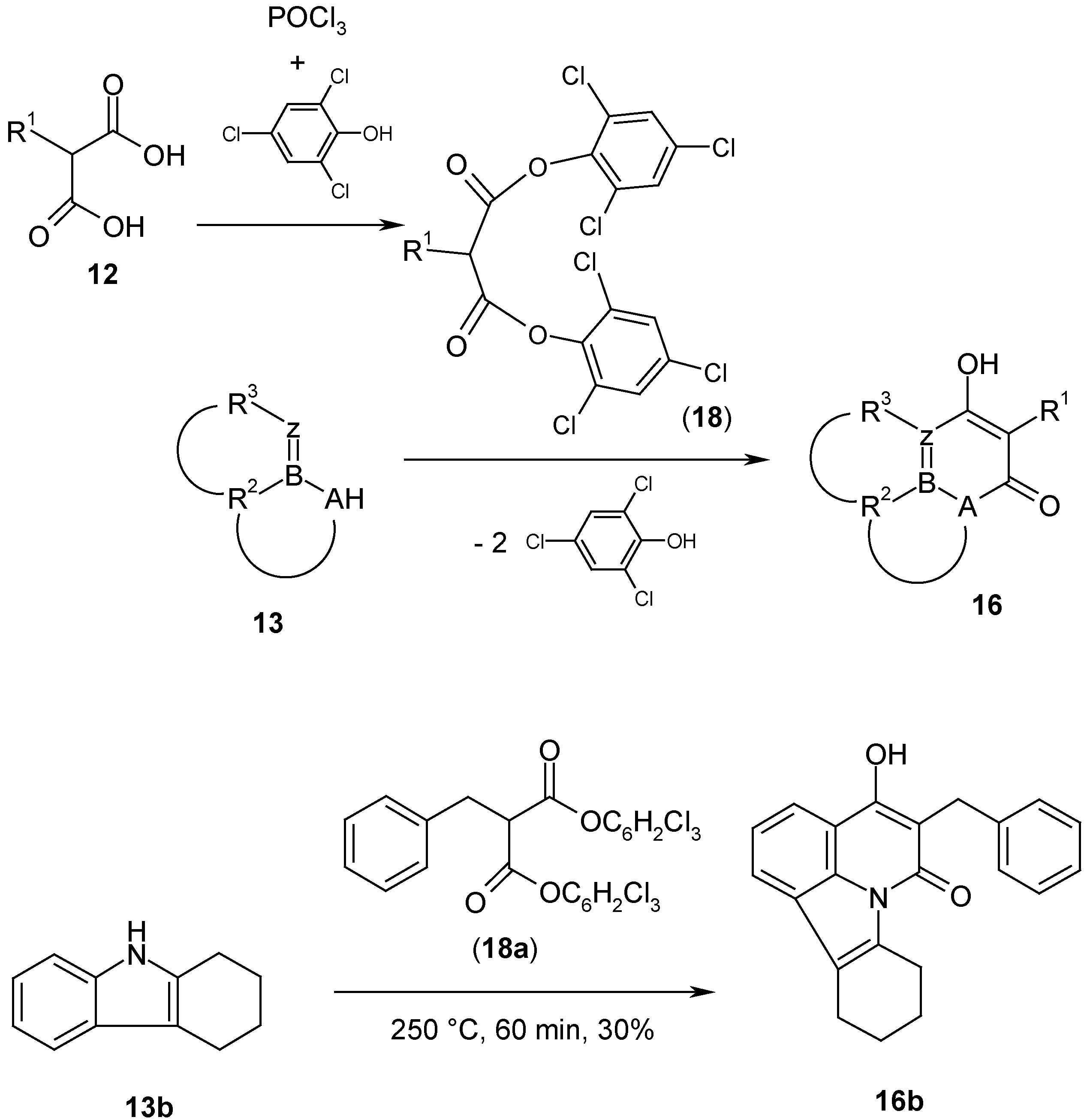
A large number of 1,3-dinucleophiles
13 has been reacted with bis(2,4,6-trichlorophenyl) malonates
18 (obtained from malonic acids
12 and 2,4,6-trichlorophenol) leading to six-membered malonyl heterocycles [
13]. Most of these condensations have been carried out thermally by fusion of
13 and
18 at 150 - 250°C in the melt; in some cases bromobenzene or diphenylether have been used as solvent. N,N'-dinucleophiles
13 (A = z = NR) can also be reacted at room temperature. As an example procedure, the synthesis of 5-benzyl-4-hydroxy-8,9,10,11-tetrahydro-pyrido[3,2,1:jk]carbazol-6-one (
16b) at 250 °C is described, which yields
16b in about 30% yield (
Scheme 7). The reported synthesis uses bis(2,4-dichlorophenyl) malonate [
23], which is avoided nowadays because of synthetic and health problems. Experiments with diethyl malonates
9 give only yields below 5%.
The reactive malonate reagents, malonyl dichlorides 14, carbon suboxide 17 and bis(2,4,6-trichlorophenyl) malonates 18 are very valuable reagents, but they are expensive and can be stored only for a limited time without decomposition. Another aspect is that malonyl dichlorides 14 and bis(2,4,6-trichlorophenyl) malonates 18 contain chlorine, an element of ill repute in Green Chemistry. Consequently we made several attempts to replace these reagents with other reactive derivatives without such disadvantages.
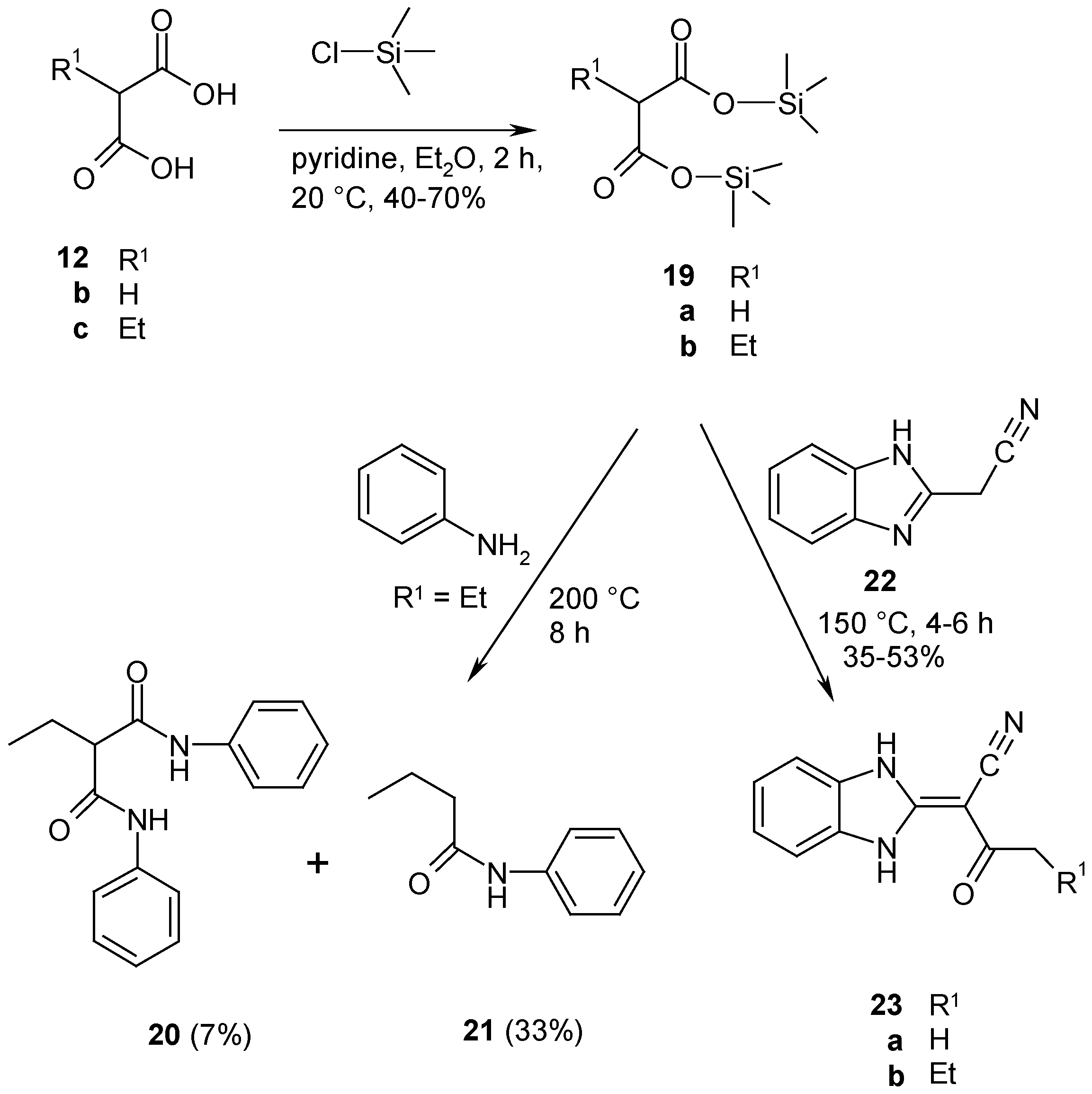
For this reason we have tried to prepare a new generation of reactive malonates such as trimethylsilyl malonates and mixed anhydrides of malonates. As test substrates we have used aromatic amines which have only weak basic nitrogens, with which suitably reactive malonates should readily form malondianilides 20 or cyclization products at low temperatures.
Bis(trimethylsilyl) malonates
19 have been prepared according to a reported method [
14], by reacting the appropriate malonic acid
12 with trimethylsilyl chloride in the presence of pyridine. The spectroscopic data of
19 confirmed their structure. The known reaction of anilines with substituted diethyl malonates
9 gives malondianilides at temperatures below 200°C, and 4-hydroxyquinolones such as
11b are obtained at higher temperatures (
Scheme 4). With bis(trimethylsilyl) 2-ethylmalonate
19b, a reaction occured at similar temperatures as observed with malonates
9, but the products were 2-ethyl-N,N'-diphenylmalondiamide
20, obtained as a minor byproduct, together with butyranilide
21, which is formed from
19b after a desilylation and decarboxylation step. 2-Cyanomethylbenzimidazole
22 was used as 1,3-dinucleophilic substrate for cyclocondensation with bis(trimethylsilyl) malonates
19. According to a previous investigation [
15] the dinucleophile
22 reacts readily with reactive bis(2,4,6-trichlorophenyl) malonates
18 in refluxing bromobenzene at 150°C to give pyrido[1,2-
a]benzimidazoles. The reaction of bis(trimethylsilyl) malonates
19 in refluxing bromobenzene, however, gave only C-acylation to yield 2-(2,3-dihydro-1
H-benzimidazol-2-yliden)-3-oxoalkane-nitriles
23 without cyclization to the nitrogen (
Scheme 8). Thus our experiments revealed that bis(trimethylsilyl) malonates
19 are no substitutes for reactive malonyl reagents
14,
15, 17 and
18. Their reactivity for synthetic purposes is too small and decomposition takes place already at rather low reaction temperatures.
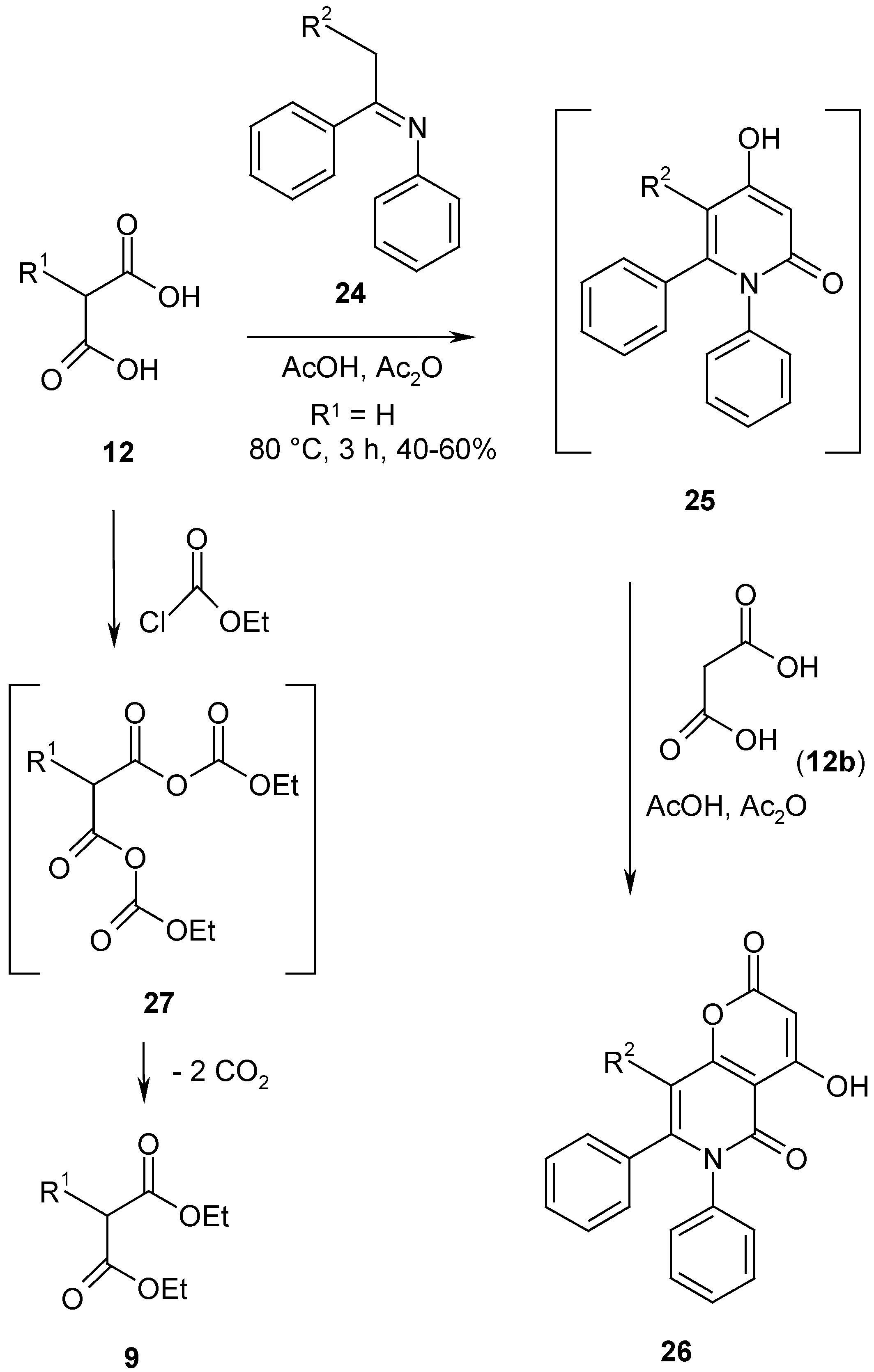
Mixed anhydrides of malonic acid
(12, R
1 = H
) with acetic acid have been used as
in situ reagents for reactive 1,3-dinucleophiles, however, the scope of this reaction has been found to be limited to a small number of 1,3-dinucleophiles such as ureas [
1]. With acetophenonaniles
24 the intermediate 4-hydroxypyridones
25 formed react with excess malonic acid (
12, R
1 = H) to give pyrono-pyridinediones
26. As an example, the synthesis of 4-hydroxy-6,7-diphenyl-pyrano[3,2-
c]pyridine-2,5(6
H)-dione (
26a, R
2 = H) is described. In order to obtain a more versatile reactive anhydride reagent, we tried to synthesize a mixed anhydride
27 of malonic acid and carbonic acid by reaction of malonic acids
12 with ethyl chloroformate in the presence of triethylamine at 0-5°C in dichloromethane using a standard procedure for benzoyl malonates [
16]. However, only 2-substituted diethyl malonates
9 have been isolated, a result which is similar to the findings reported for the mixed anhydrides of the half ester of malonic acids [
17] (
Scheme 9).
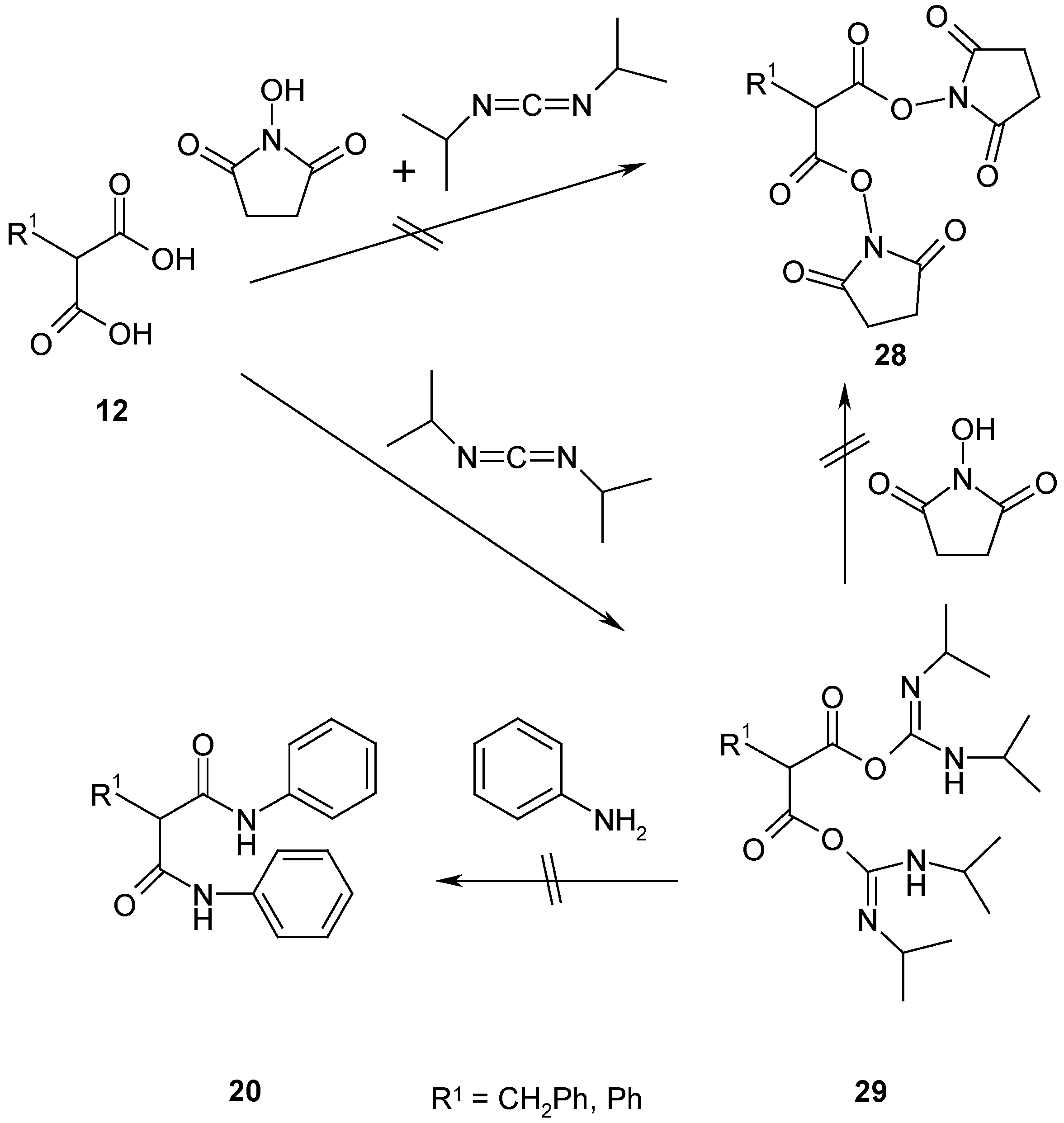
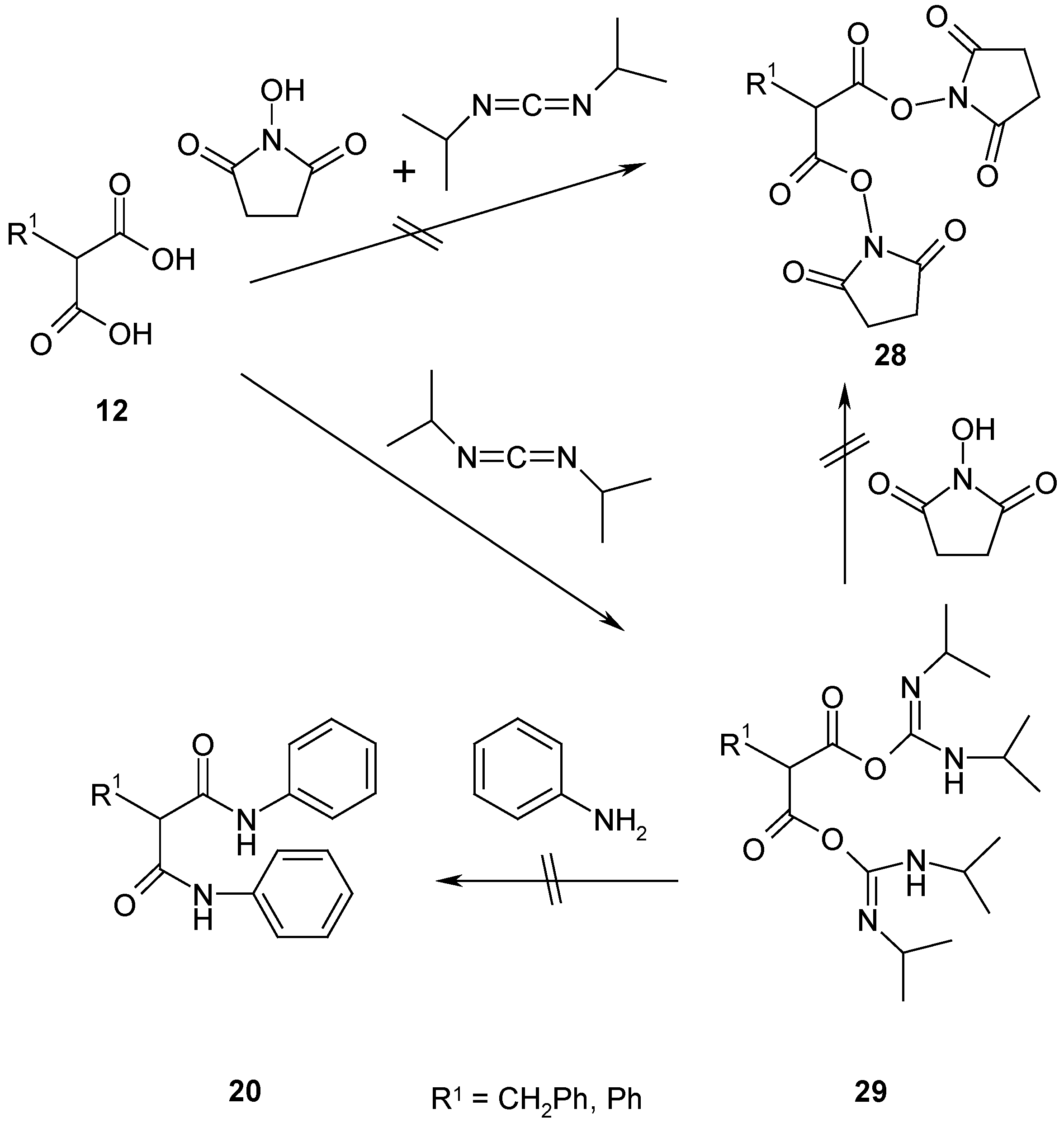
The reaction of succinimidyl- and O-acylurea esters is a well-known and widely used method for acylations and condensations of carboxylic acids in peptide synthesis [
18]. The preparation of succinimidyl esters is in general mediated by the activation with N,N'-diisopropylcarbodiimide [
18], which should give as intermediate the bis(carbamimidoyl) malonates
29. Nucleophilic attack by N-hydroxysuccinimide should give succinimidylates
28, and as byproduct N,N'-diisopropylurea. The succinimide-N-oxy group in succinimidylates is reported as a good leaving group in the reaction with nucleophiles [
18].
The reaction of malonic acids
12 with N-hydroxysuccinimide has been performed using a recently successfully applied reaction sequence for alkanoic acids [
19]. We used this procedure and dissolved the appropriate malonic acid
12 in dry dichloromethane; then a small excess of N-hydroxysuccinimide was added, the solution was cooled and a small excess of N,N'-diisopropylcarbodiimide was then added. After a reaction time of 24 hours, the reaction mixture contained a large number of compounds. The main product was N,N'-diisopropylurea, the other compounds were present in too small quantities and they have not been isolated. Other solvents such as diethylether decreased the number of compounds, however we were unable to isolate bis(succinimidoyl) malonates
28.
Similar reactive derivatives of carboxylic acids have been described in ref. [
20]. We used the reported procedure and reacted 2-benzylmalonic acid (
12a) with N,N'-diisopropylcarbodiimide in diethylether at 0°C for 2 hours and then removed the solvent. The resulting oil contained one new main product, probably bis(carbamimidoyl) 2-benzylmalonate
29a as shown by NMR data, and some byproducts in minor quantities. The crude oil was not further purified and reacted with aniline as an example nucleophile in dioxane at room temperature. However, tlc analysis did not show any reaction, which means that at low temperatures no reaction takes place between
29a and aniline (
Scheme 10). At higher temperatures only decomposition products have been observed.
Experimental
General
Melting points were determined on a Gallenkamp Melting Point Apparatus, Mod. MFB-595 in open capillary tubes. Calorimetric data were obtained on a Rheometric Scientific DSC-Plus instrument equipped with V5.42 of the differential scanning calorimetry software. The differential scanning calorimetry plots were recorded between 25 – 400°C, with a heating rate of 2-10°C/min, and 1.5-3 mg compound in sealed aluminium crucibles (11 bar). The 1H-NMR spectra were recorded on a Varian Gemini 200 instrument (200 MHz) or a Bruker AM 360 instrument (360 MHz). The 13C-NMR spectra were recorded on a Bruker AM 360 instrument (90 MHz). Chemical shifts are reported in ppm from internal tetramethylsilane standard and are given in δ-units. Infrared spectra were taken on a Perkin-Elmer 298 spectrophotometer or a Galaxy Series FTIR 7000 in potassium bromide pellets. Elemental analyses were performed on a Fisons elemental analyzer, Mod. EA 1108 and are within ±0.4 of the theoretical percentages. Mass spectra were taken on a Finnigan 4000 (EI: 70 eV, CI: 120 eV, methane). All reactions were monitored by thin layer chromatography (tlc) carried out on 0.2 mm silica gel F-254 (Merck) plates using uv light (254 and 366 nm) for detection. Common reagent-grade chemicals are either commercially available and were used without further purification or prepared by standard literature procedures.
4-Hydroxy-7-methoxy-3-(4-methoxyphenyl)-coumarin (11a).
A solution of dimethyl 2-(4-methoxyphenyl)malonate (
9a) (23.8 g, 0.1 mol) and 3-methoxyphenol (
8a) (12.4 g, 0.1 mol) in diphenylether (20 g) was heated for 3 h to 200-250°C in a metal bath. During this time methanol (about 5 mL) was liberated. When the liberation of methanol had stopped, the reaction mixture was heated to an oil bath temperature of 300°C for 2 h; during this time further 4 mL of methanol was liberated. After cooling, the reaction mixture was diluted with toluene (50 mL), filtered by suction and washed with cyclohexane (100 mL). The yield was 24.3 g (78%), yellowish prisms, mp 220.1-220.3°C (ethanol); lit. mp. 215-217°C [
6]. Spectral data are identical with the reported data in ref. [
6]. Calorimetric data of the reaction mixture of
8a and
9a: b.p. onset 157.4°C, peak maximum 160.1°C, ΔH = 0.2 mcal/mg, 1. reaction onset at 161.7°C, peak maximum 172.6°C, ΔH = -2.6 mcal/mg, 2. reaction onset at 240.5°C, peak maximum 267.2°C, ΔH = -2.3 mcal/mg, 3. reaction onset at 281.5°C, peak maximum 310.2°C, ΔH = -7.8 mcal/mg.
4-Hydroxy-1-phenyl-2(1H)-quinolone (11b).
A suspension of diphenylamine (
8b) (84.5 g, 0.5 mol) and malonic acid (
12b) (72.8 g, 0.7 mol) in phosphoryl chloride (183.6 g, 1.2 mol) was heated for 1 h to 90°Then the mixture was poured into ice/water (1 L), the precipitate filtered, dissolved in 2 N sodium hydroxide solution and filtered again. The solution was acidified with 2 N hydrochloric acid, filtered by suction, washed with water and dried. The yield was 65.3 g (55%), yellowish prisms, mp 293°C (lit. mp 294.5-295.5°C [
3]). Spectral data are identical with the reported data in ref. [
3].
5-Benzyl-2,3-diphenyl-6H-6-oxo-1,3-oxazin-3-ium-4-olate (16a).
a) 2-Benzyl-2-(chlorocarbonyl)ketene (15a): To a stirred solution of 2-benzylmalonic acid (
12a, R
1 = CH
2Ph) (39 g, 0.2 mol) in toluene (50 mL), thionyl chloride (59.5 g, 0.6 mol) was added dropwise under a nitrogen atmosphere. The reaction mixture was heated under nitrogen for 24 h under reflux and then toluene and excess thionyl chloride was removed by distillation under reduced pressure (60°C, 130 mm Hg). The residue was purified by distillation under reduced pressure (134-140°C, 15 mm Hg). The yield was 27.2 g (70%); lit. bp 110-112°C (1.5 mm Hg) [
21].
b)
5-Benzyl-2,3-diphenyl-6H-6-oxo-1,3-oxazin-3-ium-4-olate (
16a): To a solution of benzanilide (
13a) (5.9 g, 0.03 mol) in warm dry ethyl acetate (150 mL, 40-50°C) 2-benzyl-2-(chloro- carbonyl)ketene (
15a) (6.5 g, 0.033 mol) and dry triethylamine (4.5 mL) was added. The yellow reaction mixture turned red and viscous and a small amount of a dark solid precipitated. The reaction mixture was kept for 30 min at room temperature and then the precipitate was filtered off, the filtrate taken to dryness under reduced pressure and triturated with xylene. The resulting precipitate was filtered by suction and dried. The yield was 6.5 g (61%), yellow prisms, mp 160°C. Lit. mp. 161°C [
11]. Spectral data are identical with the reported data in ref. [
11].
5-Benzyl-4-hydroxy-8,9,10,11-tetrahydro-pyrido[3,2,1:jk]carbazol-6-one (16b).
a)
Bis(2,4,6-trichlorophenyl) 2-benzylmalonate (
18a): A mixture of dry 2-benzylmalonic acid (
12a, R
1 = CH
2Ph) (97.1 g, 0.5 mol), 2,4,6-trichlorophenol (157.9 g, 0.8 mol) and phosphorylchloride (161 g, 1.05 mol) was heated under reflux until the evolution of HCl gas had stopped (about 6 h). The reaction mixture was then poured onto crushed ice (1.5 l), filtered by suction, washed with ice-water, dissolved in toluene (1 L) and washed with 5% sodium bicarbonate solution and water. After drying with sodium sulfate, the solvent was removed under reduced pressure. The residue was triturated with hexane, filtered and dried at room temperature in a desiccator. The yield was 111.4 g (50.4%), beige prisms, mp 104°C (lit. mp 106-107°C [
22]).
b)
5-Benzyl-4-hydroxy-8,9,10,11-tetrahydro-pyrido[3,2,1:jk]carbazol-6-one (
16b): A mixture of 1,2,3,4-tetrahydrocarbazole (
13b) (3.75 g, 22 mmol) and bis(2,4,6-trichlorophenyl) 2-benzylmalonate (
18a, R
1 = CH
2Ph) (12.1 g, 22 mmol) was heated without solvent for 60 min to 250°C. The 2,4,6-trichlorophenol vapors were removed
via a funnel into the water pump. After cooling the dark reaction mixture was washed with hexane until the product became semi-solid. Then the product was stirred with 0.5 M sodium hydroxide solution (500 mL) overnight, filtered, the filtrate extracted with toluene (2 x 100 mL) and then the aqueous phase acidified with conc. hydrochloric acid. The precipitate was washed with water, recrystallized from glacial acetic acid/water and dried. The yield was 21.6 g (30%), mp 240°C dec. (from glacial acetic acid/water). Lit. [
23] mp. 240-245°C (dec.; no yield given); IR: 3100-2900 m, 1670 s, 1640 sh, 1610 s, 1545 s cm
-1;
1H-NMR (DMSO-d
6): δ = 1.80-1.95 (t, J = 6 Hz, 4 H, 9-CH
2, 10-CH
2), 2.60 (t, J = 6 Hz, 2 H, 11-CH
2), 2.80 (m, 2 H, 8-CH
2), 3.50 (s, benzyl-CH
2), 7.10-7.60 (m, 6 H, 5 ArH, H-2), 7.70 (d, J = 6 Hz, 1 H, H-1), 8.00 (d, J = 6 Hz, 1 H, H-3), 11.1 (s, 1 H, OH).
Bis(trimethylsilyl) malonate (19a, R1 = H).
Malonic acid (
12b, R
1 = H) (10.4 g, 100 mmol) was reacted according to the procedure described in ref. [
14] with trimethylsilylchloride (21.7 g, 200 mmol) and worked up as described. The yield was 20.0 g (81%), yellowish oil, bp 102°C (15 mm Hg); lit. bp: 97°C (12 mm Hg) [
14]. IR: 2960 s, 1720 s, 1410 w, 1340 m cm
-1;
1H-NMR (CDCl
3): δ = 0.35 (s, 18 H, 6 Me), 3.3 (s, 2 H, CH
2). Analysis: for C
9H
20O
4Si
2 (248.43) Calcd. C 43.51, H 8.11; Found C 43.30, H 8.29
Bis(trimethylsilyl) 2-ethylmalonate (19b, R1 = Et).
To a stirred suspension of 2-ethylmalonic acid (12c, R1 = Et) (13.2 g, 100 mmol) and pyridine (16 mL, 200 mmol) in dry diethylether (150 mL), trimethylsilylchloride (21.7 g, 200 mmol) was added dropwise keeping the temperature at 0°C. Then the reaction mixture was stirred at room temperature for 2 h, hexane (200 mL) was added, and the separated salt was filtered off. The filtrate was distilled under reduced pressure (105-115°C, 15 mm Hg). The yield was 20.1 g (73 %), yellowish oil, bp 112°C (20 mm Hg). IR: 2985 s, 1720 s, 1460 w, 1350 m cm –1; 1H-NMR (CDCl3): δ = 0.35 (s, 18 H, 6 Me), 1.0 (t, 3 H, Me), 1.9 (m, 2 H, CH2), 3.2 (t, 1 H, CH); Analysis: for C11H24O4Si2 (276.48) Calcd. C 47.79, H 8.75; Found C 47.88, H 8.78
2-Ethyl-N,N'-diphenylmalondiamide (20) and N-phenylbutanamide (21).
A mixture of aniline (0.93 g, 10 mmol) and bis(trimethylsilyl) 2-ethylmalonate (
19b, R
1 = Et) (2.77 g, 10 mmol) was heated in an oil bath at 200°C for 8 h. After cooling to room temperature, the reaction mixture was digested with ethanol and cooled to 5°C. The dianilide
20 precipitated and was filtered by suction. The yield of
20 was 0.2 g (7 %), colorless prisms, mp 216-219°C (ethanol); tlc and spectral data are identical with an authentic sample obtained from diethyl 2-ethylmalonate and aniline. The filtrate was concentrated to 5 mL under reduced pressure and the residue treated with hexane. The precipitate was filtered by suction to yield 0.5 g (31 %) of
21, mp 87-89°C (hexane); lit. mp. 91-92°C [
25]; tlc and spectral data are identical with an authentic sample obtained from butanoic chloride and aniline.
2-(2,3-Dihydro-1H-benzimidazol-2-yliden)-3-oxobutanenitrile (23a, R1 = H).
A solution of 2-cyanomethylbenzimidazole (22) (1.57 g, 10 mmol) and bis(trimethylsilyl) malonate (19a, R1 = H) (2.62 g, 10.6 mmol) was refluxed in bromobenzene (50 mL) for 4-6 h during which the product separated out. After cooling, the product was filtered and washed with benzene. The yield was 1.0 g (50 %), mp > 350°C (dimethylformamide). IR: 3300-2500 b, m, 2220 s, 1620 w, 1600 s, 1510 m, 1470 w cm-1; 1H-NMR (DMSO-d6): δ = 2.20 (s, 3 H, Me), 7.20 and 7.50 (2 dd, 4 H, ArH), 13.1 (s, NH); Analysis: for C11H9N3O (199.21) Calcd. C 66.32, H 4.55, N 21.09; Found C 65.97, H 4.66, N 20.86
2-(2,3-Dihydro-1H-benzimidazol-2-yliden)-3-oxohexanenitrile (23b, R1 = Et).
A solution of 2-cyanomethylbenzimidazole (22) (1.57g, 10 mmol) and bis(trimethylsilyl) 2-ethylmalonate (19b, R1 = Et) (2.76 g, 10 mmol) was reacted and worked up as described for 23a. The yield was 0.8 g (35%), mp 318-320°C (dimethylformamide). IR: 3600-2600 b, m, 2220 s, 1620 w, 1600 s, 1560 s, 1530 s cm-1; 1H-NMR (DMSO-d6): δ = 0.95 (t, 3 H, Me), 1.60 (m, 2 H, CH2), 2.45 (t, 2 H, CH2CO), 7.20 and 7.50 (2 dd, J = 7 Hz, 4 H, ArH), 12.80 (s, b, 2 NH); 13C-NMR (DMSO-d6): δ = 13.0 (Me), 18.0 (CH2), 41.1 (CH2), 64.5 (=C-CN), 112.2 (C5 and C-6), 121.1 (CN), 123.2 (C4 and C7), 130.9 (C3a and C7a), 151.0 (C2 of benzimidazole), 192 (CO); Analysis: for C13H13N3O (227.27) Calcd. C 68.71, H 5.77, N 18.49; Found C 68.23, H 5.32, N 18.40
4-Hydroxy-6,7-diphenyl-pyrano[3,2-c]pyridine-2,5(6H)dione (26a, R2 = H).
A solution of malonic acid (
12b, R
1 = H) (104 g, 1 mol, well dried over potassium hydroxide) in warm dry ethyl acetate (800 mL) was treated with acetic anhydride (190 mL, 2.0 mol) and N1,1-diphenyl-1-ethanimine (
24a, R
2 = H) (98.0 g, 0.5 mol) and heated under reflux for 3 h by exclusion of moisture. Strong stirring is necessary because sometimes the reaction product precipitated which caused bumping of the boiling reaction mixture. After cooling the mixture was allowed to stand several hours for crystallization and was then filtered by suction. The precipitate was triturated with methanol (300 mL) for 3 h to remove impurities and filtered again to give a light yellow product which was pure enough for synthetic purposes. The yield was 71 g (43%), beige prisms, mp. 294-296°C (DMF), lit. mp. 293-296°C [
24].
Bis(carbamimidoyl) 2-benzylmalonate (29a).
To a solution of 2-benzylmalonic acid (12a, R1 = CH2Ph) (1.95 g, 0.01 mol) in diethylether (50 mL) at 0°C, N,N'-diisopropylcarbodiimide (2.52 g, 0.2 mol) was added and stirred at this temperature for 2 hours. The solvent was removed i.vac. and the oily residue washed with hexane and dried. The yield was 4.2 g of a yellow oil, which contained one new main product (shown by tlc analysis) besides traces of 12a and N,N’-diisopropyl urea. The oil was used without further purification for reactions. 1H-NMR (DMSO-d6) of the crude product: δ = 0.7-0.9 (m, aliphatic H, solvent), 1.1-1.4 (m, Me of i-propyl group), 3.1-3.3 (m, benzyl-CH2), 3.6-4.0 (m, i-propyl-CH, malonyl-CH), 7.1-7.4 (m, ArH), 8.8 (m, NH, OH).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}