
Electronic Interactions Between the Receptor-Binding Domain of Omicron Variants and Angiotensin-Converting Enzyme 2: A Novel Amino Acid–Amino Acid Bond Pair Concept

Abstract
1. Introduction
2. Results and Discussion
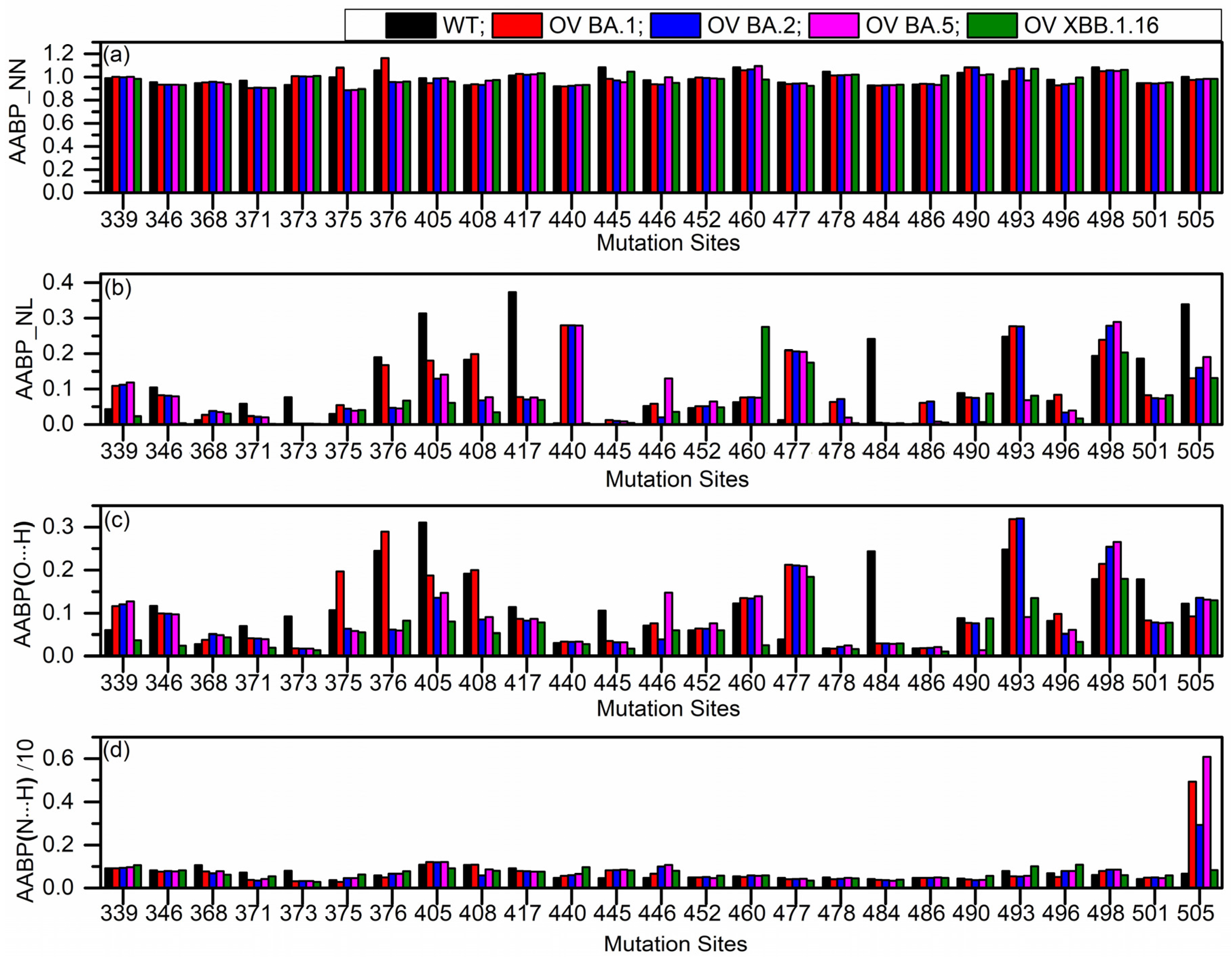
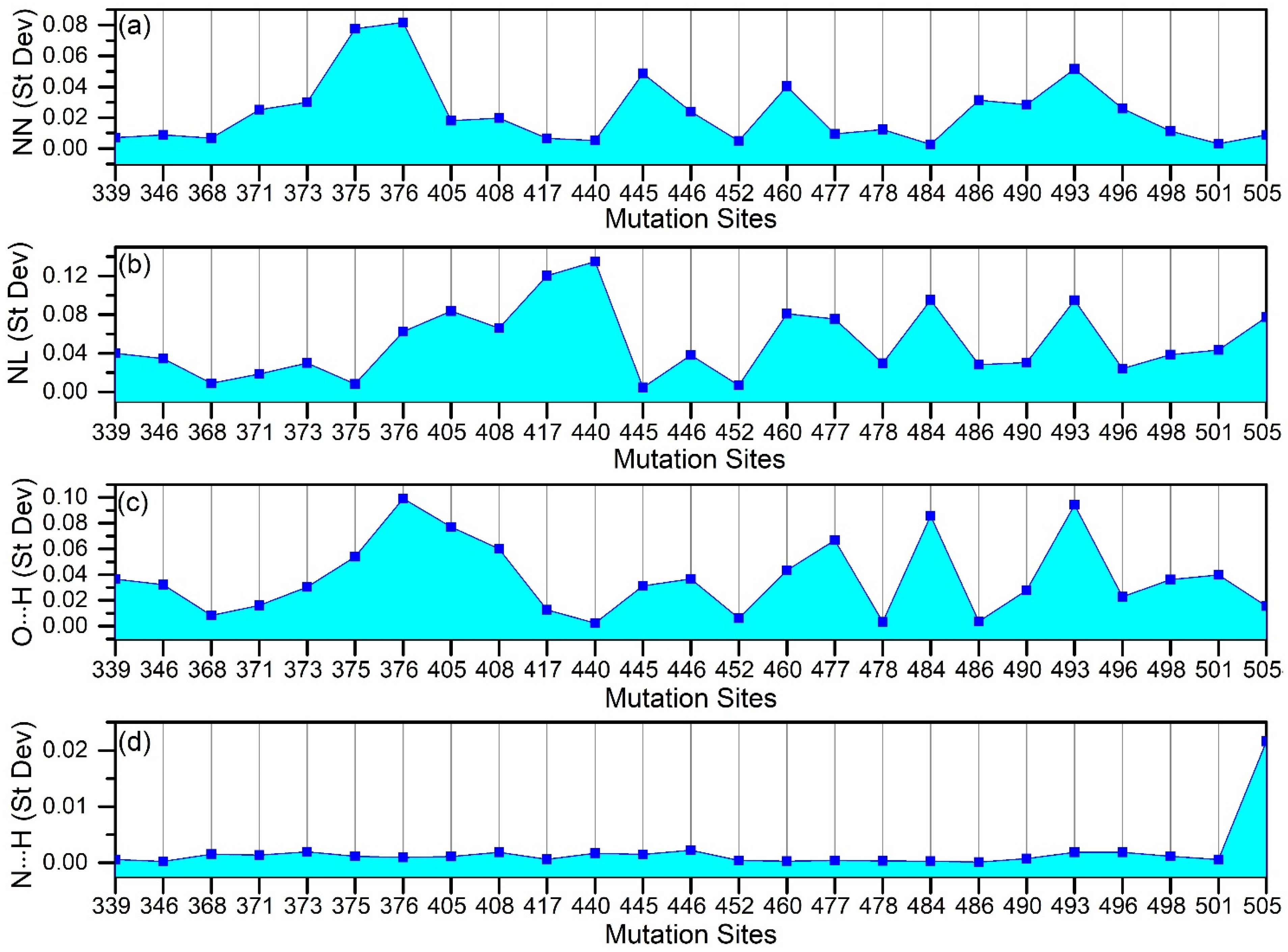
2.1. Amino Acid–Amino Acid Bond Pair (AABP) for Mutated Sites
2.2. Amino Acid–Amino Acid Bond Pair (AABP) for Entire Model
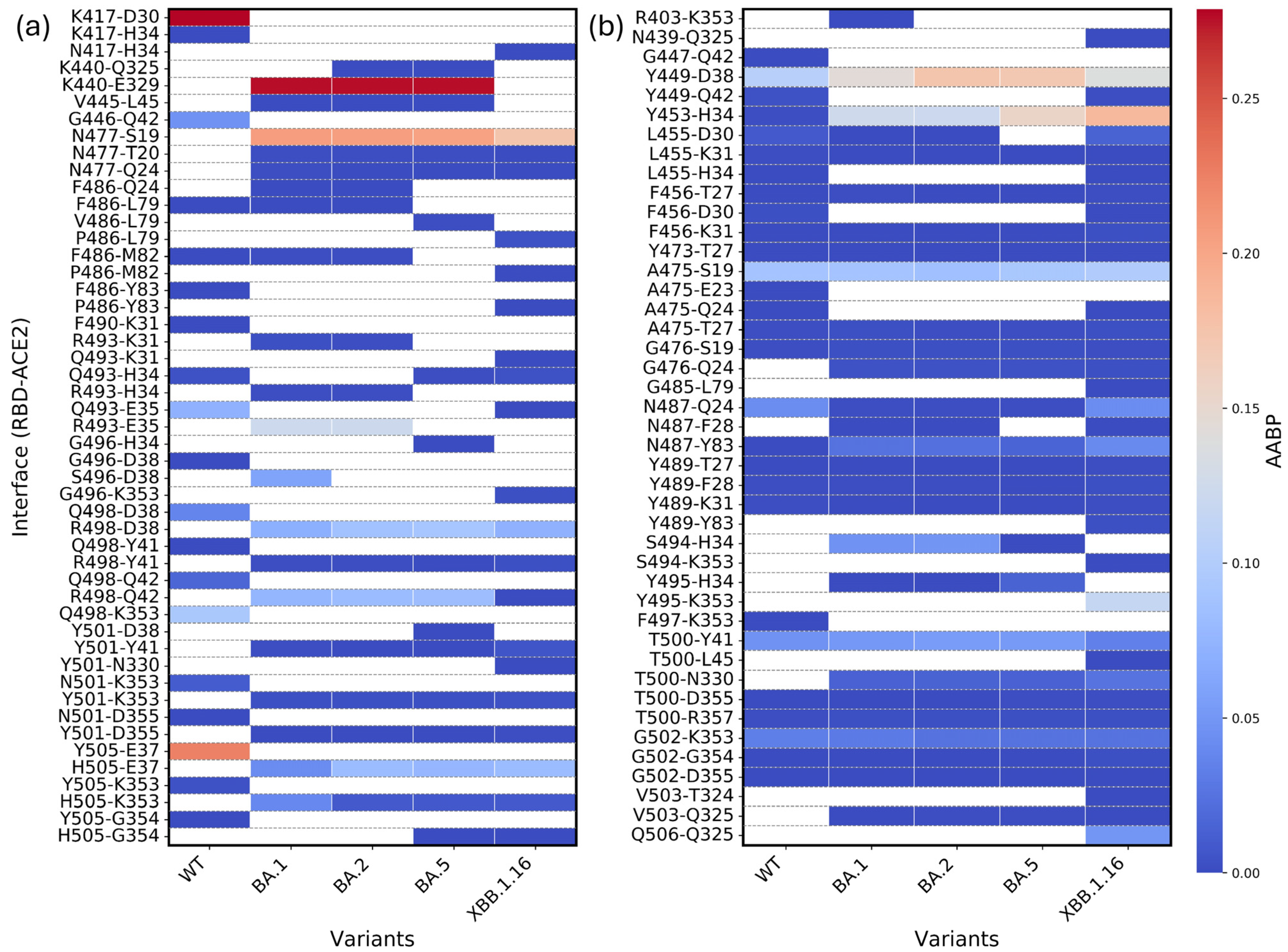
2.3. Bonding Between RBD and ACE2
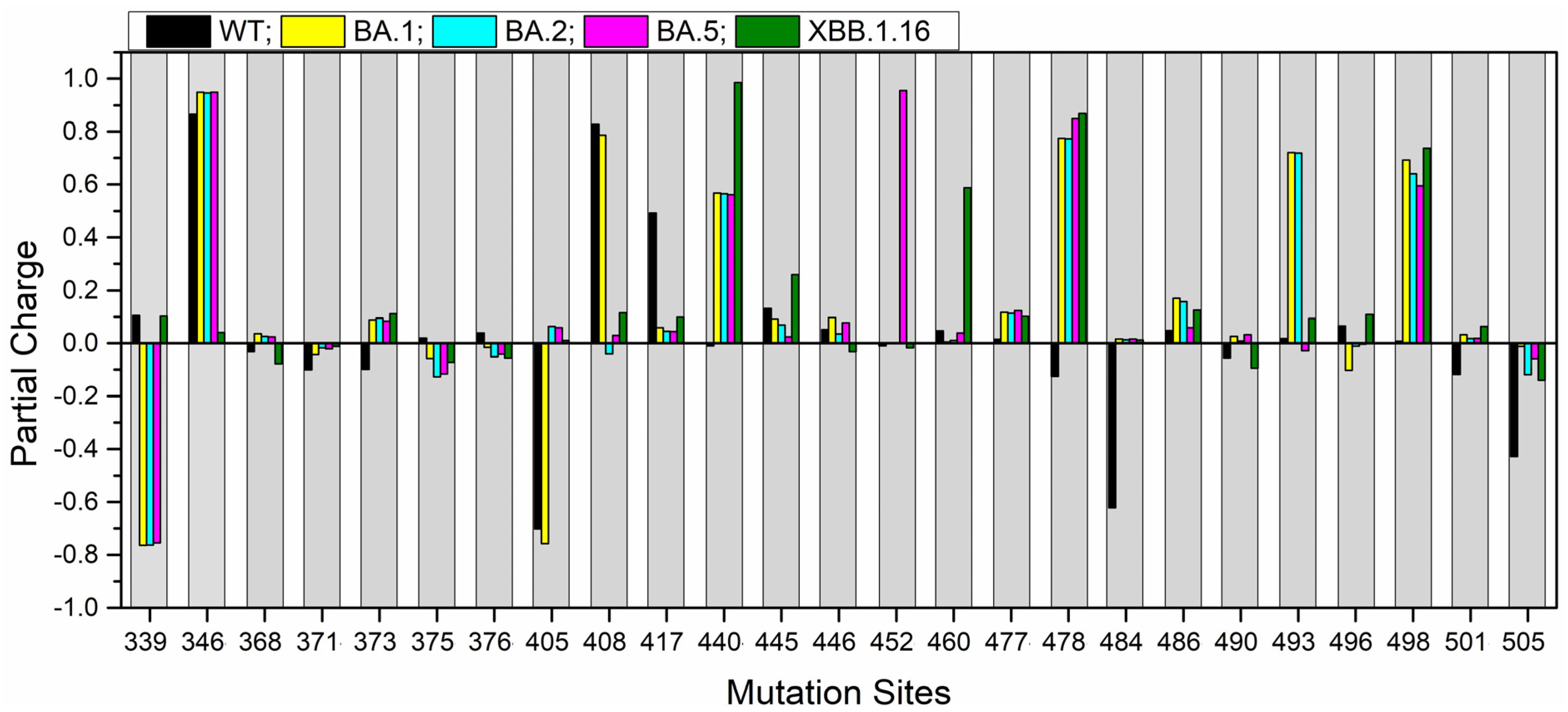
2.4. Partial Charge for the Mutation Sites
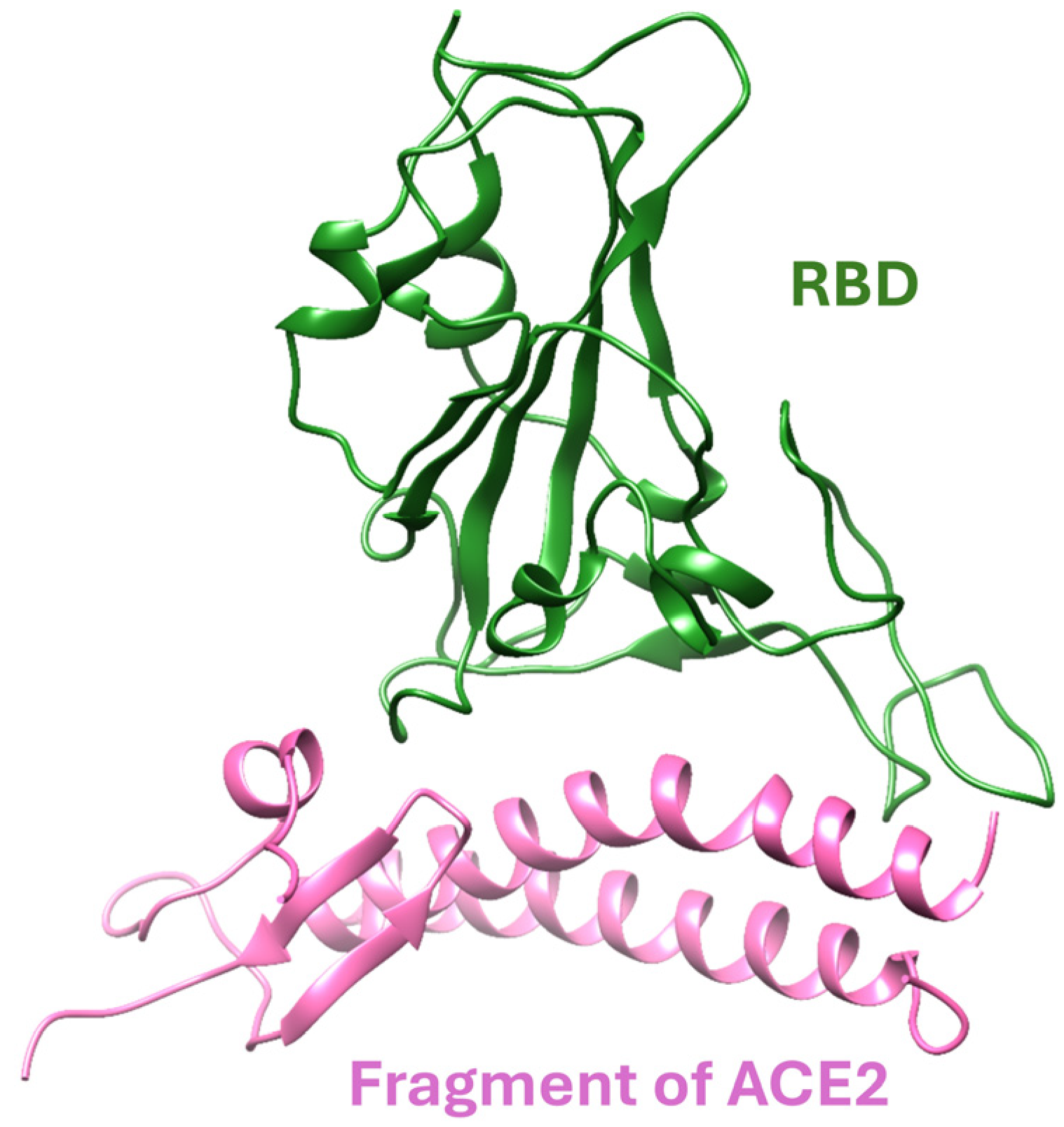
3. Models
4. Methods
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lostao, A.; Lim, K.; Pallarés, M.C.; Ptak, A.; Marcuello, C. Recent advances in sensing the inter-biomolecular interactions at the nanoscale–A comprehensive review of AFM-based force spectroscopy. Int. J. Biol. Macromol. 2023, 238, 124089. [Google Scholar] [CrossRef] [PubMed]
- Ding, Z.; Kihara, D. Computational methods for predicting protein-protein interactions using various protein features. Curr. Protoc. Protein Sci. 2018, 93, e62. [Google Scholar] [CrossRef] [PubMed]
- Skrabanek, L.; Saini, H.K.; Bader, G.D.; Enright, A.J. Computational prediction of protein–protein interactions. Mol. Biotechnol. 2008, 38, 1–17. [Google Scholar] [CrossRef]
- Grigoriev, A. A relationship between gene expression and protein interactions on the proteome scale: Analysis of the bacteriophage T7 and the yeast Saccharomyces cerevisiae. Nucleic Acids Res. 2001, 29, 3513–3519. [Google Scholar] [CrossRef]
- Consortium, G.O. Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res. 2017, 45, D331–D338. [Google Scholar] [CrossRef]
- Chen, J.; Hsu, W.; Lee, M.L.; Ng, S.-K. Increasing confidence of protein interactomes using network topological metrics. Bioinformatics 2006, 22, 1998–2004. [Google Scholar] [CrossRef]
- Zhang, Q.C.; Petrey, D.; Deng, L.; Qiang, L.; Shi, Y.; Thu, C.A.; Bisikirska, B.; Lefebvre, C.; Accili, D.; Hunter, T. Structure-based prediction of protein–protein interactions on a genome-wide scale. Nature 2012, 490, 556–560. [Google Scholar] [CrossRef]
- Adebiyi, A.; Adhikari, P.; Rao, P.; Ching, W.-Y. Bond strength between receptor binding domain of spike protein and human angiotensin converting enzyme-2 using machine learning. BME Horiz. 2024, 2, 110. [Google Scholar] [CrossRef]
- Lan, J.; Ge, J.; Yu, J.; Shan, S.; Zhou, H.; Fan, S.; Zhang, Q.; Shi, X.; Wang, Q.; Zhang, L. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020, 581, 215–220. [Google Scholar] [CrossRef]
- Beyerstedt, S.; Casaro, E.B.; Rangel, É.B. COVID-19: Angiotensin-converting enzyme 2 (ACE2) expression and tissue susceptibility to SARS-CoV-2 infection. Eur. J. Clin. Microbiol. Infect. Dis. 2021, 40, 905–919. [Google Scholar] [CrossRef]
- World Health Organization. Tracking SARS-CoV-2 Variants. Available online: https://www.who.int/activities/tracking-SARS-CoV-2-variants (accessed on 31 March 2025).
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.-Y. Impact of BA.1, BA.2, and BA.4/BA.5 Omicron mutations on therapeutic monoclonal antibodies. Comput. Biol. Med. 2023, 167, 107576. [Google Scholar] [CrossRef] [PubMed]
- O’Toole, Á.; Scher, E.; Underwood, A.; Jackson, B.; Hill, V.; McCrone, J.T.; Colquhoun, R.; Ruis, C.; Abu-Dahab, K.; Taylor, B. Assignment of epidemiological lineages in an emerging pandemic using the pangolin tool. Virus Evol. 2021, 7, veab064. [Google Scholar] [CrossRef] [PubMed]
- Shuai, H.; Chan, J.F.-W.; Hu, B.; Chai, Y.; Yuen, T.T.-T.; Yin, F.; Huang, X.; Yoon, C.; Hu, J.-C.; Liu, H. Attenuated replication and pathogenicity of SARS-CoV-2 B.1.1.529 Omicron. Nature 2022, 603, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Yuan, S.; Ye, Z.-W.; Liang, R.; Tang, K.; Zhang, A.J.; Lu, G.; Ong, C.P.; Man Poon, V.K.; Chan, C.C.-S.; Mok, B.W.-Y. Pathogenicity, transmissibility, and fitness of SARS-CoV-2 Omicron in Syrian hamsters. Science 2022, 377, 428–433. [Google Scholar] [CrossRef]
- Suzuki, R.; Yamasoba, D.; Kimura, I.; Wang, L.; Kishimoto, M.; Ito, J.; Morioka, Y.; Nao, N.; Nasser, H.; Uriu, K. Attenuated fusogenicity and pathogenicity of SARS-CoV-2 Omicron variant. Nature 2022, 603, 700–705. [Google Scholar] [CrossRef]
- Meng, B.; Abdullahi, A.; Ferreira, I.A.; Goonawardane, N.; Saito, A.; Kimura, I.; Yamasoba, D.; Gerber, P.P.; Fatihi, S.; Rathore, S. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 2022, 603, 706–714. [Google Scholar] [CrossRef]
- Hui, K.P.; Ho, J.C.; Cheung, M.-C.; Ng, K.-C.; Ching, R.H.; Lai, K.-L.; Kam, T.T.; Gu, H.; Sit, K.-Y.; Hsin, M.K. SARS-CoV-2 Omicron variant replication in human bronchus and lung ex vivo. Nature 2022, 603, 715–720. [Google Scholar] [CrossRef]
- Evans, J.P.; Zeng, C.; Qu, P.; Faraone, J.; Zheng, Y.-M.; Carlin, C.; Bednash, J.S.; Zhou, T.; Lozanski, G.; Mallampalli, R. Neutralization of SARS-CoV-2 Omicron sub-lineages BA.1, BA.1.1, and BA.2. Cell Host Microbe 2022, 30, 1093–1102.e3. [Google Scholar] [CrossRef]
- Xia, H.; Zou, J.; Kurhade, C.; Cai, H.; Yang, Q.; Cutler, M.; Cooper, D.; Muik, A.; Jansen, K.U.; Xie, X. Neutralization and durability of 2 or 3 doses of the BNT162b2 vaccine against Omicron SARS-CoV-2. Cell Host Microbe 2022, 30, 485–488.e3. [Google Scholar] [CrossRef]
- Planas, D.; Saunders, N.; Maes, P.; Guivel-Benhassine, F.; Planchais, C.; Buchrieser, J.; Bolland, W.-H.; Porrot, F.; Staropoli, I.; Lemoine, F. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature 2022, 602, 671–675. [Google Scholar] [CrossRef]
- Zeng, C.; Evans, J.P.; Qu, P.; Faraone, J.; Zheng, Y.-M.; Carlin, C.; Bednash, J.S.; Zhou, T.; Lozanski, G.; Mallampalli, R. Neutralization and stability of SARS-CoV-2 Omicron variant. biorxiv 2021. [Google Scholar] [CrossRef]
- Wang, X.; Zhao, X.; Song, J.; Wu, J.; Zhu, Y.; Li, M.; Cui, Y.; Chen, Y.; Yang, L.; Liu, J. Homologous or heterologous booster of inactivated vaccine reduces SARS-CoV-2 Omicron variant escape from neutralizing antibodies. Emerg. Microbes Infect. 2022, 11, 477–481. [Google Scholar] [CrossRef] [PubMed]
- Schmidt, F.; Muecksch, F.; Weisblum, Y.; Da Silva, J.; Bednarski, E.; Cho, A.; Wang, Z.; Gaebler, C.; Caskey, M.; Nussenzweig, M.C. Plasma neutralization of the SARS-CoV-2 Omicron variant. N. Engl. J. Med. 2022, 386, 599–601. [Google Scholar] [CrossRef] [PubMed]
- Pérez-Then, E.; Lucas, C.; Monteiro, V.; Miric, M.; Brache, V.; Cochon, L.; Vogels, C.; Malik, A.; De la Cruz, E.; Jorge, A. Neutralizing antibodies against the SARS-CoV-2 Delta and Omicron variants following heterologous CoronaVac plus BNT162b2 booster vaccination. Nat. Med. 2022, 28, 481–485. [Google Scholar] [CrossRef]
- Jacobsen, H.; Strengert, M.; Maaß, H.; Ynga Durand, M.A.; Katzmarzyk, M.; Kessel, B.; Harries, M.; Rand, U.; Abassi, L.; Kim, Y. Diminished neutralization responses towards SARS-CoV-2 Omicron VoC after mRNA or vector-based COVID-19 vaccinations. Sci. Rep. 2022, 12, 19858. [Google Scholar] [CrossRef]
- Shrestha, L.B.; Foster, C.; Rawlinson, W.; Tedla, N.; Bull, R.A. Evolution of the SARS-CoV-2 omicron variants BA.1 to BA.5: Implications for immune escape and transmission. Rev. Med. Virol. 2022, 32, e2381. [Google Scholar] [CrossRef]
- Yamasoba, D.; Kimura, I.; Nasser, H.; Morioka, Y.; Nao, N.; Ito, J.; Uriu, K.; Tsuda, M.; Zahradnik, J.; Shirakawa, K. Virological characteristics of the SARS-CoV-2 Omicron BA.2 spike. Cell 2022, 185, 2103–2115.e19. [Google Scholar] [CrossRef]
- Cao, Y.; Yisimayi, A.; Jian, F.; Song, W.; Xiao, T.; Wang, L.; Du, S.; Wang, J.; Li, Q.; Chen, X. BA.2.12.1, BA.4 and BA.5 escape antibodies elicited by Omicron infection. Nature 2022, 608, 593–602. [Google Scholar] [CrossRef]
- Cele, S.; Jackson, L.; Khoury, D.S.; Khan, K.; Moyo-Gwete, T.; Tegally, H.; San, J.E.; Cromer, D.; Scheepers, C.; Amoako, D.G. Omicron extensively but incompletely escapes Pfizer BNT162b2 neutralization. Nature 2022, 602, 654–656. [Google Scholar] [CrossRef]
- Rössler, A.; Riepler, L.; Bante, D.; von Laer, D.; Kimpel, J. SARS-CoV-2 Omicron variant neutralization in serum from vaccinated and convalescent persons. N. Engl. J. Med. 2022, 386, 698–700. [Google Scholar] [CrossRef]
- VanBlargan, L.A.; Errico, J.M.; Halfmann, P.J.; Zost, S.J.; Crowe, J.E., Jr.; Purcell, L.A.; Kawaoka, Y.; Corti, D.; Fremont, D.H.; Diamond, M.S. An infectious SARS-CoV-2 B.1.1.529 Omicron virus escapes neutralization by therapeutic monoclonal antibodies. Nat. Med. 2022, 28, 490–495. [Google Scholar] [CrossRef] [PubMed]
- Richardson, S.I.; Madzorera, V.S.; Spencer, H.; Manamela, N.P.; van der Mescht, M.A.; Lambson, B.E.; Oosthuysen, B.; Ayres, F.; Makhado, Z.; Moyo-Gwete, T. SARS-CoV-2 Omicron triggers cross-reactive neutralization and Fc effector functions in previously vaccinated, but not unvaccinated, individuals. Cell Host Microbe 2022, 30, 880–886.e884. [Google Scholar] [CrossRef] [PubMed]
- Tamura, T.; Ito, J.; Uriu, K.; Zahradnik, J.; Kida, I.; Anraku, Y.; Nasser, H.; Shofa, M.; Oda, Y.; Lytras, S. Virological characteristics of the SARS-CoV-2 XBB variant derived from recombination of two Omicron subvariants. Nat. Commun. 2023, 14, 2800. [Google Scholar] [CrossRef]
- World Health Organization. TAG-VE Statement on Omicron Sublineages BQ.1 and XBB. 2022. Available online: https://www.who.int/news/item/27-10-2022-tag-ve-statement-on-omicron-sublineages-bq.1-and-xbb (accessed on 30 June 2024).
- Karyakarte, R.P.; Das, R.; Rajmane, M.V.; Dudhate, S.; Agarasen, J.; Pillai, P.; Chandankhede, P.M.; Labhshetwar, R.S.; Gadiyal, Y.; Kulkarni, P.P. Chasing SARS-CoV-2 XBB.1.16 recombinant lineage in India and the clinical profile of XBB.1.16 cases in Maharashtra, India. Cureus 2023, 15, 1–22. [Google Scholar] [CrossRef]
- World Health Organization. XBB.1.16 Initial Risk Assessment, 17 April 2023. Available online: https://www.who.int/docs/default-source/coronaviruse/21042023xbb.1.16ra-v2.pdf (accessed on 30 June 2024).
- CDC Adds New XBB.1.16 Omicron Subvariant to Variant Tracker. Available online: https://www.usnews.com/news/health-news/articles/2023-04-14/cdc-adds-new-xbb-1-16-omicron-subvariant-to-variant-tracker (accessed on 30 June 2024).
- Qu, P.; Faraone, J.N.; Evans, J.P.; Zheng, Y.-M.; Carlin, C.; Anghelina, M.; Stevens, P.; Fernandez, S.; Jones, D.; Panchal, A.R. Enhanced evasion of neutralizing antibody response by Omicron XBB.1.5, CH.1.1, and CA.3.1 variants. Cell Rep. 2023, 42, 112443. [Google Scholar] [CrossRef]
- Wang, Q.; Iketani, S.; Li, Z.; Liu, L.; Guo, Y.; Huang, Y.; Bowen, A.D.; Liu, M.; Wang, M.; Yu, J. Alarming antibody evasion properties of rising SARS-CoV-2 BQ and XBB subvariants. Cell 2023, 186, 279–286.e8. [Google Scholar] [CrossRef]
- Zhang, X.; Chen, L.-L.; Ip, J.D.; Chan, W.-M.; Hung, I.F.-N.; Yuen, K.-Y.; Li, X.; To, K.K.-W. Omicron sublineage recombinant XBB evades neutralising antibodies in recipients of BNT162b2 or CoronaVac vaccines. Lancet Microbe 2023, 4, e131. [Google Scholar] [CrossRef]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, function, and antigenicity of the SARS-CoV-2 spike glycoprotein. Cell 2020, 181, 281–292.e6. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.S.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Chen, J.; Wang, R.; Gilby, N.B.; Wei, G.-W. Omicron variant (B.1.1.529): Infectivity, vaccine breakthrough, and antibody resistance. J. Chem. Inf. Model. 2022, 62, 412–422. [Google Scholar] [CrossRef]
- Jawad, B.; Adhikari, P.; Cheng, K.; Podgornik, R.; Ching, W.-Y. Computational design of miniproteins as SARS-CoV-2 therapeutic inhibitors. Int. J. Mol. Sci. 2022, 23, 838. [Google Scholar] [CrossRef]
- Podoly, E.; Hanin, G.; Soreq, H. Alanine-to-threonine substitutions and amyloid diseases: Butyrylcholinesterase as a case study. Chem.-Biol. Interact. 2010, 187, 64–71. [Google Scholar] [CrossRef]
- Rehman, S.; Mahmood, T.; Aziz, E.; Batool, R. Identification of novel mutations in SARS-CoV-2 isolates from Turkey. Arch. Virol. 2020, 165, 2937–2944. [Google Scholar] [CrossRef] [PubMed]
- Barton, M.I.; MacGowan, S.A.; Kutuzov, M.A.; Dushek, O.; Barton, G.J.; Van Der Merwe, P.A. Effects of common mutations in the SARS-CoV-2 Spike RBD and its ligand, the human ACE2 receptor on binding affinity and kinetics. eLife 2021, 10, e70658. [Google Scholar] [CrossRef] [PubMed]
- Mannar, D.; Saville, J.W.; Zhu, X.; Srivastava, S.S.; Berezuk, A.M.; Tuttle, K.S.; Marquez, A.C.; Sekirov, I.; Subramaniam, S. SARS-CoV-2 Omicron variant: Antibody evasion and cryo-EM structure of spike protein–ACE2 complex. Science 2022, 375, 760–764. [Google Scholar] [CrossRef] [PubMed]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.-Y. Binding interactions between receptor-binding domain of spike protein and human angiotensin converting enzyme-2 in omicron variant. J. Phys. Chem. Lett. 2022, 13, 3915–3921. [Google Scholar] [CrossRef]
- Tegally, H.; Moir, M.; Everatt, J.; Giovanetti, M.; Scheepers, C.; Wilkinson, E.; Subramoney, K.; Makatini, Z.; Moyo, S.; Amoako, D.G. Emergence of SARS-CoV-2 omicron lineages BA.4 and BA.5 in South Africa. Nat. Med. 2022, 28, 1785–1790. [Google Scholar] [CrossRef]
- Aljohani, M.S.; Bhatta, P.; Hu, X. Molecular Determinants for the Binding of the Highly Infectious SARS-CoV-2 Omicron (BA.1) Variant to the Human ACE2 Receptor. Physchem 2025, 5, 8. [Google Scholar] [CrossRef]
- Hristova, S.H.; Zhivkov, A.M. Omicron coronavirus: pH-dependent electrostatic potential and energy of association of spike protein to ACE2 receptor. Viruses 2023, 15, 1752. [Google Scholar] [CrossRef]
- Hristova, S.H.; Zhivkov, A.M. Three-dimensional structural stability and local electrostatic potential at point mutations in spike protein of SARS-CoV-2 coronavirus. Int. J. Mol. Sci. 2024, 25, 2174. [Google Scholar] [CrossRef]
- Giron, C.C.; Laaksonen, A.; Barroso da Silva, F.L. Differences between Omicron SARS-CoV-2 RBD and other variants in their ability to interact with cell receptors and monoclonal antibodies. J. Biomol. Struct. Dyn. 2023, 41, 5707–5727. [Google Scholar] [CrossRef] [PubMed]
- Ching, W.-Y.; Adhikari, P.; Jawad, B.; Podgornik, R. Effect of Delta and Omicron mutations on the RBD-SD1 domain of the spike protein in SARS-CoV-2 and the Omicron mutations on RBD-ACE2 interface complex. Int. J. Mol. Sci. 2022, 23, 10091. [Google Scholar] [CrossRef] [PubMed]
- Ching, W.-Y.; Adhikari, P.; Jawad, B.; Podgornik, R. Towards quantum-chemical level calculations of SARS-CoV-2 spike protein variants of concern by first principles density functional theory. Biomedicines 2023, 11, 517. [Google Scholar] [CrossRef] [PubMed]
- French, R.H.; Parsegian, V.A.; Podgornik, R.; Rajter, R.F.; Jagota, A.; Luo, J.; Asthagiri, D.; Chaudhury, M.K.; Chiang, Y.-m.; Granick, S. Long range interactions in nanoscale science. Rev. Mod. Phys. 2010, 82, 1887–1944. [Google Scholar] [CrossRef]
- Kim, S.H.; Kearns, F.L.; Rosenfeld, M.A.; Votapka, L.; Casalino, L.; Papanikolas, M.; Amaro, R.E.; Freeman, R. SARS-CoV-2 evolved variants optimize binding to cellular glycocalyx. Cell Rep. Phys. Sci. 2023, 4, 101346. [Google Scholar] [CrossRef]
- Lauster, D.; Haag, R.; Ballauff, M.; Herrmann, A. Balancing stability and function: Impact of the surface charge of SARS-CoV-2 Omicron spike protein. npj Viruses 2025, 3, 23. [Google Scholar] [CrossRef]
- Jawad, B.; Adhikari, P.; Podgornik, R.; Ching, W.-Y. Key interacting residues between RBD of SARS-CoV-2 and ACE2 receptor: Combination of molecular dynamics simulation and density functional calculation. J. Chem. Inf. Model. 2021, 61, 4425–4441. [Google Scholar] [CrossRef]
- Adhikari, P.; Jawad, B.; Podgornik, R.; Ching, W.-Y. Mutations of Omicron variant at the interface of the receptor domain motif and human angiotensin-converting enzyme-2. Int. J. Mol. Sci. 2022, 23, 2870. [Google Scholar] [CrossRef]
- Han, P.; Li, L.; Liu, S.; Wang, Q.; Zhang, D.; Xu, Z.; Han, P.; Li, X.; Peng, Q.; Su, C. Receptor binding and complex structures of human ACE2 to spike RBD from omicron and delta SARS-CoV-2. Cell 2022, 185, 630–640.e10. [Google Scholar] [CrossRef]
- Case, D.A.; Aktulga, H.M.; Belfon, K.; Ben-Shalom, I.; Brozell, S.R.; Cerutti, D.S.; Cheatham III, T.E.; Cruzeiro, V.W.D.; Darden, T.A.; Duke, R.E. Amber 2021 Reference Manual; University of California, San Francisco: San Francisco, CA, USA, 2021. [Google Scholar]
- Shapovalov, M.V.; Dunbrack, R.L. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011, 19, 844–858. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera—A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Liao, H.; Meng, Y.; Li, W.; Han, P.; Liu, K.; Wang, Q.; Li, D.; Zhang, Y.; Wang, L. Structural basis of human ACE2 higher binding affinity to currently circulating Omicron SARS-CoV-2 sub-variants BA.2 and BA.1.1. Cell 2022, 185, 2952–2960.e10. [Google Scholar] [CrossRef] [PubMed]
- VASP—Vienna Ab Initio Simulation Package. Available online: https://www.vasp.at/ (accessed on 1 May 2020).
- Ching, W.-Y.; Rulis, P. Electronic Structure Methods for Complex Materials: The Orthogonalized Linear Combination of Atomic Orbitals; Oxford University Press: Oxford, UK, 2012. [Google Scholar]
- Adhikari, P.; Jawad, B.; Podgornik, R.; Ching, W.-Y. Quantum chemical computation of omicron mutations near cleavage sites of the spike protein. Microorganisms 2022, 10, 1999. [Google Scholar] [CrossRef]
- Adhikari, P.; Li, N.; Shin, M.; Steinmetz, N.F.; Twarock, R.; Podgornik, R.; Ching, W.-Y. Intra-and intermolecular atomic-scale interactions in the receptor binding domain of SARS-CoV-2 spike protein: Implication for ACE2 receptor binding. Phys. Chem. Chem. Phys. 2020, 22, 18272–18283. [Google Scholar] [CrossRef]
- Adhikari, P.; Ching, W.-Y. Amino acid interacting network in the receptor-binding domain of SARS-CoV-2 spike protein. RSC Adv. 2020, 10, 39831–39841. [Google Scholar] [CrossRef]
- Ching, W.-Y.; Adhikari, P.; Jawad, B.; Podgornik, R. Ultra-large-scale ab initio quantum chemical computation of bio-molecular systems: The case of spike protein of SARS-CoV-2 virus. Comput. Struct. Biotechnol. J. 2021, 19, 1288–1301. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized gradient approximation made simple. Phys. Rev. Lett. 1996, 77, 3865. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. I. J. Chem. Phys. 1955, 23, 1833–1840. [Google Scholar] [CrossRef]
- Mulliken, R.S. Electronic population analysis on LCAO–MO molecular wave functions. II. Overlap populations, bond orders, and covalent bond energies. J. Chem. Phys. 1955, 23, 1841–1846. [Google Scholar] [CrossRef]
- Dharmawardhana, C.; Misra, A.; Ching, W.-Y. Quantum mechanical metric for internal cohesion in cement crystals. Sci. Rep. 2014, 4, 7332. [Google Scholar] [CrossRef]
- Adhikari, P.; Li, N.; Rulis, P.; Ching, W.-Y. Deformation behavior of an amorphous zeolitic imidazolate framework–from a supersoft material to a complex organometallic alloy. Phys. Chem. Chem. Phys. 2018, 20, 29001–29011. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| OV BA.1 | OV BA.2 | OV BA.5 | OV XBB.1.16 |
|---|---|---|---|
| G339D | G339D | G339D | G339H |
| - | - | - | R346T |
| - | - | - | L368I |
| S371L | S371F | S371F | S371F |
| S373P | S373P | S373P | S373P |
| S375F | S375F | S375F | S375F |
| - | T376A | T376A | T376A |
| - | D405N | D405N | D405N |
| - | R408S | R408S | R408S |
| K417N | K417N | K417N | K417N |
| N440K | N440K | N440K | N440K |
| - | - | - | V445P |
| G446S | - | - | G446S |
| - | - | L452R | - |
| - | - | N460K | |
| S477N | S477N | S477N | S477N |
| T478K | T478K | T478K | T478R |
| E484A | E484A | E484A | E484A |
| F486V | F486P | ||
| - | - | - | F490S |
| Q493R | Q493R | - | - |
| G496S | - | - | |
| Q498R | Q498R | Q498R | Q498R |
| N501Y | N501Y | N501Y | N501Y |
| Y505H | Y505H | Y505H | Y505H |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Adhikari, P.; Jawad, B.; Ching, W.-Y. Electronic Interactions Between the Receptor-Binding Domain of Omicron Variants and Angiotensin-Converting Enzyme 2: A Novel Amino Acid–Amino Acid Bond Pair Concept. Molecules 2025, 30, 2061. https://doi.org/10.3390/molecules30092061
Adhikari P, Jawad B, Ching W-Y. Electronic Interactions Between the Receptor-Binding Domain of Omicron Variants and Angiotensin-Converting Enzyme 2: A Novel Amino Acid–Amino Acid Bond Pair Concept. Molecules. 2025; 30(9):2061. https://doi.org/10.3390/molecules30092061
Chicago/Turabian StyleAdhikari, Puja, Bahaa Jawad, and Wai-Yim Ching. 2025. "Electronic Interactions Between the Receptor-Binding Domain of Omicron Variants and Angiotensin-Converting Enzyme 2: A Novel Amino Acid–Amino Acid Bond Pair Concept" Molecules 30, no. 9: 2061. https://doi.org/10.3390/molecules30092061
APA StyleAdhikari, P., Jawad, B., & Ching, W.-Y. (2025). Electronic Interactions Between the Receptor-Binding Domain of Omicron Variants and Angiotensin-Converting Enzyme 2: A Novel Amino Acid–Amino Acid Bond Pair Concept. Molecules, 30(9), 2061. https://doi.org/10.3390/molecules30092061