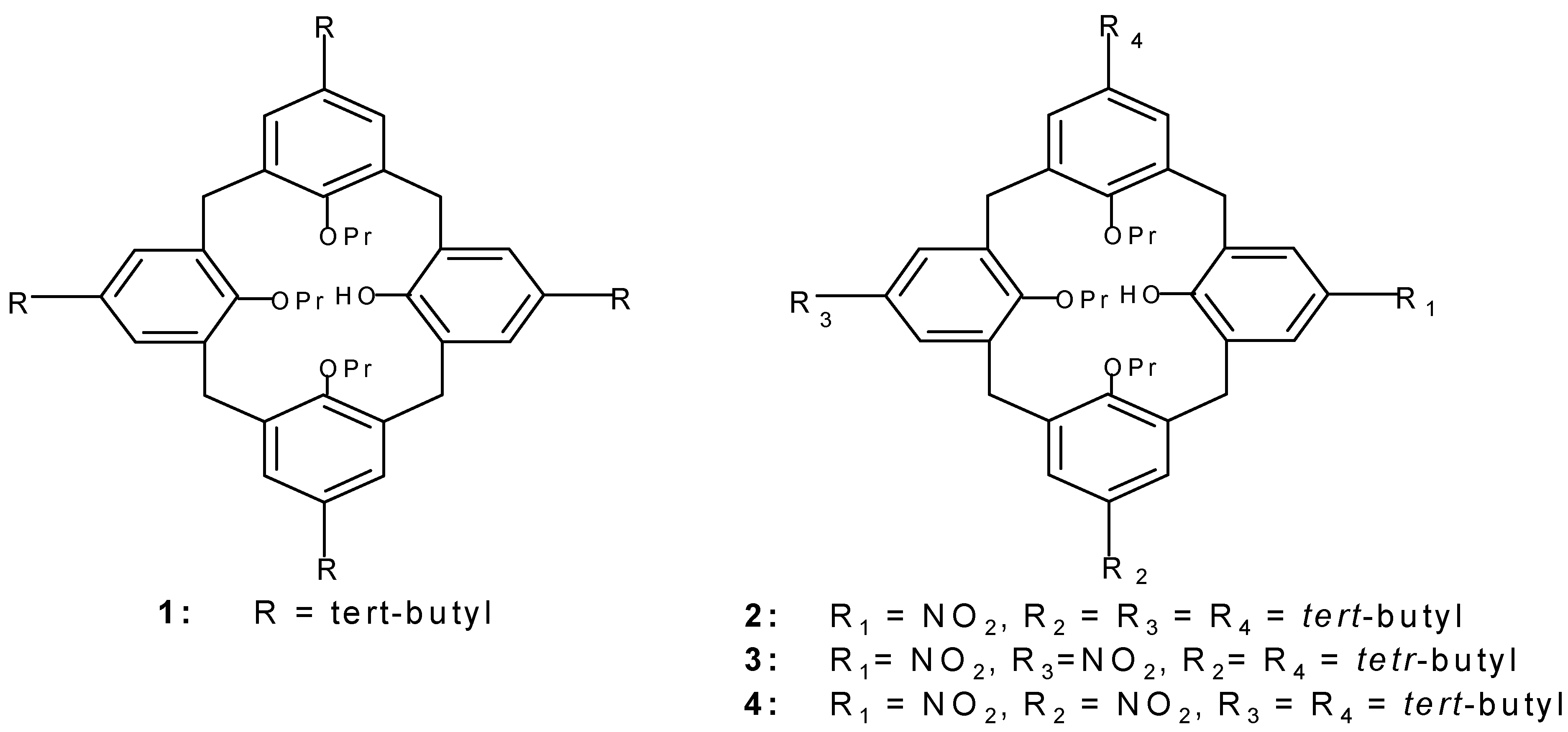
General
Melting points are taken on a Büchi SMP-20 apparatus and are uncorrected.
1H-NMR and
13C-NMR spectra were recorded on a Bruker AM-400 MHz in CDCl
3 with Me
4Si as an internal standard. Ele-mental analysis were carried out on Carlo-Erba-Analyser Model 1104. IR spectra were recorded on a Bruker IFS-25 spectrophotometer. The calix(4)arene
1 was prepared according to the published proce-dure [
10].
Procedure for the preparation of 2
To a solution of 10g (12.9 mmol) 1 in dichloromethane (50mL) and glacial acetic acid (30 mL), 63% HNO3 (5 mL) was added dropwise at -15°C in 2 min and the reaction mixture was then stirred at room temperature for 4 min and poured into water (250 mL). The organic layer was extracted with di-chloromethane (2x100mL), the solvent was removed and solid residue was dissolved in methanol (100 mL), concentration of solution to a volume of ca. 50 mL led to the appearance of a pale yellow pre-cipitate (yield 85%).The product thus obtained was pure enough for subsequent reactions but could be further purified by recrystallization from a mixture of dichloromethane and methanol; mp 182-184°C.
IR ν max (KBr)/cm-1 3456, 1592, 1472, 1334, 1201, 1006; δ H( 400 MHz; CDCl3 ) 0.83[18H, s, C(CH3 )3 )], 0.95[3H, t, CH3 ], 1.10 [6H, t, CH3 ], 1.35[9H, s, C(CH3)3 ], 1.93[4H, m, CH2 ], 2.2[2H, m, CH2 ], 3.19 and 4.34[4H, d of d, J=12.6 Hz, ArCH2 Ar], 3.75[2H, t, OCH2 ], 3.81[ 4H, t, O CH2 ], 3.39 and 4.31[4H, d of d, J=13.9 Hz, ArCH2 Ar], 6.45[ 2H, d, J=2.3 Hz, ArH], 6.60[2H, d, J=2.3Hz, ArH], 7.16[2H, s, ArH], 7.22[1H, s, OH], 8.06[2H, s, ArH]; δ C( 100 MHz) 9.56, 10.70, 22.55, 23.37, 31.10, 31.36, 31.47, 31.68, 33.74, 34.15, 58.46, 76.15, 78.01, 124.31, 125.68, 125.70,129.80, 132.65, 135.76, 139.34, 145.82, 151.75, 153.78, 159.98; M/Z (FD) 764(m+ , 100%). Anal. Calcd for C49 H65 NO6 : C, 77.06%; H, 8.52%; N, 1.84%. Found C, 76.8%, H, 8.7%; N, 2.2%.
Procedure for the preparation of 3 and 4
To a solution of 2g (2.58 mmol) 1 in dichloromethane (25mL) and acetic anhydride (4mL) at -10°C, 63% HNO3 (1 mL) was added in 2 min and the reaction mixture was then stirred at room temperature for 5 min and poured into water (250mL). The organic layer was extracted by dichloromethane (2x100mL), the solvent was removed and the solid residue was dissolved in methanol (100mL), con-centration of solution to ca. 50 mL led to a white precipitate. 3 is obtained as a white crystals in 63% separated yield (crystallized from a mixture of CH2Cl2 and CH3OH). mp, 214-217°C; 4 was purified by column chromatography of the mother liquors (silicagel, CH2Cl2 , n-hexane, 1:4); yield 22% mp, 225-228°C.
Spectral data for 3
IR ν max (KBr)/cm-1 3510, 1598, 1513, 1343 and 1014. δ H( 400 MHz; CDCl3 ) 0.86[18H, s, C(CH3)3 ], 0.97[3H, t, CH3 ], 1.11 [6H, t, CH3 ],1.94 [4H, m, CH2 ], 2.22[2H, m, CH2 ], 3.35 and 4.42[4H, d of d, J=12.9 Hz, ArCH2Ar], 3.78[4H, t, OCH2 ], 3.96[2H, t, OCH2 ], 3.43, 4.29[4H, d of d, J=13.8 Hz, ArCH2Ar], 6.56[4H, s, ArH], 7.37[1H, s, OH], 8.07[2H, s, ArH], 8.10[2H, s, ArH]; δ C( 100 MHz) 9.48, 10.66, 22.44, 23.40, 33.01, 31.13, 31.43, 33.87, 76.67, 78.15, 124.12, 124.46, 125.28, 125.46, 129.43, 130.30, 130.98, 138.06, 140.10, 143.05, 146.64; 151.83, 159.70, 164.05; M/Z (FD) 753(m+, 100%). Anal. Calcd for C45H56N2O8 : C, 71.79%; H, 7.50%; N, 3.72%. Found C, 71.5%, H, 7.5%; N, 3.1%.
Spectral data for 4
IR ν max (KBr)/cm-1 3508, 1592, 1521, 1341 and 1005. δ H( 400 MHz; CDCl3 ) 0.74[9H, s, C(CH3)3], 0.92[3H, t, CH3 ], 1.12 [3H, t, CH3 ], 1.13[3H, t, CH3 ], 1.40[9H, s, C(CH3)3 ], 1.90 [2H, m, CH2 ], 2.08[2H, m, CH2 ], 2.23[2H, m,CH2 ], 3.21 and 4.40[2H, d of d, J=13.1 Hz, ArCH2Ar], 3.25 and 4.30[2H, d of d, J=13.0 Hz, ArCH2Ar], 3.42 and 4.46[2H, d of d, J=13.7 Hz, ArCH2Ar], 3.51 and 4.20[2H, d of d, J=14.5 Hz, ArCH2Ar], 3.77[6H, m, OCH2 ], 6.17[1H, s, OH], 6.45[1H, d, J=2.1 Hz, ArH], 6.51[1H, d, J=2.1 Hz, ArH], 7.21[1H, d, J=2.3 Hz, ArH], 7.22[1H, d, J=2.6 Hz, ArH], 7.24[1H, d, J=2.2 Hz, ArH], 7.27[1H, d, J=2.6 Hz, ArH], 8.11[1H, d, J=2.5 Hz, ArH], 8.14[1H, d, J=2.5 Hz, ArH]; δ C( 100 MHz ) 9.43, 10.69, 10.71, 22.52, 23.34, 23.46, 30.63, 30.80, 31.05, 31.08, 31.63, 34.01, 34.30, 76.45, 77.63, 78.24, 122.31, 123.51, 124.30, 124.66, 124.77, 125.88, 126.72, 126.77,128.64, 128.77, 129.94, 133.56, 133.78, 134.80, 135.08, 136.00, 139.93, 142.81, 146.78, 147.54, 150.68, 153.79, 159.31, 161.73; M/Z (FD) 753(m+, 100%). Anal. Calcd for C45H56N2O8: C, 71.79%; H, 7.50%; N, 3.72%. Found C, 71.6%, H, 7.5%; N, 3.1%.

{kind=link}