1. Introduction
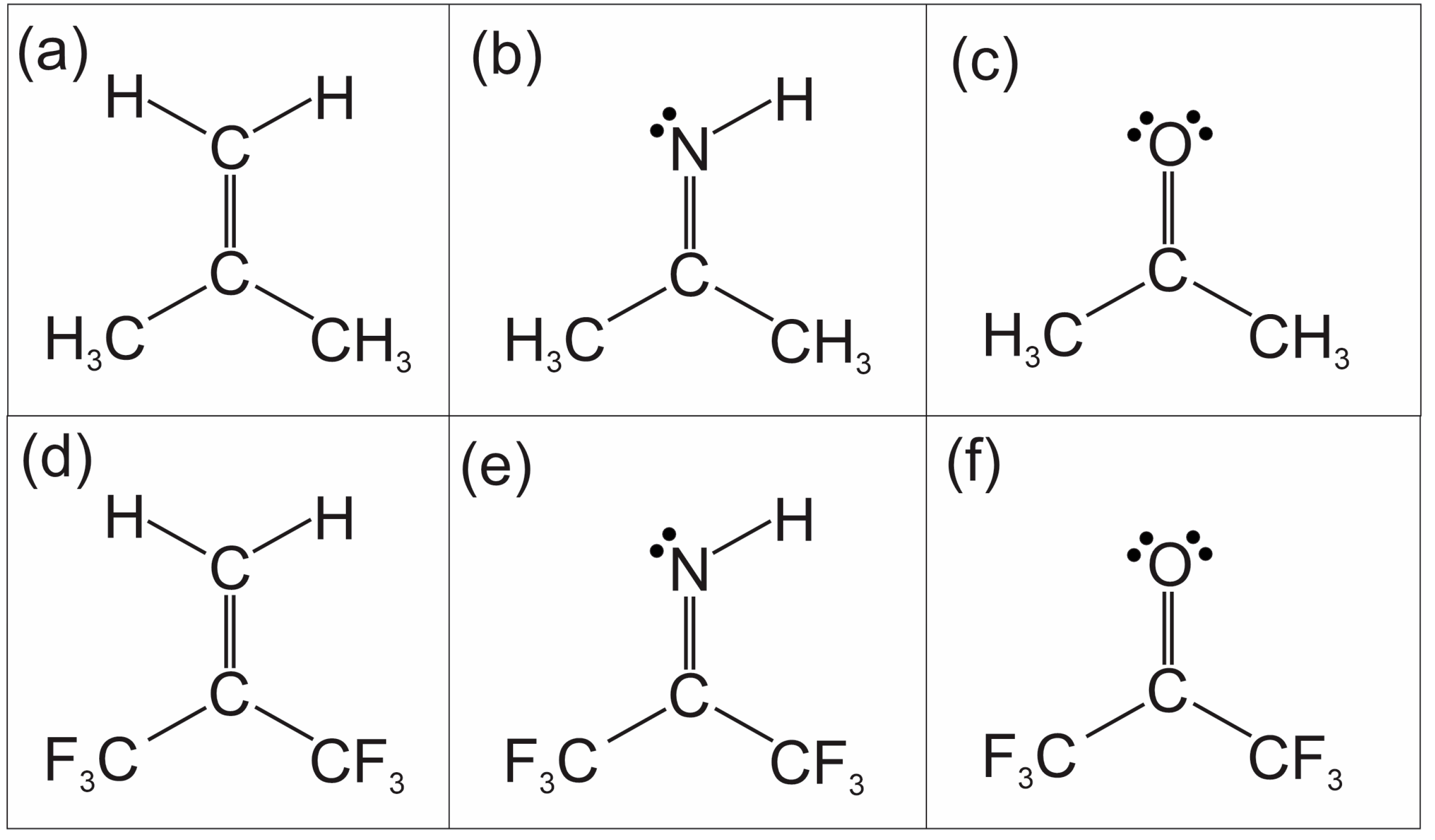
Hexafluoroacetone imine may be considered a member of a grouping of six molecules shown in
Figure 1. This grouping consists of two isoelectronic series, the first being isobutene, acetone imine, and lastly acetone, and the second being the hexafluorinated analogs of the first series. Except for hexafluoroacetone imine, all these molecules have had their rotational spectra recorded at high resolution. These spectra are reported, or most recently discussed, in the following references [
1,
2,
3,
4,
5]. Isobutene and acetone are well-known examples of rotational spectra in which the effects of the internal rotation of two equivalent methyl tops are manifested. At high resolution, rotational transitions from these types of molecules will appear as quartets owing to transitions occurring from within the
AA,
EE,
AE, and
EA torsional sub-levels.
These spectra have been interpreted to yield effective CH
3 internal rotation tunneling barriers of 761(1) cm
−1 for isobutene [
1] and 251.4(26) cm
−1 for acetone [
3]. One would correctly assume that the tunneling barrier to CH
3 internal rotation in acetone imine would be intermediate between isobutene and acetone. However, the spectra are more complex owing to the two CH
3 rotors being non-equivalent. In that case, the effects of the two non-equivalent CH
3 rotors result in five torsional substrates, one with
A1 symmetry and four doubly degenerate states with
E symmetry. Hence, rotational transitions from within these substates present to the observer as quintets. An analysis of these spectra by Zou et al. [
2] has resulted in internal rotation barrier heights of 531.956(64) cm
−1, attributed to the CH
3 furthest from the imine hydrogen, and 465.013(26) cm
−1, attributed to the CH
3 nearest to the imine hydrogen. Taken as a series, then we observe that the CH
3 barriers to internal rotation increase in the order of increasing hydrogenation of the apex atom, i.e., in the order O, N-H, H-C-H. From a purely classical mechanics standpoint, this is rational in the sense that the apex hydrogen(s) clearly impede the CH
3 internal rotations.
We then consider the hexafluorinated analogs. The rotational spectrum of hexafuoroacetone has been recorded at high resolution with spectral line widths on the order of 7 kHz at full width, half maximum height [
5]. However, none of the transitions observed for hexafluoroacetone were observed as multiplets, or broadened as they were for acetone. Now, it is generally true that, the higher the barrier heights to internal motion the closer together in energy will be the torsional substates and, hence, the rotational transitions from within those different torsional substates will appear closer together in frequency. Given that the barrier heights to CF
3 internal rotation are significantly higher than those for CH
3 internal rotation, together with the very small internal rotational constant for a CF
3 group,
F ≈ 10 GHz (compared to that for a CH
3 group,
F ≈ 160 GHz), then effects due to CF
3 internal rotation are most often unresolvable. A helpful discussion on this topic is presented in reference [
6]. Accordingly, the rotational spectrum of hexafluoroacetone presents like that of an “ordinary” semirigid rotor, with no evidence of any internal motions.
Given that isobutene has a higher barrier to CH
3 internal rotation compared to that of acetone, it may be assumed that the barrier to CF
3 internal rotation in hexaflouroisobutene would be higher than that in hexafluoroacetone and, therefore, would have a rotational spectrum with no observable evidence of internal motion. However, the recorded rotational spectrum of hexafluoroisobutene presented as being doubled with spacings between the rotational transitions being on the order of tens of megahertz. The spectral analyses revealed that the bis-trifluoromethyl groups of hexafluoroisobutene are staggered in the equilibrium configuration, and that a novel, out-of-phase rotation through an F-CCC-F planar configuration with a low barrier (<100 cm
−1), leads to the observed doubled rotational spectra [
4]. This bistrifluoromethy effect has subsequently been explored in related molecules [
7,
8].
In this article, we present the measurement and analyses of rotational spectra for the remaining member of the molecules in
Figure 1, hexafluoroacetone imine. We ask the question of how well the spectra are present, like that of hexafluoroacetone or that of hexafluoroisobutene. And further, how do we interpret the spectra?
3. Discussion
The pure rotational spectrum of hexafluoroacetone imine presents as consisting of two nearly-equivalent conformers. However, it is undoubtedly the case that these two sets of measured rotational transitions arise from two torsional substates, State I and State II, resulting from a relatively low barrier between two equivalent F-CCC-F staggered configurations. The torsional substates most likely arise from an out-of-phase internal rotation of the two CF
3 groups which is governed by an unusual potential energy function with six minima, three high barriers (V1) and three low barriers (V2) as observed in the related molecule hexafluoroisobutene [
4]. The substrates arise via tunneling through the low barrier. An example of the potential energy function is given in
Figure 5. For the hexafluorinated species, the global minimum corresponds to a staggered F
1-CCC-F
2 configuration exemplified in
Figure 3.
Careful calculations by Shahi and Arunan [
8] have revealed that for the isoelectronic sequence of molecules hexafluoroisobutene (HFIB), hexafluoroacetone imine (HFAI), and hexafluoacetone (HFA) the barrier height V1 varies as HFIB > HFAI > HFA, whereas the V2 barrier height varies as HFA > HFAI > HFIB. In all three cases, the V1 barrier height is too high for tunneling effects to manifest in the observed spectra consistent with the absence of groupings of rotational transitions from within four or five torsional substates. The V2 tunneling barrier height is more nuanced. With the molecule unable to tunnel through V1, we are left with a tunneling motion through a much lower barrier separating two equivalent F-CCC-F staggered configurations, more akin to a double minimum potential often observed in ring-puckering problems. The barrier V2 is lowest for hexafluoroisobutene consistent with rotational transitions from the two torsional states being separated by tens of MHz. Whereas the barrier V2 is highest for hexafluoroacetone consistent with no observed “doublets” in the rotational spectrum. Hexafluoroacetone imine is intermediate, again consistent with the observation of paired rotational transitions separated by tens of kHz.
The above trends in both V1 and V2 may be rationalized through an appeal to the molecular geometries. In regards to the trend in V1, we note that all three molecules possess a C
1-C
3-C
2 structural component, see
Figure 3. It is found that the distance C
1…C
2 decreases in the order HFIB > HFAI > HFA. A large value of C
1…C
2 means that the two CF
3-groups are further away from one another, and thus easier to rotate, compared to a small value of C
1…C
2 which corresponds to the CF
3-groups being closer together. The correspondence between V1 and C
1…C
2 is shown in
Figure 5. In regards to V2 in which the top of the smaller barrier corresponds to a planar F
1-CCC-F
2 (see
Figure 5), it is useful to compare the C=X distance where X = CH
2, NH, and O. This C=X distance decreases in the order HFIB > HFAI > HFA. The larger C=X distance in hexafluoroisobutene corresponds to a small V2, i.e., it is easier for F-CCC-F to pass through a planar configuration, compared to the longer C=X distance in hexafluoroacetone where the planar F-CCC-F configuration is more greatly interfered with by the oxygen lone pairs. The correspondence between V2 and C=X is also shown in
Figure 5.
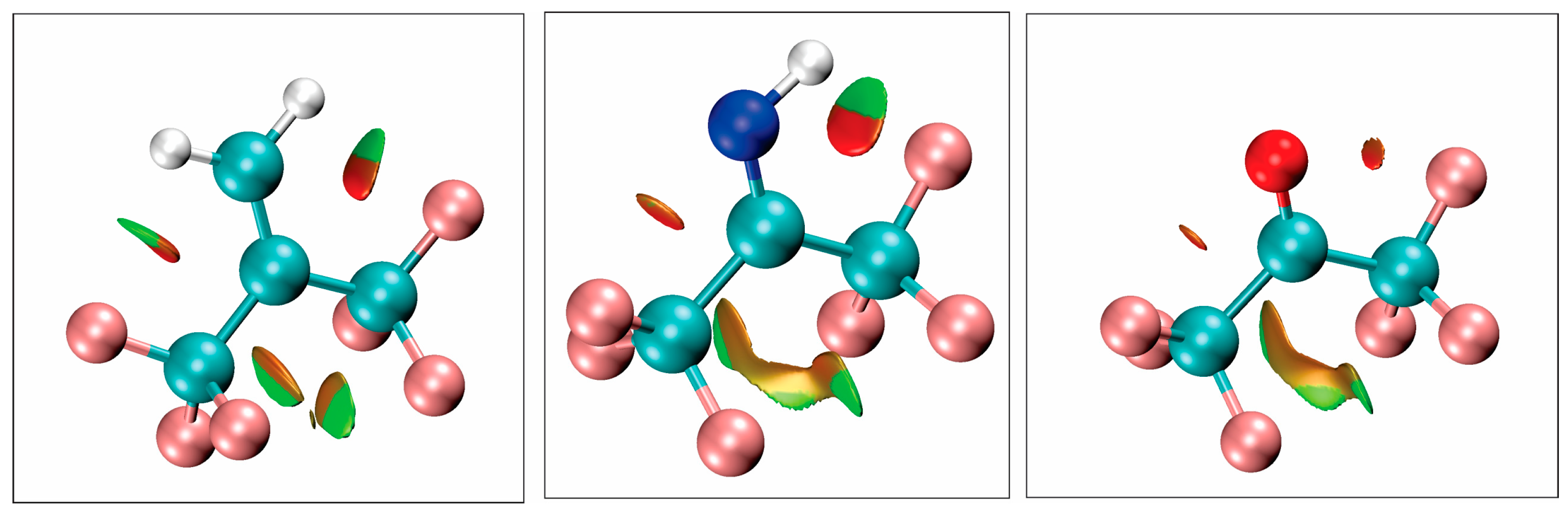
The above arguments are supported and augmented by calculations that reveal non-covalent interactions. Following the work of Johnson et al. [
30] non-covalent interactions based on the electron density and its derivatives have been located for hexafluoroisobutene, hexafluoroacetone imine, and hexafluoroacetone. The results are presented in
Figure 6. In this figure, green indicates weak attractive interactions whereas the orange and reddish colors indicate repulsive interactions. To obtain these plots, the electron density at the B3LYP level was analyzed using the Multiwfn [
31,
32] program and then visualized using VMD [
33].
Within the figure, it is observed that in going from hexafluoroacetone-to-hexafluoroacetone imine to hexafluorisobutene the attractive interactions increase between the CF3 groups and O, NH and CH2, which in turn explains why the rotation of the CF3 groups becomes easier in that series. It is also observed that interactions with the lone pairs are purely repulsive whereas interactions with the NH and CH2 are partially attractive.



 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}