Results and Discussion
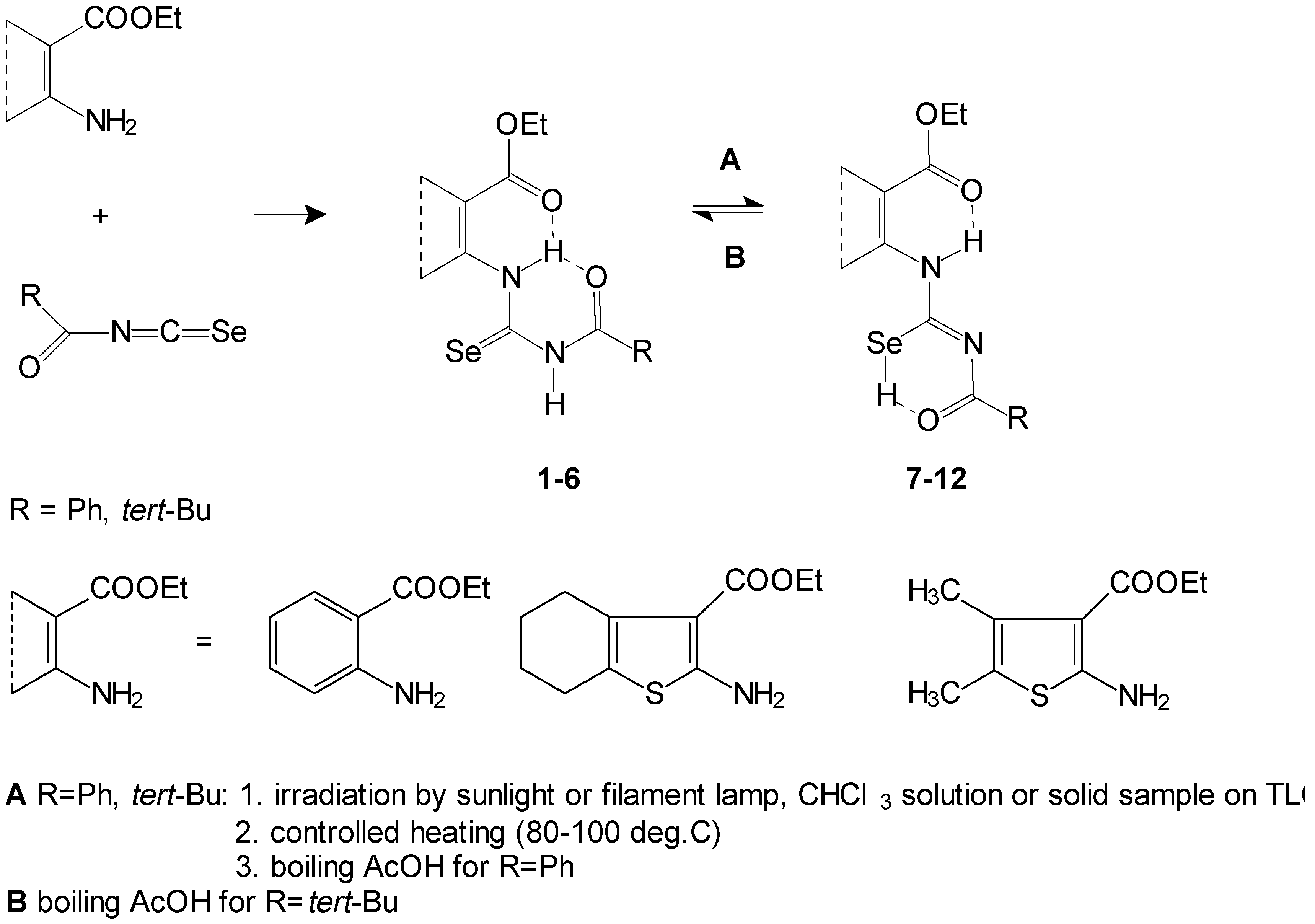
Starting with acylselenoureido
1-
6 and acylisoselenoureido
7-
12, derivatives were prepared as mentioned in ref. [
1] (
Scheme 1).
Sulfur analogues of the title compounds were cyclized in acid medium to 4
H-1,3-thiazine skeleton formation as mentioned in the introduction. This cyclization proceeded by attack of sulfur on the carbon atom of the alkoxycarbonyl group. Fused 2-acylamino-4
H-1,3-thiazine-4-ones were formed after neutralization of the reaction mixture also in the cases of the benzene and thiophene skeleton. The products of deacylation,
i.e. the corresponding 2-amino compounds were formed at 100°C [
2,
3,
4,
5].
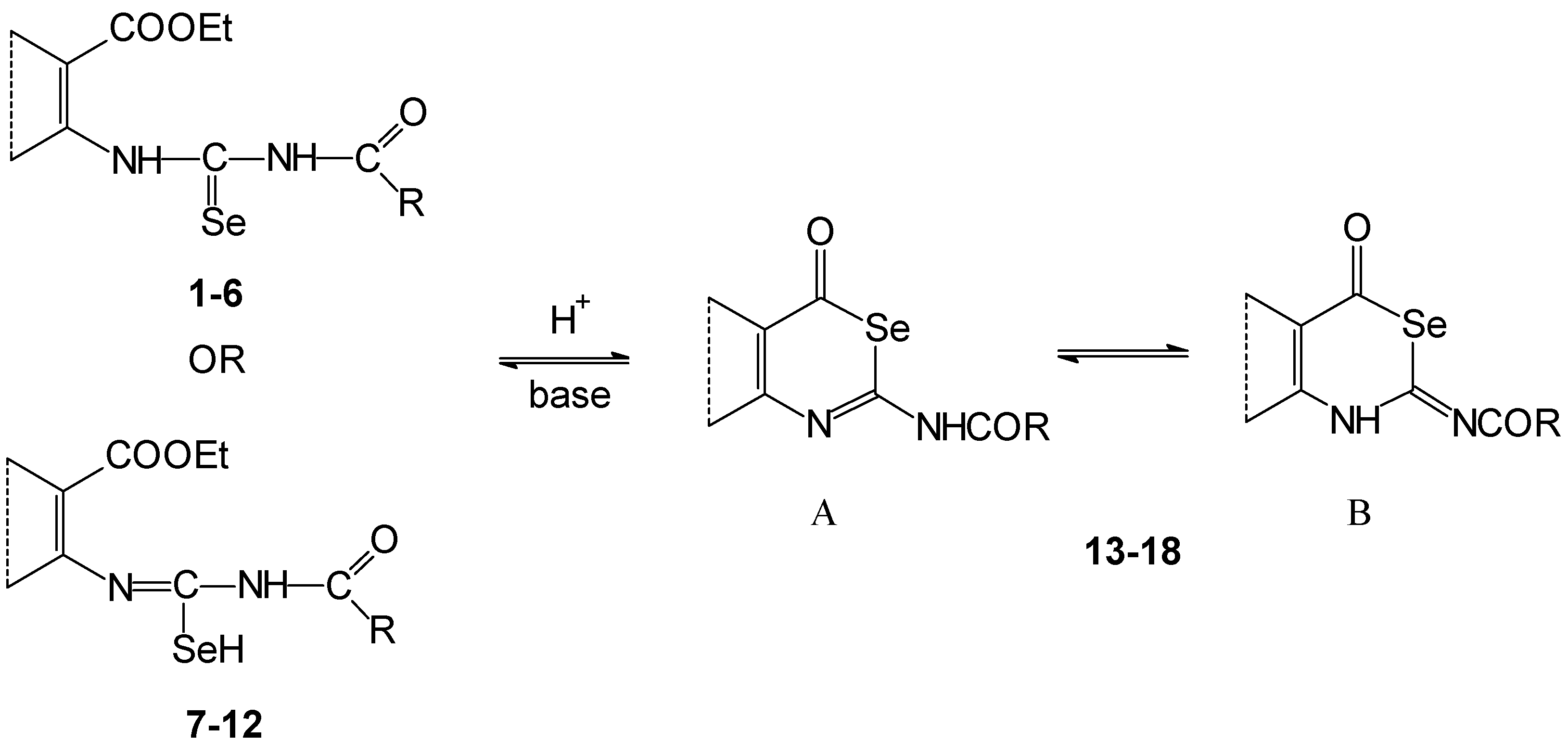
We prepared the corresponding 1,3-selenazine compounds with the aim of comparing the properties and reactivity of the 1,3-thiazine and 1,3-selenazine skeletons. Acylselenourea
1-
6 or acylisoselenourea
7-
12 underwent reaction in 94-96% sulfuric acid at room temperature (
Scheme 2). Fused 2-acylamino-4
H-1,3-selenazine-4-ones
13-
18 were isolated after mixing with ice as free bases. We attempted deacylation of
13-
18 by increasing the temperature of the reaction mixture but oxidative destruction gave elimination of elementary selenium. We also attempted to deacylation of compounds
13-
18 by treating with hydrogen chloride in refluxing methanolic solution. Destruction of these compounds was also observed during this operation. Selane was eliminated and oxidized by oxygen in the air to the red modification of selenium. Selane was identified in the vapors over the reaction mixture by a saturated solution of silver nitrate. The conversion of starting compounds
1-
12 to
13-
18 occurred considerably quickly in comparison to sulfur analogues.
Identification of 1,3-selenazine-4-ones 13-18 was supported by C, H, N, Se elemental analysis, FTIR, 1H- and 13C-NMR spectra and by comparison of data with simulated values and with data of sulfur analogues.
FTIR and NMR spectra of compounds
13-
18 showed that they may exist in two tautomeric forms (A and B). This was also observed for sulfur analogues: 2-acylamino-4
H-1,3-selenazine-4-ones (A) and 2-acylimino-1
H,4
H-selenazine-4-ones (B) (
Scheme 2). The tautomeric form B is favored in the case of benzoyl derivatives. The tautomeric form of
14 is in the ratio 1:5 in favor (B) and
17 are in the ratio 7:1 in favor (A). The ratio of tautomers was measured on the basis of NMR experiments. This form is stabilized probably by a hydrogen bond between the hydrogen atom of N1 and the oxygen atom of the benzoyl group. A similar case occurs acylselenoureas
1-
6.
Our attention was also concentrated on the cyclization of both types of the title compounds,
i.e. selenoureas
1-
6 and isoselenoureas
7-
12 in the presence of base. Cyclization of acylthioureas similar to selenoureas
1-
6 occurred by an attack of the nitrogen atom of a functionalized carboxyl group on the pyrimidine skeleton, provided by the action of a base (ammonium, sodium or potassium carbonate, hydroxide in water or water-alcoholic solution)[
4,
6,
7,
8]. Cyclization proceeded in independently of either pH or reaction temperature either with deacylation or without elimination of the acyl group.
Application of the mentioned methods for the cyclization of acylselenoureas 1-6 and acylisoselenoureas 7-12 was not successful because the conditions evoked their destruction and selenium elimination. The destruction proceeded also in the atmosphere of an inert gas at -20°C.
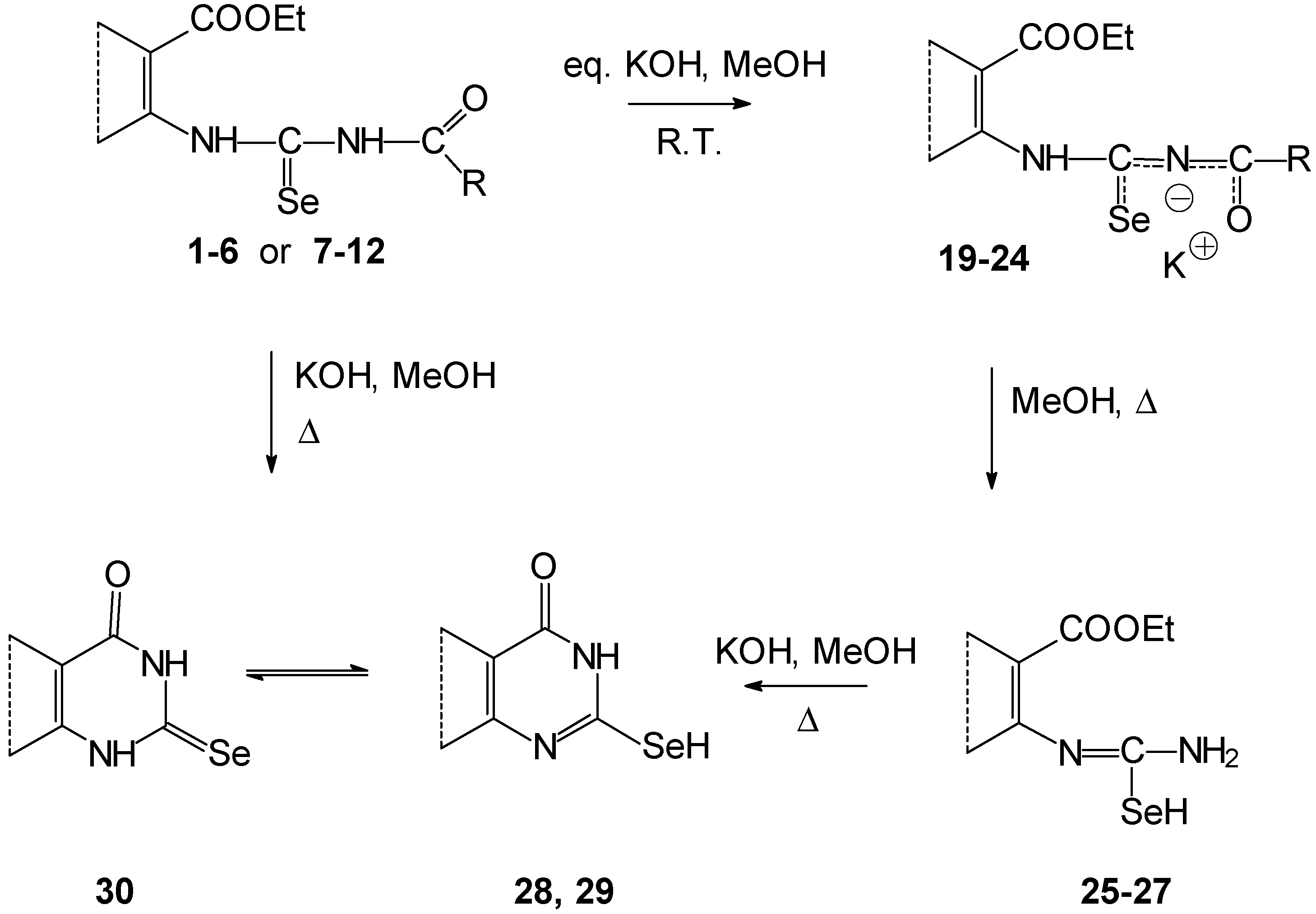
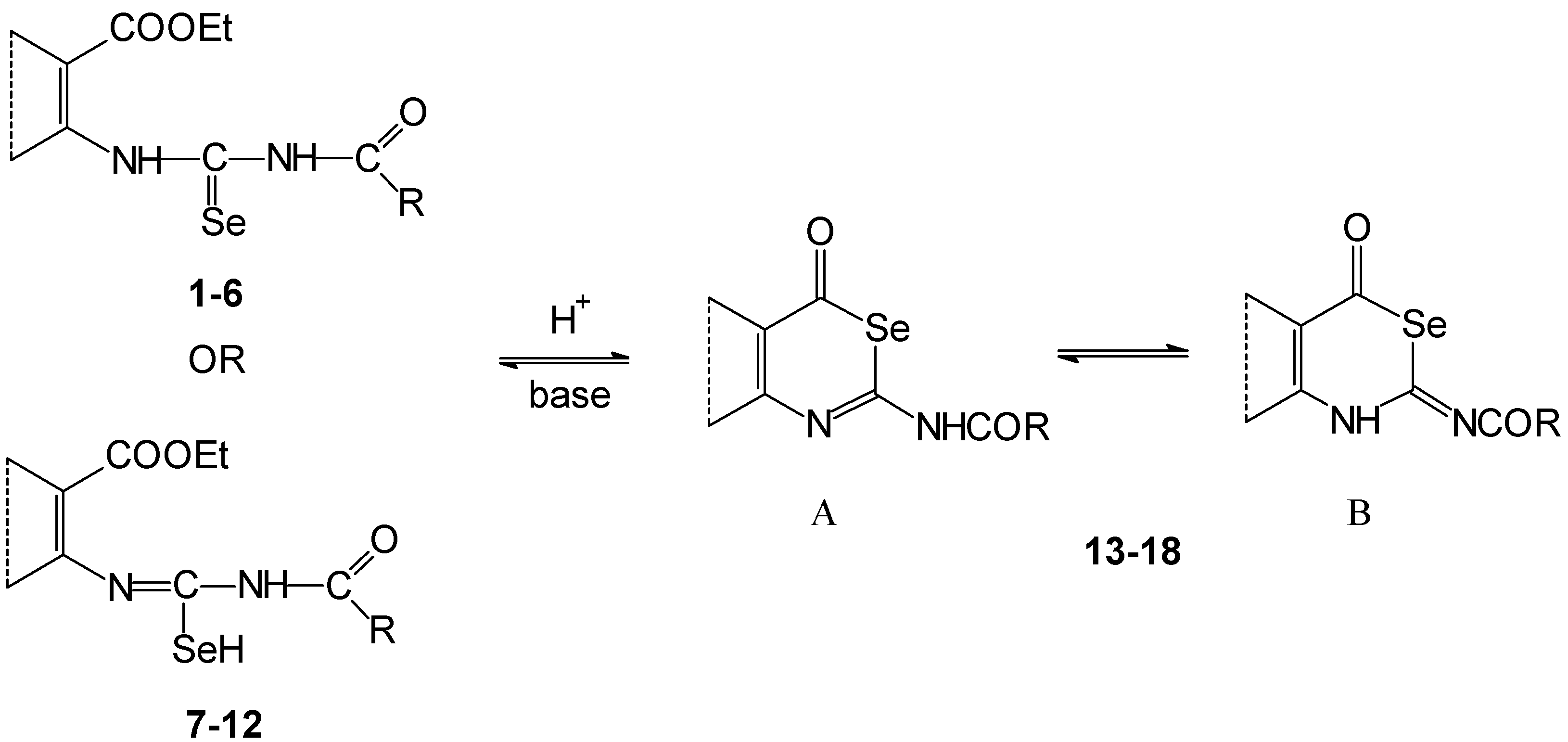
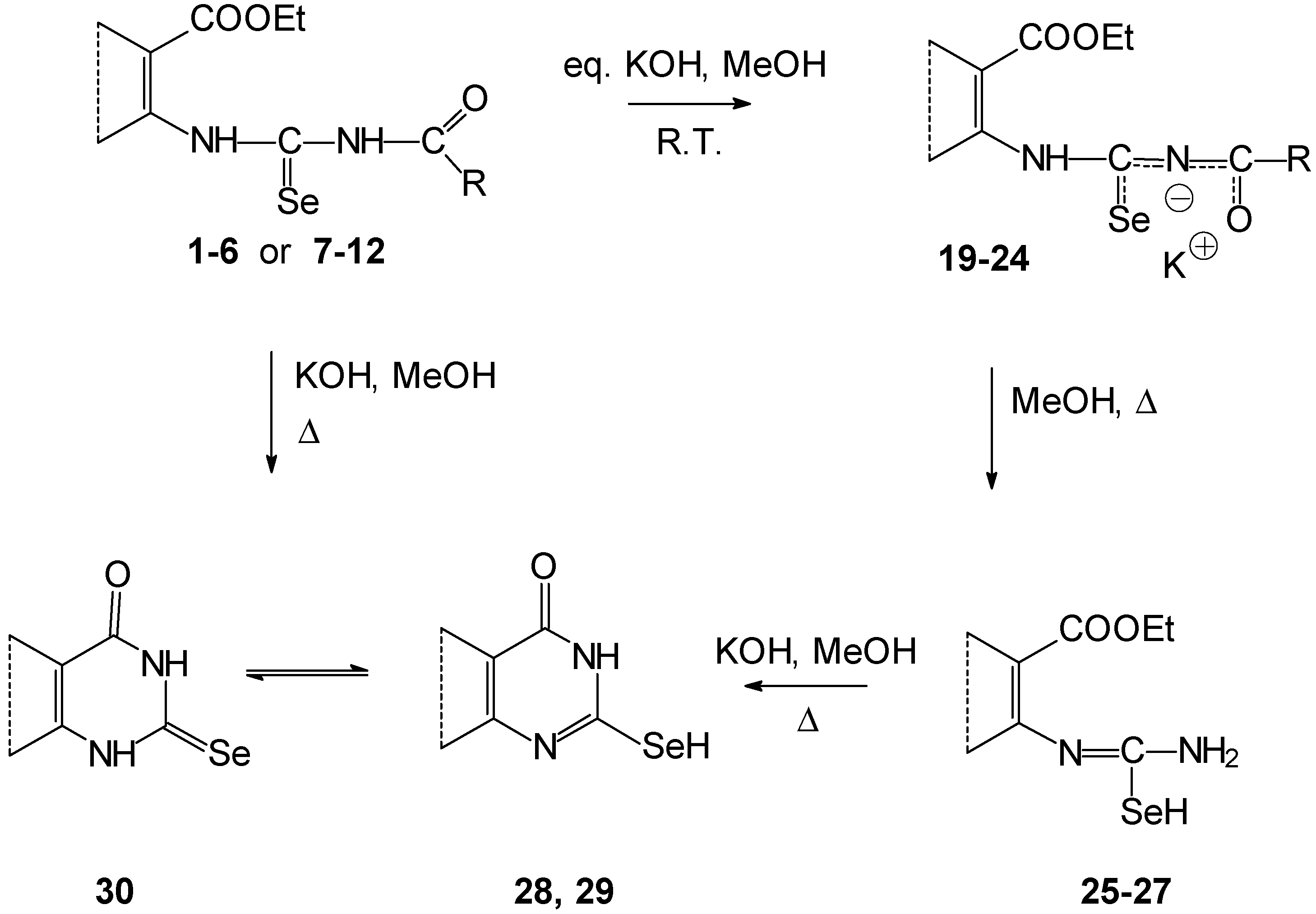
Deprotonation of the compounds
1-
12, treated with anhydrous methanolic potassium hydroxide evoked formation of their potassium salts (
Scheme 3). We have assumed that deprotonation is predominately on the nitrogen atom of the acylamino group. The created anion may be thermodynamically more stable than in the case of deprotonation of the amino group in the vicinity of an aryl skeleton. This is in consequence of delocalization of the negative charge in the [C(Se)NC(O)]
- fragment. A similar situation occurs for example on deprotonation of the 1,3-dioxo compounds.
If the deprotonations were accomplished with an equivalent of potassium hydroxide at room temperature, potassium salts 20, 21, 23, 24 were isolated in the thiophene series in comparison to the sulfur analogues because their solubility is lower.
Potassium salts 19 and 22 were not obtained in crystalline form nor eliminated from solution by addition of solvents such as benzene, dichlormethane, and their mixtures with petroleum ether, respectively. The products 19 and 22 were obtained after precipitation as a tar substance.
Identification of potassium salts 20, 21, 23, 24 was confirmed by C, H, N, Se elemental analysis, FTIR, 1H- and 13C-NMR (in the case of 20 and 23) spectroscopies. The 13C-NMR of salts 21 and 24 were not measured because of their low solubility and stability in hexadeuterodimethyl sulfoxide. The corresponding vibration bands of NHCO or NHCSe groups were not found in the FTIR spectra of potassium salts. On the other hand two very intense bands were observed at the wave number 1460 and 1340 cm-1. We have assumed that these are vibration bands of the deprotoned [C(Se)NC(O)]- group.
If the reaction mixture containing salts
20,
21,
23 and
24 in a methanolic suspension or salts
19,
22 prepared
in situ in methanolic solution were refluxed in the presence of an excess of potassium hydroxide, cyclization and deacylation proceeded to the corresponding pyrimidine-4-ones. After acidification of the reaction mixture, they crystallized as free bases (fused 2-selanyl-pyrimidine-4-one
28,
29 and 2-selenoxo compounds
30) (
Scheme 3).
Cyclization of salts
19,
22 (prepared
in situ) proceeded in methanolic solution with an equivalent of potassium hydroxide. 2-Selanyl-3,4-dihydroquinazoline-4-one
28 was formed by standing of the reaction mixture at room temperature or by short reflux. Compound
28 was obtained directly as free base. We assume deacylation is the first process. Then the formed 2-selenoureidobenzoate
25 thermally and spontaneously cyclizes. Similar results were observed by the addition of ammonia to 2-isoselenocyanatobenzonitrile [
9] or its sulfur analogue[
10]. The adducts formed
in situ already cyclized below 0°C. The corresponding selenoureas or thioureas were not observed.
Isolated potassium salts
20,
21,
23, and
24 did not cyclize because of their lower reactivity under the same conditions. The lower reactivity of thiophene
vs. benzoderivatives was also found during cyclocondensation of thioureas[
11]. The deacylated isoselenoureas
26 and
27 were isolated as the finished products. Their cyclizations were by base action only. The lower reactivity of cyclization in the thiophene series in comparison with the benzene series may be explained by a more favorable distribution of electron density on the reaction centers of benzo derivatives. Another explanation may be by due to the higher internal strain of the thienopyrimidine skeleton as a consequence of the higher external bond angles of the thiophene ring compared to the benzoanalogue [
11].
Our results indicate a different course of base initiated cyclization in the comparison with acylselenoureas and their sulfur analogues. In the case of the latter, reaction courses were preparative [
7] and by kinetic methods[
12] confirmed that deacylation proceeded after pyrimidine skeleton formation. The title compounds were converted in the reverse order during this process.
The identity of synthesized isoselenoureas 26, 27 was supported by elemental analysis, FTIR, 1H- and 13C-NMR spectroscopy. In FTIR spectra there were observed intensive C=N vibration, but vibration bands of selenoamide group I, III were not found. The chemical shift of 13C(Se) signal with a lower value than 160 ppm and value of coupling 13C-77Se showed that this carbon is bonded to the selenium atom by a single bond as was observed in acylisoselenoureas1. This fact shows that the compounds 26 and 27 exist mainly in the tautomeric 3-isoselenoureido form, as was confirmed by 1H-NMR spectra in which two different protons were not observed. Existence of the tautomeric 1-isoselenourea form was not found. The proton signal of the imino group would be at a higher value of the chemical shift than was found, in implication of magnetic anisotropy of C=N bond.
The structure of fused 4-pyrimidinones 28-30 were supported by elemental analysis, FTIR, 1H- and 13C- NMR spectroscopy. FTIR and 13C- NMR spectra of products showed that compound 28 and 29 were stable under the experimental conditions in tautomeric form as fused 2-selanylpyrimidine-4-one but 30 as selenoxo compounds. NMR spectra of 28-30 compounds was measured in deuterotrifluoroacetic acid solution because of their low solubility in other solvents.
Experimental
General
Chemicals and reagents were purchased from Fluka Chemie Co. and used without further purification. The title selenoureas derivatives were prepared according to paper [
1]. Melting points of prepared the compounds were measured on Boetius Rapido PHMK 79/2106 (Wägetechnik) instruments and were not corrected. Purity of all compounds was determined by C, H, N elemental analyses on an instrument 1102 (Erba) and by determinations of selenium on spectrometer ICP AES 7500 (Unicam). TLC was carried out on Silufol UV 254 plates (Kavalier, Votice) and the detection with Fluotes Universal (Quartzlampen, Hanau) and with iodine vapors, respectively. Chloroform, diethyl ether or acetonitrile was used as eluent in a container saturated with vapors of the used solvent. FTIR spectra were taken on a spectrometer Genesis ( Unicam) in potassium bromide pellets or in chloroform solution.NMR spectra were measured in deuterochloroform, hexadeuterodimethyl sulfoxide or in deuterotrifluoroacetic acid on a Bruker Avance DRX-500 spectrometer. The
13C- and
1H-NMR spectra were referenced to tetramethylsilane as internal standard or to the solvent signals of CDCl
3 and of residual CHCl
3 at 77.00 ppm (
13C) and 7.27 ppm (
1H), respectively. Spectral width : 9000 Hz for
1H, 27500 Hz for
13C. The measured
13C - and
1H-NMR spectra were correlated with those obtained by on-line simulation (Advanced Chemistry Development, INC., Toronto, Canada).
Fused 2-acylamino-4H-1,3-selenazine-4-ones 13-18
Acylselenourea 1-6 or acylisoselenourea 10-12 (10 mmol) was suspended under stirring in concentrated sulfuric acid (ca 96%, 50 ml) at a temperature of under 20°C. The formed reaction solution was stirred at room temperature for 2-3 h, and subsequently mixed with 100 g crushed ice. The suspension of products (13-18) was separated by suction. Crystals were washed with cold water, methanol and diethyl ether. Drying was in vacuo at room temperature.
2-Benzoylimino-1H,4H-benzo[d][3,1]selenazine-4-one 13B
M.w. 329.22.
C15H10N2O2Se
M.p. 188-189°C.
Elemental analysis (%calc./%found.) C 54.73/54.55, H 3.06/3.08 N5.51/8.49, Se 23.98/23.99.
Yield (from 1) 3.2 g (96%), (from 7) 3.0 g (91%).
FTIR (KBr pellets) cm-1: 3195 (NH), 1670 (Se-C=O), 1689, 1560 (NHCO), 1634 (C=N).
1H NMR (CDCl3, B) δ: 7.26-8.15 (m, 9H, C6H4 and C6H5), 12.75 (s, 1H, NH).
13C NMR (CDCl3, B) δ: 120.93 (CH), 124.35 (CH), 124.22 (CH), 127.28 (CH), 127.52 (CH), 127.99 (CH), 133.22 (CH), 135.33 (C), 142.41 (C), 142.39 (C), 143.41 (N-C-Se), 175.97 (C=O, COC6H5), 191.88 (Se-C=O).
2-Benzoylamino-5,6,7,8-tetrahydro-4H-benzo[1]thieno[2,3-d][1,3]selenazine-4-one 14A and 2-Benzoylimino-5,6,7,8-tetrahydro-4H-benzo[1]thieno[2,3-d][1,3]selenazine-4-one 14B (in ratio 1:5)
M.w. 389.33.
C17H14N2O2SSe
M.p. 204-205°C.
Elemental analysis (%calcd/%found) C 52.54/52.42, H 3.62/3.63, N 7.20/7.12, Se 20.49/20.53.
Yield (from 2) 3.8 g (98%), (from 8) 3.7 g (95%).
FTIR (KBr pellets) cm-1: 3190 (NH), 1670 (Se-C=O), 1695, 1540 (NHCO), 1620 (C=N).
1H NMR (CDCl3, A) δ: 1.82-1.84 (m, 4H, 5-CH2 and 6-CH2), 2.74-2.76 (m, 2H, 4-CH2), 2.94-2.96 (m, 2H, 7-CH2), 7.55-8.07 (m, 5H, C6H5CO), 9.30 (s, 1H, NHCO).
1H NMR (CDCl3, B) δ: 1.44-1.45 (m, 4H, 5-CH2 and 6-CH2), 2.68-2.70 (m, 2H, 4-CH2), 2.77-2.79 (m, 2H, 7-CH2), 7.65-7.94 (m, 5H, C6H5CO), 13.28 (s, 1H, NH).
13C NMR (CDCl3, A) δ: 22.24 (CH2), 22.68 (CH2), 24.81 (CH2), 26.46 (CH2), 128.23 (CH), 127.45 (C), 129.16 (C), 131.31 (C), 133.36 (CH), 133.48 (CH), 140.95 (C), 163.07 (N-C-Se), 167.01 (C=O, COC6H5), 183.54 (Se-C=O).
13C NMR (CDCl3, B) δ: 22.24 (CH2), 22.68 (CH2), 24.81 (CH2), 26.46 (CH2), 128.23 (CH), 127.45 (C), 129.16 (C), 131.31 (C), 133.36 (CH), 133.48 (CH), 140.95 (C), 157.52 (N-C-Se), 165.64 (C=O, COC6H5), 183.54 (Se-C=O).
2-Benzoylimino -5,6-dimethyl-4H-thieno[2,3-d][1,3]selenazine-4-one 15B
M.w. 363.29.
C15H12N2O2SSe
M.p. 217-220°C.
Elemental analysis (%calcd/%found) C 49.59/49.12, H 3.33/3.17, N 7.71/7.26, Se 21.96/21.34.
Yield (from 3) 3.5 g (96%), (from 9) 3.5 g (96%).
FTIR (KBr pellets) cm-1: 3300, 3190 (NH), 1680 (Se-C=O), 1662, 1534 (NHCO), 1624 (C=N).
1H NMR (CDCl3, B) δ: 2.37 (s, 3H, CH3), 2.45 (s, 3H, CH3), 7.52-8.04 (m, 5H, C6H5CO), 11.92 (s, 1H, NH).
13C NMR (CDCl3, B) δ: 12.63 (CH3), 14.24(CH3), 120.56 (C), 127.42 (C), 128.22 (CH), 128.38 (C), 129.15 (C), 129.24 (CH), 131.69 (CH), 133.49 (C), 157.21 (N-C-Se), 165.65 (C=O, COC6H5), 183.45 (Se-C=O).
2-(2,2-dimethylpropanoyl)amino-4H-[3,1]-benzoselenazine-4-one 16A
M.w. 309.23.
C13H14N2O2Se
M.p. 118.120°C.
Elemental analysis (%calcd/%found) C 50.49/50.42, H 4.56/4.48, N 9.06/8.98, Se 25.78/25.69.
Yield (from 4) 2.8 g (91%), (from 10) 2.6 g (84%).
FTIR (KBr pellets) cm-1: 3190 (NH), 1677 (Se-C=O), 1651, 1630 (NHCO), 1620 (C=N).
1H NMR (CDCl3, A) δ: 1.21 (s, 9H, (CH3)3CO), 8.22-8.87 (m, 4H, C6H4), 9.24 (s, 1H, NHCO).
13C NMR (CDCl3, A) δ: 27.61 (CH3, (CH3)C), 42.15 (C), 128.65 (C), 134.25 (C), 142.83 (C), 145.87 (C), 157.26 (N-C-Se), 163.15 (C=O, COC6H5), 186.32 (Se-C=O).
2-(2,2-dimethylpropanoyl)amino-5,6,7,8-tetrahydro-4H-benzo[1]thieno[2,3-d][1,3]selenazine-4-one 17A and 2-(2,2-dimethylpropanoyl)imino-5,6,7,8-tetrahydro-4H-benzo[1]thieno[2,3-d][1,3]selenazine-4-one 17B
(in ratio 7:1)
M.w. 369.34.
C15H18N2O2SSe
M.p. 158-161°C.
Elemental analysis (%calcd/%found) C 48.78/48.63, H 4.91/4.94, N 7.58/7.64, Se 21.60/21.62.
Yield (from 5) 3.6 g (97%), (from 11) 3.4 g (92%).
FTIR (KBr pellets) cm-1: 3280 (NH), 1685 (Se-C=O), 1650, 1540 (NHCO), 1630 (C=N).
1H NMR (CDCl3, A) δ: 1.37 (s, 9H, (CH3)3CO), 1.51-1.64 (m, 4H, 5-CH2 and 6-CH2), 2.72-2.79 (m, 2H, 4-CH2), 2.92-2.97 (m, 2H, 7-CH2), 8.72 (s, 1H, NH).
1H NMR (CDCl3, B) δ: 1.81 (s, 9H, (CH3)3CO), 1.59-1.78 (m, 4H, 5-CH2 and 6-CH2), 2.69-2.73 (m, 2H, 4-CH2), 2.91-2.96 (m, 2H, 7-CH2), 11.95 (s, 1H, NH).
13C NMR (CDCl3) δ: 23.22 (CH2), 25.15 (CH2), 26.48 (CH2), 27.85 (CH3, (CH3)3C), 28.65 (CH2), 42.32 (C), 131.15 (C), 131.52 (C), 134.25 (C), 142.67 (C), 157.22 (N-C-Se), 164.12 (C=O, COC6H5), 186.34 (Se-C=O).
5,6-dimethyl-2-(2,2-dimethylpropanoyl)amino-4H-thieno[2,3-d][1,3]selenazine-4-one 18A
M.w. 343.30.
C13H16N2O2SSe
M.p. 207-208°C.
Elemental analysis (%calcd/%found) C 45.48/45.59, H 4.70/4.67, N 8.16/8.19, Se 23.23/23.25.
Yield (from 6) 3.2 g (93%), (from 12) 3.0 g (87%).
FTIR (KBr pellets) cm-1: 3257 (NH), 1674 (Se-C=O), 1721, 1546 (NHCO), 1616 (C=N).
1H NMR (CDCl3, A) δ: 1.37 (s, 9H, (CH3)3CO), 2.65 (s, 3H, CH3), 2.92 (s, 3H, CH3), 8.64 (s, 1H, NH).
13C NMR (CDCl3, A) δ: 13.51 (CH3), 15.51(CH3), 27.45 (CH3, (CH3)3C), 42.11 (C), 126.05 (C), 126.55 (C), 130.05 (C), 138.40 (C), 154.21 (N-C-Se), 163.25 (C=O, COC6H5), 184.19(Se-C=O).
Retrocyclizations of fused 2-acylamino-4H-1,3-selenazine-4-ones 13-18
Compound
13-
18 was dissolved under stirring in an argon atmosphere in potassium ethoxide solution, prepared by reaction of potassium
tert.butoxid (1.12 g, 10 mmol) with 25 ml of dry ethanol - dried by azeotropic distillation with benzene and after by distillation with magnesium. The reaction mixture was stirred at room temperature for 50 - 90 min, the conversion of the reaction was monitored by TLC after acidification of the reaction mixture with acetic acid. With the reactions of thiophene derivatives
14, 15, 16 and
17 potassium salts of the title acylselenoureas
20, 21, 23, 24 were formed, in the case of benzoderivatives
13, 16, corresponding salts
19, 22 were dissolved in the reaction mixtures. The produts
1-6 were obtained after acidification of the reaction mixture by acetic acid (ca 1 ml) at 5-10°C, removing of the solvent on an evaporator at 25-30°C. The evaporated residue was suspended in chloroform (50 ml) and filtered with charcoal. The filtrate was concentrated to 1/5 original volume and mixed with an equivalent volume of petroleum ether. The precipitated crystals were filtered, washed with petroleum ether and cold methanol (5-10°C). The pure products
1-
6, dried
in vacuo, were identical to standard
1-
6 by TLC, m. p. and FTIR [
1].
Potassium salts of title acylselenoureas 20, 21, 23, 24
Retrocyclization of compounds 14, 15, 16 and 17 (a)
Suspensions of salts 20, 21, 23, 24, were prepared as mentioned above and were filtered by suction. The precipitate was washed with methanol, diethyl ether and dried in vacuo at room temperature.
Deprotonation of compounds 2, 3, 5 and 6 or 8, 9, 11 and 12 by action of potassium hydroxide in equivalent (b)
Acylselenourea 2, 3, 5 and 6 or corresponding acylisoselenourea 8, 9, 11 and 12 (5 mmol) was dissolved under stirring in argon atmosphere in 50 ml methanol containing potassium hydroxide (340 mg, 6 mmol). After 30 min product was formed separated by suction, washed with methanol, diethyl ether and dried in vacuo at room temperature.
Ethyl 2-(3-benzoylselenoureido)-4,5,6,7-tetrahydrobenzo[1]thiophene-3-carboxylate potassium salt 20
M.w. 473.49.
C19H19N2O3SSeK
M.p. 292-293°C (decomp.).
Elemental analysis (%calcd/%found) C 48.20/47.86, H 4.04/3.95, N 5.92/5.86, Se 16.68/16.68.
Yield (a) 3.7 g (78%), (b) 2.0 g (86%).
FTIR (KBr pellets) cm-1: 3363, 3158 (NH), 1677 (O-C=Ο), 1467, 1365 (C(Se)N-C(O)).
1H NMR (d6-DMSO) δ: 1.38 (t, 3H, CH2CH3, J = 7.0 Hz), 1.78-1.83 (m, 4H, 5-CH2 and 6-CH2), 2.79-2.81 (m, 2H, 4-CH2), 2.82-2.86 (m, 2H, 7-CH2), 4.29 (q, 2H, CH2CH3, J = 7.0 Hz), 7.23-7.96 (m, 5H, C6H5), 8.46 (s, 1H, NH).
13C NMR (d6-DMSO) δ: 14.71 (CH2CH3), 22.45 (C-5), 24.21 (C-6), 25.62 (C-4), 26.47 (C-7), 59.22 (CH2CH3), 104.30 (C-3), 111.60 (C-4, thiophene), 126.83 (C-2’and C-6’, C6H5), 128.78 (C-3’and C-5’, C6H5), 132.27 (C-5, thiophene), 134.18 (C-1’, C6H5), 136.43 (C-4’, C6H5), 157.64 (C-2, thiophene), 164.43 (C=O, COOCH2CH3), 173.10 (C=O, COC6H5), 193.10 (C=Se, 1JC, Se = 200 Hz).
Ethyl 2-(3-benzoylselenoureido)-4,5-dimethylthiophene-3-carboxylate - potassium salt 21
M.w. 447.45
C17H17N2O3SSeK
M.p. 188-189°C (decomp.)
Elemental analysis (%calcd/%found) C 45.63/45.16, H 3.83/3.75, N 6.26/6.14, Se 17.65/17.69.
Yield (a) 3.3 g (74%), (b) 1.8 g (82%).
FTIR (KBr pellets) cm-1: 3265, 3134 (NH), 1664 (O-C=Ο), 1414, 1359 (C(Se)N-C(O)).
1H NMR (d6-DMSO) δ: 1.28 (t, 3H, CH2CH3, J = 7.1 Hz), 2.15 (s, 3H, CH3, C-4 thiophene), 2.24 (s, 3H, CH3, C-5 thiophene), 4.34 (q, 2H, CH2CH3, J = 7.1 Hz), 7.32-7.95 (m, 5H, C6H5), 8.39 (s, 1H, NH).
Ethyl 2-[3-(2,2-dimethylpropanoyl)selenoureido]-4,5,6,7-tetrahydrobenzo[1]thiophene-3-carboxylate potassium salt 23
M.w. 453.50.
C17H23N2O3SSeK
M.p. 265-267°C (decomp.)
Elemental analysis (%calcd/%found) C 45.02/44.91, H 5.11/ 5.03, N 6.18/6.17, Se 17.41/17.46.
Yield (a) 3.7 g (82%), (b) 2.1 g (92%).
FTIR (KBr pellets) cm-1: 3347, 3133 (NH), 1682 (O-C=Ο), 1458, 1361 (C(Se)N-C(O)).
1H NMR (d6-DMSO) δ: 1.12 (s, 9H, CH3, C(CH3)3), ) 1.23 (t, 3H, CH2CH3, J = 7.0 Hz), 1.76-1.79 (m, 4H, 5-CH2 and 6-CH2), 2.74-2.78 (m, 2H, 4-CH2), 2.96-3.01 (m, 2H, 7-CH2), 4.35 (q, 2H, CH2CH3, J = 7.1 Hz), 8.96 (s, 1H, NH).
13C NMR (d6-DMSO) δ: 14.65 (CH2CH3), 22.48 (C-5), 24.26 (C-6), 25.59 (C-4), 26.52 (C-7), 27.81 (CH3, C(CH3)3), 52.83 (CH2, C(CH3)3), 59.28 (CH2CH3), 104.36 (C-3), 111.57 (C-4, thiophene), 133.24(C-5, thiophene), 157.62 (C-2, thiophene), 166.01(C=O, COOCH2CH3), 173.56 (C=O, COC(CH3)3), 181.94 (C=Se, 1JC, Se = 200 Hz).
Ethyl 2-[3-(2,2-dimethylpropanoyl)selenoureido]-4,5-dimethylthiophene-3-carboxylate potassium salt 24
M.w. 427.46.
C15H21N2O3SSeK
M.p. 184-185°C (decomp.)
Elemental analysis (%calcd/%found) C 42.15/41.38, H 4.95/4.87, N 6.55/6.54, Se 18.47/18.59.
Yield (a) 3.3 g (77%), (b) 1.8 g (84%).
FTIR (KBr pellets) cm-1: 3324, 3166 (NH), 1669 (O-C=Ο), 1443, 1371 (C(Se)N-C(O)).
1H NMR (d6-DMSO) δ: 1.20 (t, 3H, CH2CH3, J 7.1 Hz), 1.26 (s, 9H, CH3, (CH3)3C), 2.04 (s, 3H, CH3, C-4 thiophene), 2.18 (s, 3H, CH3, C-5 thiophene), 4.31 (q, 2H, CH2CH3, J 7.1 Hz), 7.99 (s, 1H, NH).
Isoselenoureas 26, 27
Potassium salts 20, 21, 23 and 24 (2.5 mmol) were suspended in methanol (30 ml) and heated for 10 min on a steam bath under reflux. The product 26, 27, which was crystallized from the reaction solution by holding overnight in a freezing box, was separated by suction and recrystallized from methanol.
Ethyl 2-(3-isoselenoureido)-4,5,6,7-tetrahydrobenzo[1]thiophene-3-carboxylate 26
M.w. 331.23
C12H16N2O2SSe
M.p. 218-222°C
Elemental analysis (%calcd/%found) C 43.51/42.16, H 4.87/4.73, N8.46/8.46, Se 23.82/23.94.
Yield (from 20) 0.7 g (85%), (from 23) 0.7 g (85%).
FTIR (KBr pellets) cm-1: 3248, 3188 (NH), 1746 (C=Ο), 1650 (C=N).
1H NMR (CDCl3) δ: 1.27 (t, 3H, CH3, J = 7.1 Hz), 1.54 (s, 1H, SeH), 1.76-1.84 1.56 (m, 4H, 5-CH2 and 6-CH2), 3.05-3.13 (m, 4H, 4-CH2 and 7-CH2), 4.33 (q, 2H, CH2, J = 7.1 Hz), 6.45 (s, 2H, NH2).
13C NMR (CDCl3) δ: 13.85 (CH3), 21.15 (C-6), 24.24 (C-5), 26.64 (C-7), 26.98 (C-4), 60.02(CH2), 98.12 (C-3), 128.24 (C-5 thiophene), 132.08 (C-4 thiophene), 159.12 (C-2), 163.28 (N=C-Se, 1JC, Se 134 Hz), 169.45 (C=Ο).
Ethyl 2-(3-isoselenoureido)-4,5-dimethylthiophene-3-carboxylate 27
M.w. 305.19
C10H14N2O2SSe
M.p. 195-196°C
Elemental analysis (%calcd/%found) C 39.36/39.12, H 4.62/4.61, N9.18/9.03, Se 25.85/25.79.
Yield (from 21) 0.7 g (92%), (from 24) 0.6 g (77%).
FTIR (KBr pellets) cm-1: 3249, 3197 (NH), 1736 (C=Ο), 1652 (C=N).
1H NMR (CDCl3) δ: 1.29 (t, 3H, CH3, J = 7.1 Hz), 1.44 (s, 1H, SeH), 2.41(s, 3H, CH3, C-4 thiophene), 2.45 (s, 3H, CH3, C-5 thiophene), 4.24 (q, 2H, CH2, J = 7.1 Hz), 6.14 (s, 2H, NH2).
13C NMR (CDCl3) δ: 12.26 (CH3, C-4 thiophene), 13.84 (CH3), 14.87 (CH3, C-5 thiophene), 59.85 (CH3), 111.10 (C-3, thiophene), 127.35 (C-5, thiophene), 132.74 (C-4, thiophene), 152.68 (C-2, thiophene), 159.56 (N=C-Se, 1JC, Se 138 Hz). 168.32 (C=Ο).
Fused 3,4-dihydropyrimidinones 28-30
A: Acylselenourea 1-6 (5 mmol) was suspended in a 50 ml 5% methanolic solution of potassium hydroxide and refluxed until a colorless solution (5-10 min) was obtained. The solution was filtered with charcoal. After cooling to 5-10°C it was neutralized by glacial acetic acid. The precipitated product 28-30 was filtered by suction, washed with methanol and dried in vacuo.
B: Isoselenoureas 26, 27 (2 mmol) were suspended in a 20 ml 5% methanolic solution of potassium hydroxide and refluxed until a colorless solution (5-10 min) was obtained. Then the solution was treated by method A.
2-Selanyl-3,4-dihydroquinazoline-4-one 28
M.w. 225.05
C8H6N2 OSe
M.p. 282-284°C
Elemental analysis (%calcd/%found) C 42.70/ 42.48, H 2.69/2.62, N12.45/12.48, Se 35.06/35.50.
Yield (A, from 1) 0.9 g (80%), (A, from 4) 0.9 g (80%).
FTIR (KBr pellets) cm-1: 1682 (C=O).
1H NMR (d-TFA) δ: 7.48-7.79 (m, 4H, C6H4).
13C NMR (d-TFA) δ: 122.07 (C-5, pyrimidine), 126.18 (C-6), 127.98 (C-8), 128.17 (C-5), 137.42 (C-7), 143.64 (C-6, pyrimidine), 151.46 (C-2), 158.26 (C-4).
2-Selanyl-1,2,5,6,7,8-hexahydrobenzo[1]thieno[2,3-d]pyrimidine-4-one 29
M.w. 285.16
C10H10N2OSSe
M.p. 310-313°C
Elemental analysis (%calcd/%found) C 42.12/41.83, H 3.53/3.51, N 9.82/9.74, Se 27.67/27.63.
Yield (A, from 2) 1.30 g (91%), (A, from 5) 1.20 g (84%), (B, from 26) 0.40 g (70%).
FTIR (KBr pellets) cm-1: 1655 (C=O).
1H NMR (d-TFA) δ: 1.63-1.82 (m, 4H, 6-CH2 and 7-CH2), 2.79-2.85 (m, 4H, 5-CH2 and 8-CH2).
13C NMR (d-TFA) δ: 22.76 (C-7), 23.49 (C-6), 24.15 (C-8), 29.47 (C-5), 113.85 (C-5, pyrimidine), 116.85 (C-10, cyclohexane), 129.74 (C-9, cyclohexane), 139.01(C-6, pyrimidine), 153.49 (C-4).
5,6-Dimethyl-2-selenoxo-1,2-dihydrothieno[2,3-d]pyrimidine-4-one 30
M.w. 259.12
C8H8N2OSSe
M.p. 315-318°C
Elemental analysis (%calcd/%found) C 37.07/36.79, H 3.50/3.49, N16.22/16.16, Se 30.56/30.42.
Yield (A, from 3) 1.10 g (85%), (A, from 6) 1.10 g (85%), (B, from 27) 0.35 g (67%).
FTIR (KBr pellets) cm-1: 3440, 3293 (NH), 1622 (C=N), 1527, 954 (NHCSe, selenoamide III, I).
1H NMR (d-TFA) δ: 2.82 (s, 3H, CH3), 2.83 (s, 3H, CH3).
13C NMR (d-TFA) δ: 11.12 (CH3, C-5), 11.80 (CH3, C-6), 126.51 (C-5), 132.91 (C-6), 152.48 (C-5, pyrimidine), 155.16 (C-4), 169.71 (C-6, pyrimidine), 181.29 (C=Se, 1JC, Se = 220 Hz).

{kind=link}
{kind=link}
{kind=link}