Efficient Deprotection of Phenol Methoxymethyl Ethers Using a Solid Acid Catalyst with Wells-Dawson Structure

Abstract
:Introduction
Results and Discussion
Reactions using bulk catalyst
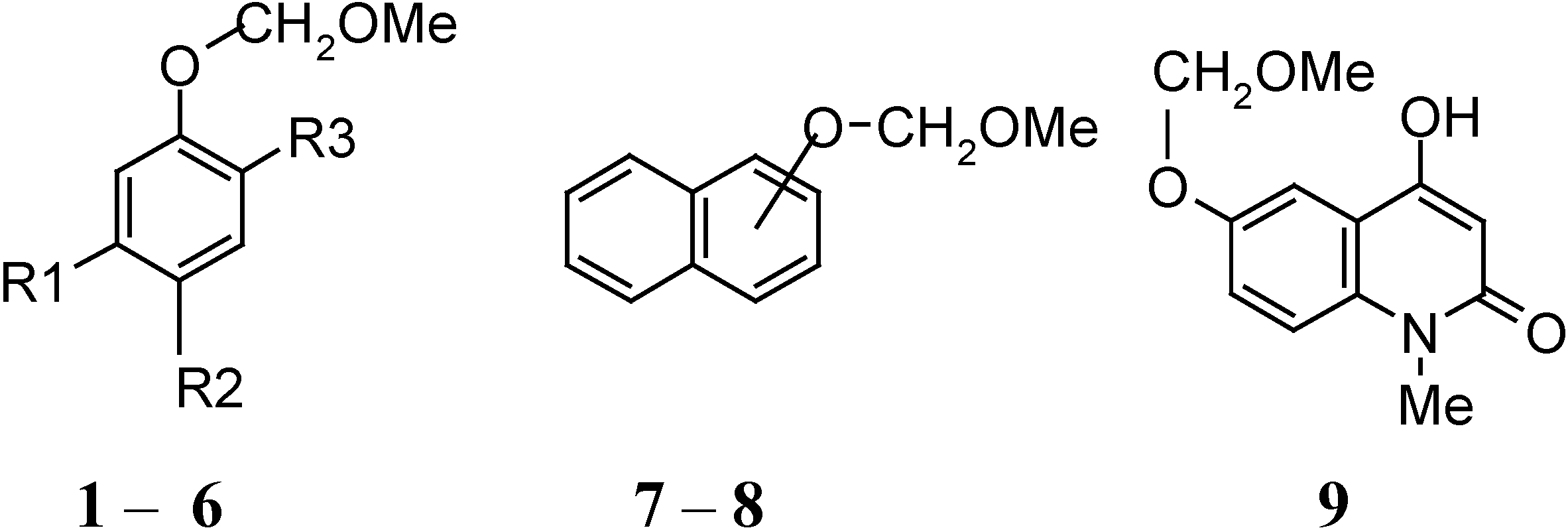
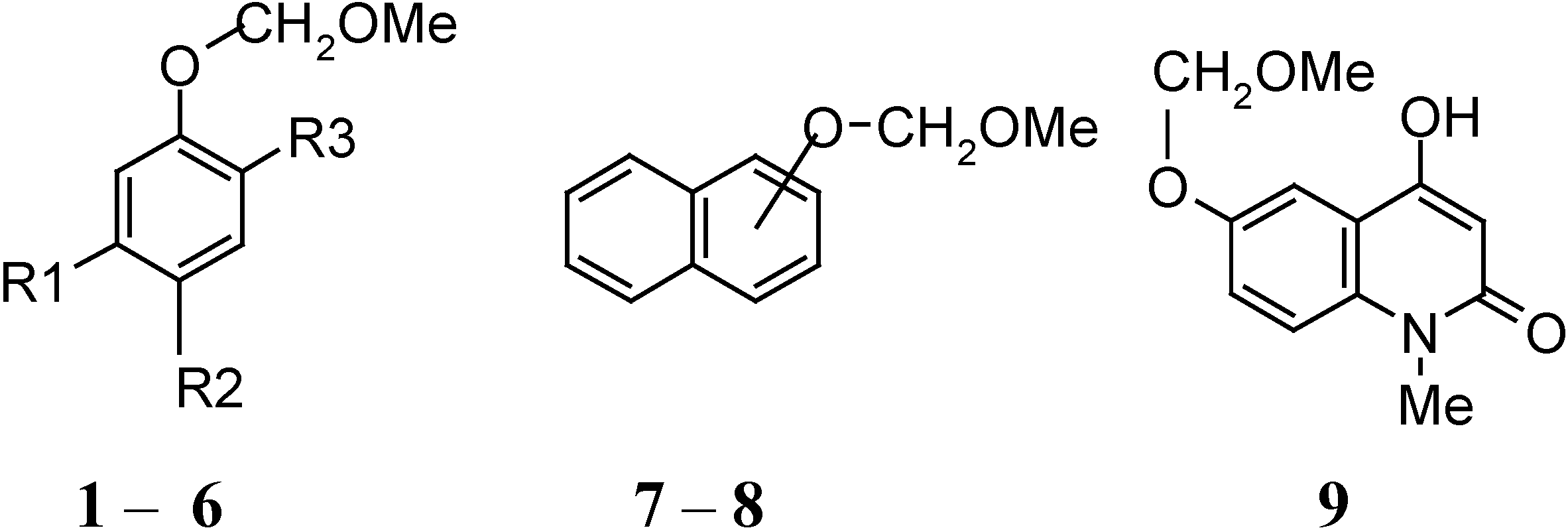
| Compound | R1 | R2 | R3 |
|---|---|---|---|
| 1 | OH | Ac | H |
| 2 | H | H | H |
| 3 | OCOC10H7-β | Ac | H |
| 4 | OH | COCH2COC10H7-β | H |
| 5 | CHO | H | OMe |
| 6 | OMe | H | Pr i- |
| 7 | α |
| 8 | β |

{kind=link}
| Run | Solvent | T (°C) | t (min) | % yield |
|---|---|---|---|---|
| 1 | Cl2(CH2)2 | 80 | 300 | 80 |
| 2 | MeOH | 65 | 45 | 98 |
| 3 | Cl2(CH2)2-MeOH 5% | 70 | 45 | 95 |
| 4 | Cl2(CH2)2-MeOH 1% | 70 | 45 | 96 |
| 5 | THF | 70 | 180 | 53 |
| 6 | THF-MeOH 1% | 65 | 40 | 98 |
Reactions using supported catalyst
| Run | Substrate | Catalyst | Catalyst (mol %) | Solvent | T (°C) | t (min) | Dilution | % yield |
|---|---|---|---|---|---|---|---|---|
| 1 | 1 | b | 1 | c | 80 | 30 | --- | 70 |
| 2 | 1 | b | 1 | c | 70 | 45 | --- | 95 |
| 3 | 1 | b | 1 | d | 70 | 45 | --- | 100 |
| 4 | 1 | b | 1 | d | 50 | 105 | --- | 97 |
| 5 | 1 | b | 1 | d | 75 | 45 | --- | 70 |
| 6 | 1 | b | 1 | d | 65 | 90 | x 2 | 100 |
| 7 | 1 | b | 1 | d | 65 | 120 | x 4 | 95 |
| 8 | 1 | b | 1 | d | 65 | 45 | x 1/2 | 90 |
| 9 | 1 | b | 5 | d | 65 | 60 | --- | 99 |
| 10 | 1 | b | 10 | d | 65 | 45 | --- | 95 |
| 11 | 1 | a | 1 | d | 65 | 50 | --- | 100 |
| 12 | 1 | a | 1 | d | 70 | 40 | --- | 90 |
| 13 | 1 | a | 5 | d | 65 | 55 | --- | 95 |
| 14 | 2 | b | 1 | d | 65 | 60 | --- | 100 |
| 15 | 2 | a | 1 | d | 65 | 55 | --- | 100 |
| 16 | 2 | a | 1 | d | 65 | 45 | x 1/2 | 90 |
| 17 | 3 | b | 1 | d | 65 | 60 | --- | 100 |
| 18 | 3 | a | 1 | d | 65 | 60 | --- | 95 |
| 19 | 4 | b | 1 | d | 65 | 60 | --- | 85 |
| 20 | 4 | a | 1 | d | 65 | 60 | --- | 80 |
| 21 | 5 | b | 1 | d | 65 | 60 | --- | 95 |
| 22 | 6 | b | 1 | d | 65 | 60 | --- | 90 |
| 23 | 7 | b | 1 | d | 65 | 60 | --- | 92 |
| 24 | 8 | b | 1 | d | 65 | 60 | --- | 95 |
| 25 | 9 | b | 1 | d | 65 | 60 | --- | 72 |
Conclusions
Acknowledgements
Experimental
General
Typical procedure
References
- Yardley, J.P.; Fletcher, H., III. Synthesis 1976, 244.
- Wiesner, K. Pure Appl. Chem. 1979, 51, 689.
- Fieser, L.F.; Fieser, M. Reagents for Organic Synthesis; J. Wiley and Sons, Inc.: New York, NY, 1967; Volume 1, p. 132. [Google Scholar]
- Jios, J.L.; Autino, J.C.; Pomilio, A.B. An. Asoc. Quim. Argent. 1995, 83, 183.
- Corral, R.A.; Orazi, O.O.; Autino, J.C. Tetrahedron Lett. 1983, 2359.
- Baronetti, G.T.; Briand, L.; Sedran, U.; Thomas, H. Appl. Catal. A: General 1998, 172, 265. [CrossRef]
- Sample Availability: Samples of compounds 1 and 3 are available from MDPI.
© 2001 by MDPI (http://www.mdpi.org). Reproduction is permitted for non commercial purposes.
Share and Cite
Romanelli, G.; Autino, J.C.; Baronetti, G.; Thomas, H. Efficient Deprotection of Phenol Methoxymethyl Ethers Using a Solid Acid Catalyst with Wells-Dawson Structure. Molecules 2001, 6, 1006-1011. https://doi.org/10.3390/61201006
Romanelli G, Autino JC, Baronetti G, Thomas H. Efficient Deprotection of Phenol Methoxymethyl Ethers Using a Solid Acid Catalyst with Wells-Dawson Structure. Molecules. 2001; 6(12):1006-1011. https://doi.org/10.3390/61201006
Chicago/Turabian StyleRomanelli, G., J. C. Autino, G. Baronetti, and H. Thomas. 2001. "Efficient Deprotection of Phenol Methoxymethyl Ethers Using a Solid Acid Catalyst with Wells-Dawson Structure" Molecules 6, no. 12: 1006-1011. https://doi.org/10.3390/61201006
APA StyleRomanelli, G., Autino, J. C., Baronetti, G., & Thomas, H. (2001). Efficient Deprotection of Phenol Methoxymethyl Ethers Using a Solid Acid Catalyst with Wells-Dawson Structure. Molecules, 6(12), 1006-1011. https://doi.org/10.3390/61201006