Facile Synthesis of 1,6-Bis(2-furyl)-2,5-bis(2-hydroxy-3-formyl-5-methylbenzyl)-2,5-diazahexane: a New Dinucleating Ligand

{kind=link}
Abstract
:Introduction
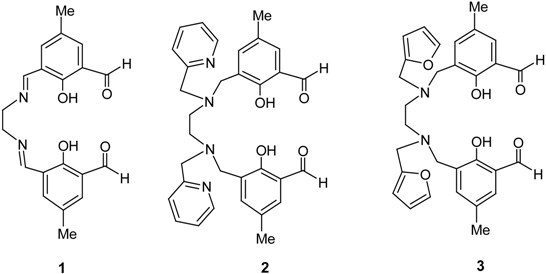
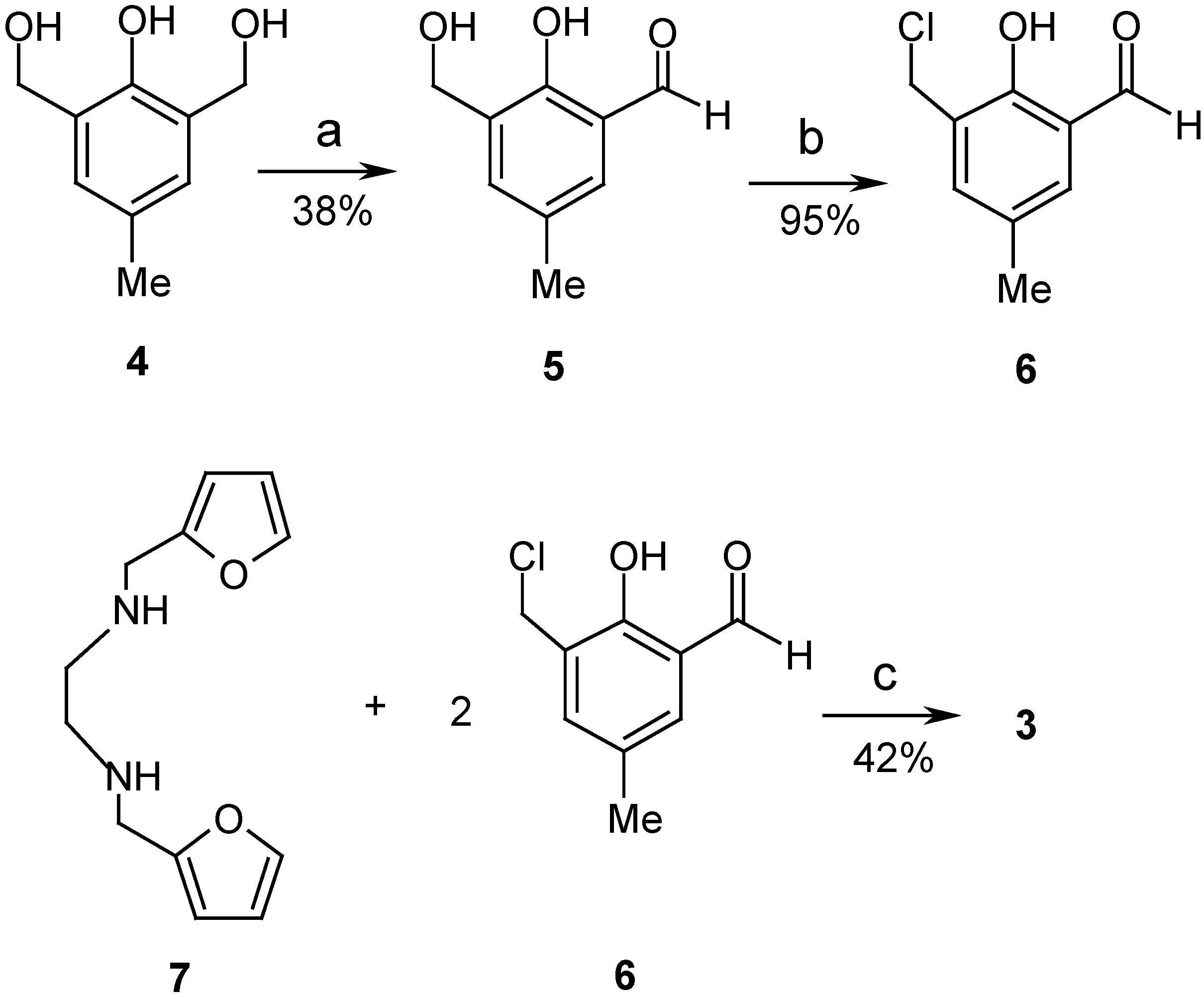
Results and Discussion
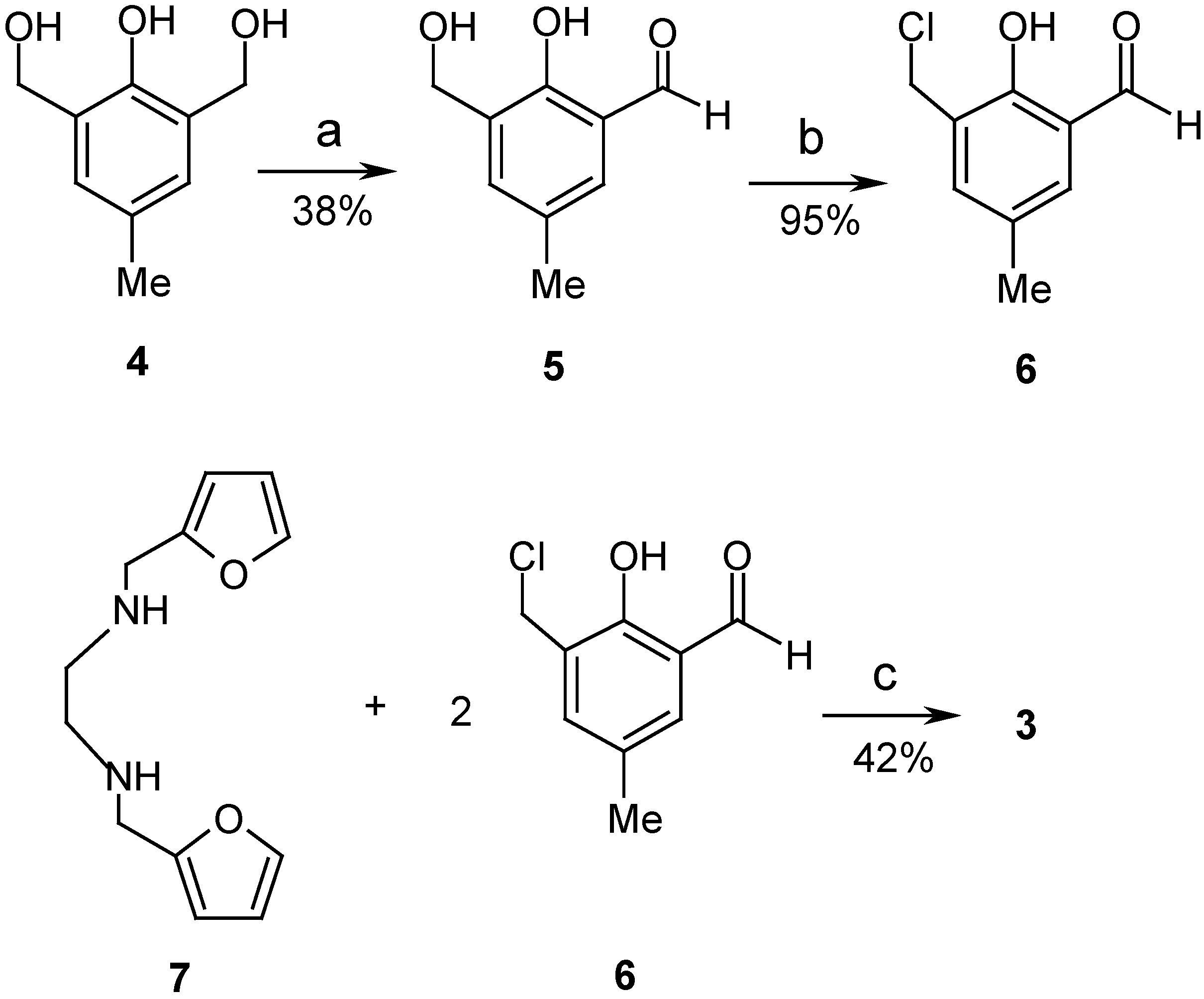
Experimental
General
2-Hydroxy-3-hydroxymethyl-5-methylbenzaldehyde (5).
2-Hydroxy-3-chloromethyl-5-methylbenzaldehyde (6).
1,6-Bis(2-furyl)-2,5-bis(2-hydroxy-3-formyl-5-methylbenzyl)-2,5-diazahexane (3).
Acknowledgements
References
- Coughlin, P.K.; Lippard, S.J. J. Am. Chem. Soc. 1984, 106, 2328.
- Lehn, J.M. Pure Appl. Chem. 1980, 52, 2441.
- Zanello, P.; Tamburini, S.; Vigato, P.A.; Mazzocchin, G.A. Coord. Chem. Rev. 1987, 77, 165.
- Okawa, H.; Kida, S. Bull. Chem. Soc. Jpn. 1972, 45, 1759.
- Tadokori, M.; Okawa, H.; Matsumoto, N.; Koikawa, M.; Kida, S. J. Chem. Soc. Dalton Trans. 1991, 1657.
- Fraser, C.; Johnston, L.; Rheingold, A.L.; Haggerty, B.S.; Williams, G.K.; Whelan, J.; Bosnich, B. Inorg. Chem. 1992, 31, 1835.
- Hu, Y.-F.; Hu, H.-W. Synthesis 1991, 325.
- Koenig, K.E.; Lein, G.M.; Stuckler, P.; Kaneda, T.; Cram, D.J. J. Am. Chem. Soc. 1979, 101, 3553.
- Rameau, J.Th.L.B. Rec. Trav. Chim. 1938, 57, 194.
- Woźniak, K.; Grech, E.; Szady-Chelmieniecka, A. Polish J. Chem. 2000, 74, 717.
- Corden, J.P.; Errington, W.; Moore, P.; Wallbridge, M.G.H. Acta Cryst. 1997, C53, 486.
- Sample Availability: Compounds 3, 5, 6, 7 are available from MDPI and the authors
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for noncommercial purposes.
Share and Cite
Sun, G.-C.; He, Z.-H.; Li, Z.-J.; Yuan, X.-D.; Yang, Z.-J.; Wang, G.-X.; Wang, L.-F.; Liu, C.-R. Facile Synthesis of 1,6-Bis(2-furyl)-2,5-bis(2-hydroxy-3-formyl-5-methylbenzyl)-2,5-diazahexane: a New Dinucleating Ligand. Molecules 2001, 6, 1001-1005. https://doi.org/10.3390/61201001
Sun G-C, He Z-H, Li Z-J, Yuan X-D, Yang Z-J, Wang G-X, Wang L-F, Liu C-R. Facile Synthesis of 1,6-Bis(2-furyl)-2,5-bis(2-hydroxy-3-formyl-5-methylbenzyl)-2,5-diazahexane: a New Dinucleating Ligand. Molecules. 2001; 6(12):1001-1005. https://doi.org/10.3390/61201001
Chicago/Turabian StyleSun, Gang-Chun, Zhan-Hang He, Zhong-Jun Li, Xiao-Dong Yuan, Zhi-Juan Yang, Guo-Xi Wang, Liu-Fang Wang, and Chang-Rang Liu. 2001. "Facile Synthesis of 1,6-Bis(2-furyl)-2,5-bis(2-hydroxy-3-formyl-5-methylbenzyl)-2,5-diazahexane: a New Dinucleating Ligand" Molecules 6, no. 12: 1001-1005. https://doi.org/10.3390/61201001
APA StyleSun, G.-C., He, Z.-H., Li, Z.-J., Yuan, X.-D., Yang, Z.-J., Wang, G.-X., Wang, L.-F., & Liu, C.-R. (2001). Facile Synthesis of 1,6-Bis(2-furyl)-2,5-bis(2-hydroxy-3-formyl-5-methylbenzyl)-2,5-diazahexane: a New Dinucleating Ligand. Molecules, 6(12), 1001-1005. https://doi.org/10.3390/61201001