
Unraveling the Specific Recognition Between PD-L1 and Engineered CLP002 Functionalized Gold Nanostructures: MD Simulation Studies

 ,
,  , , , and
, , , and
Abstract

1. Introduction
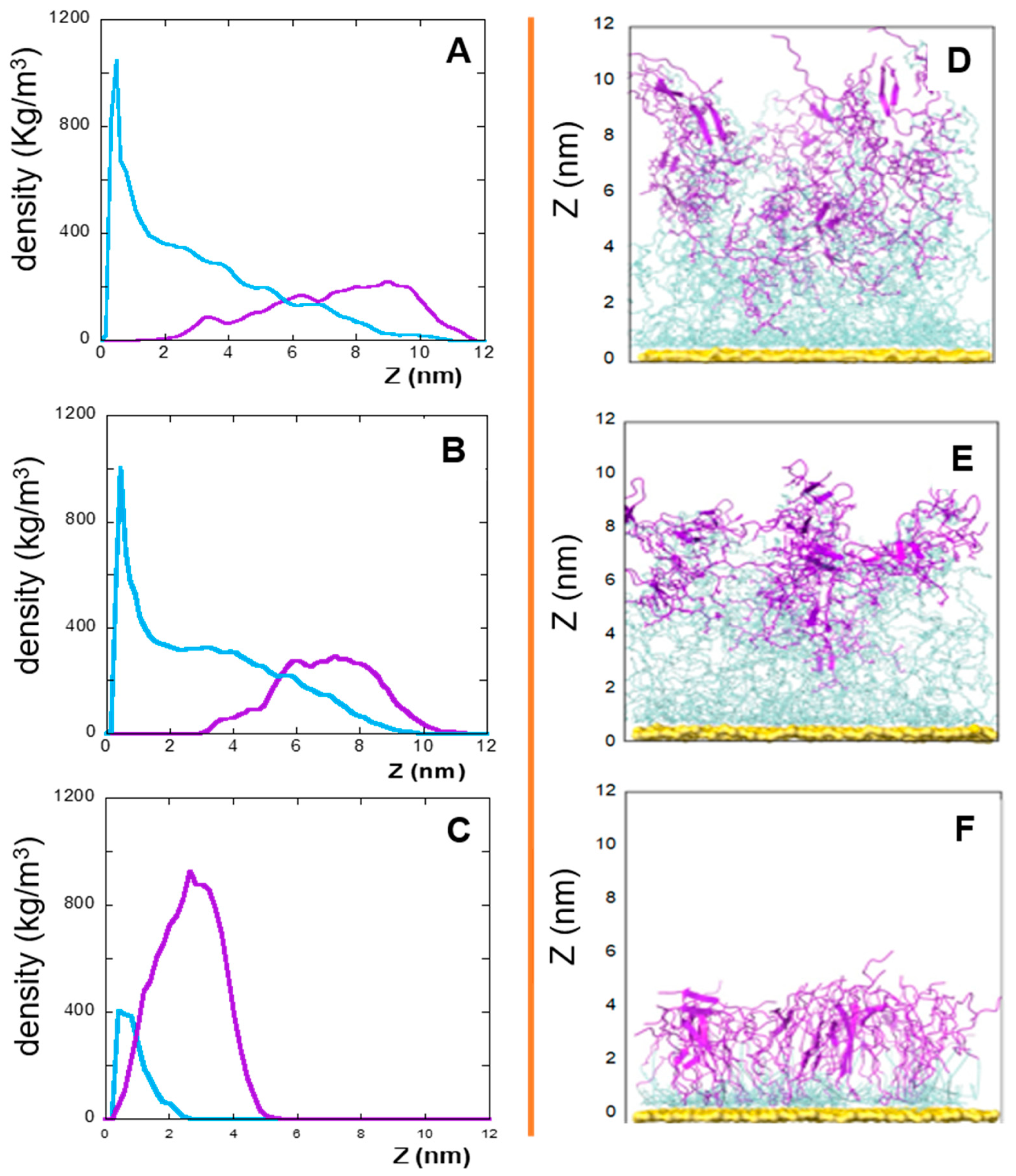
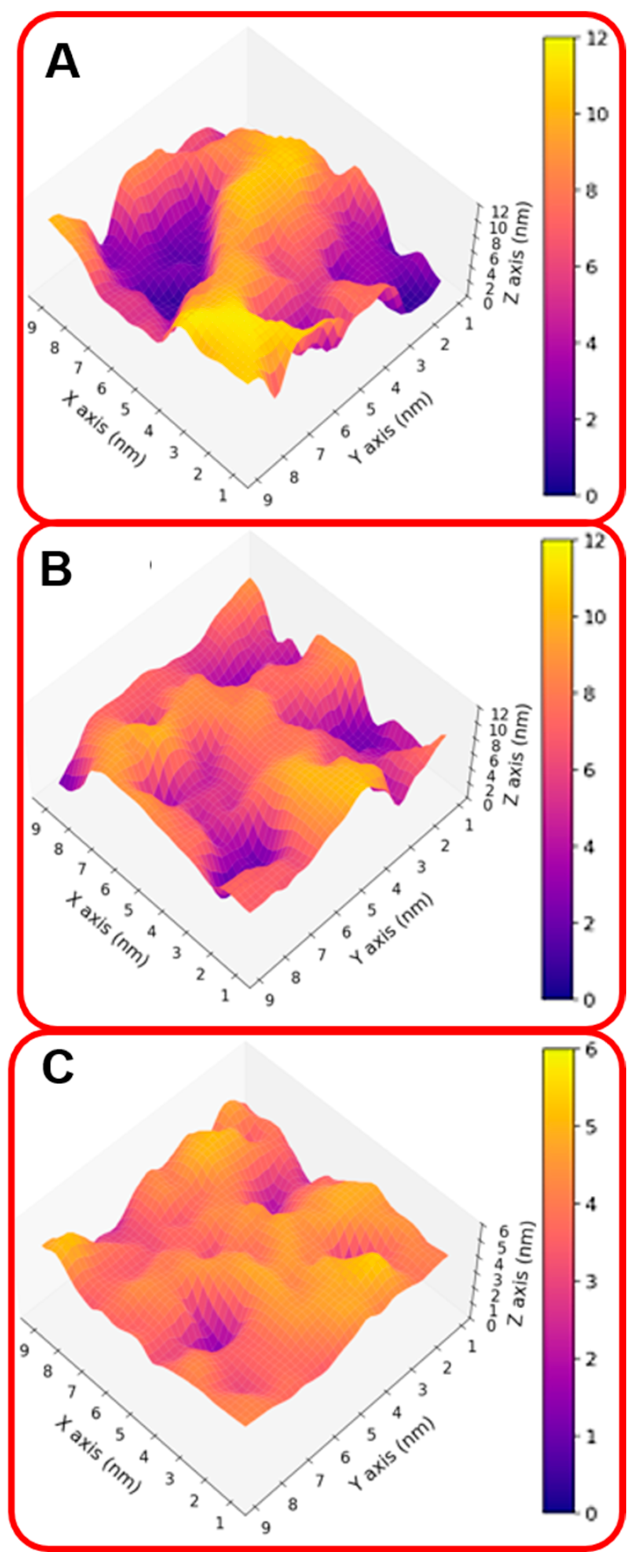
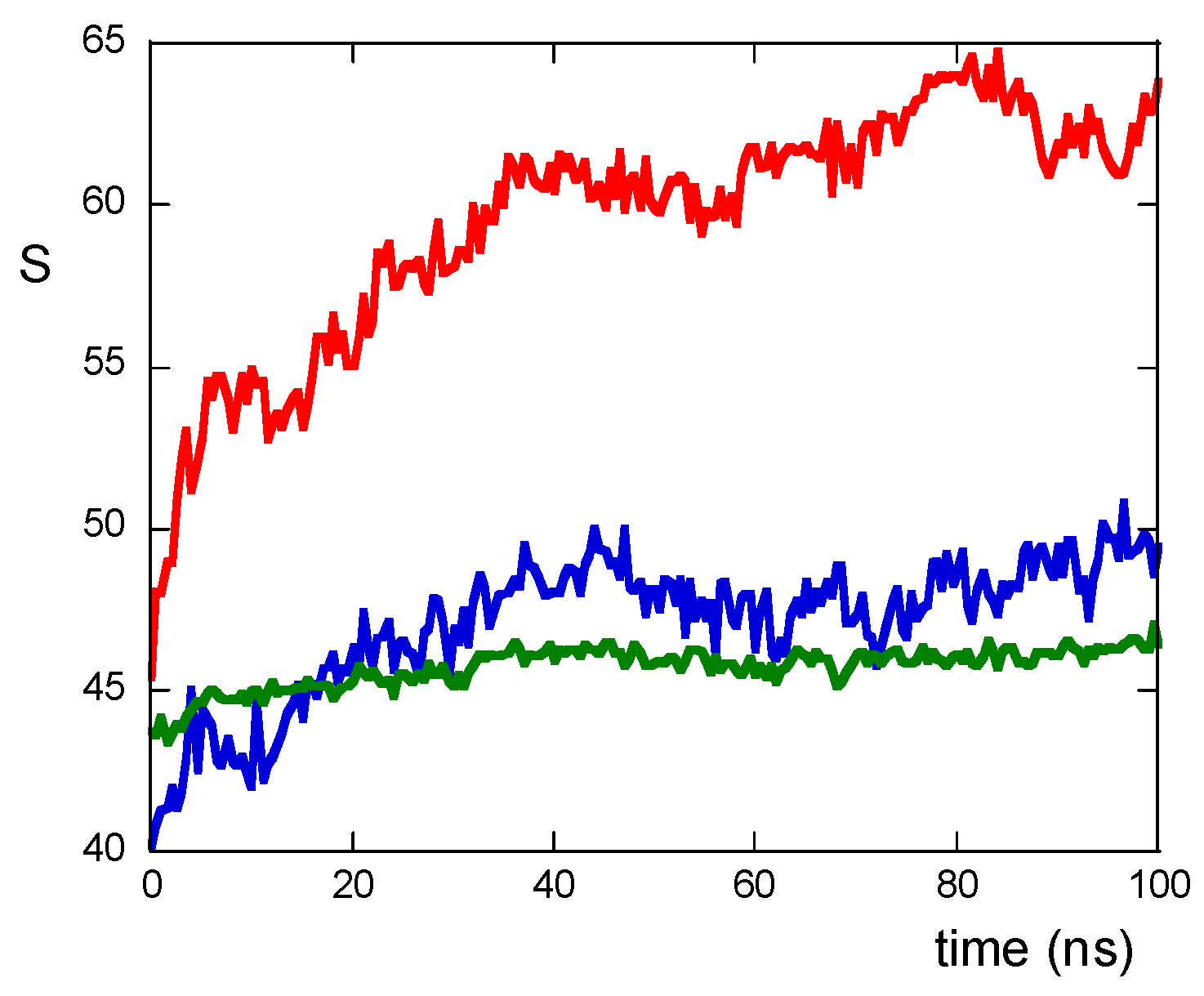
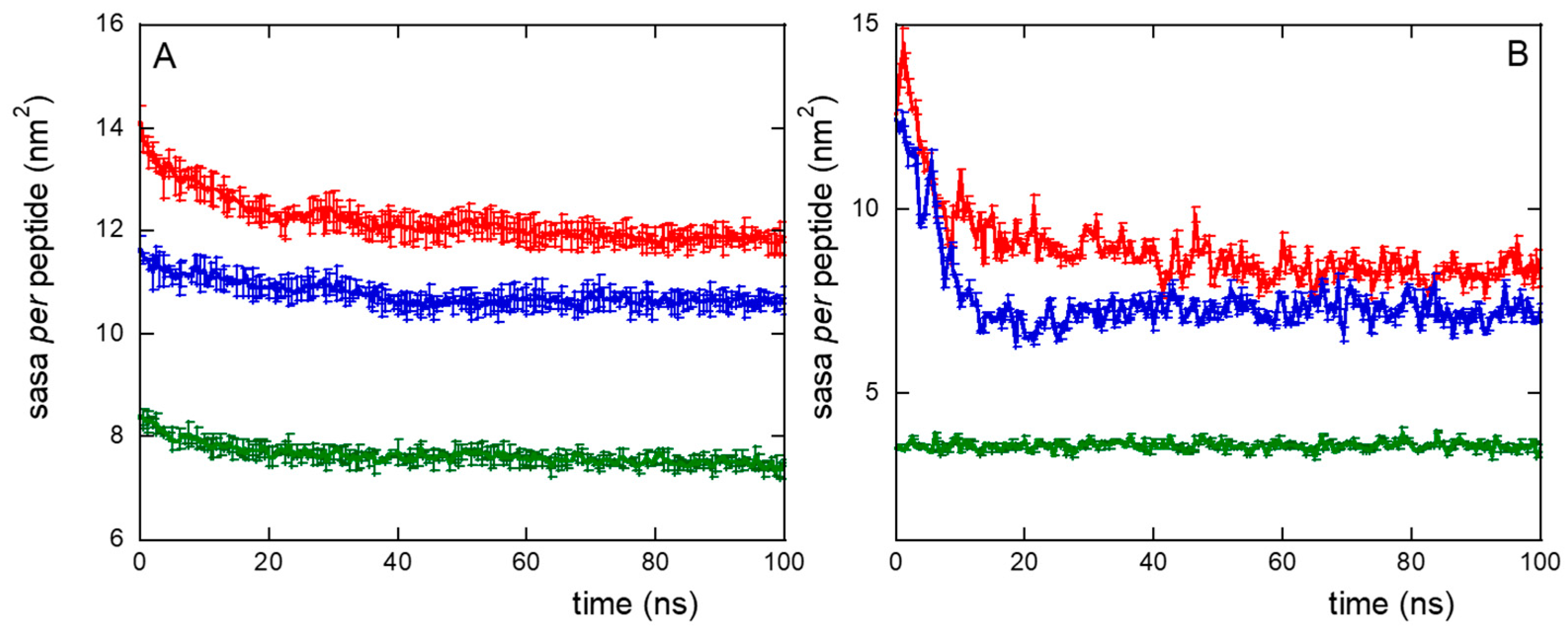
2. Results and Discussion
2.1. The Role of the Primary Structure
2.2. The Role of the Linker
3. Materials and Methods
3.1. Functionalized Nanostructures Synthesis and Characterization by Transmission Electron Microscopy Measurements
3.2. MDs Simulations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van den Boogaard, W.M.; Komninos, D.S.; Vermeij, W.P. Chemotherapy side-effects: Not all DNA damage is equal. Cancers 2022, 14, 627. [Google Scholar] [CrossRef] [PubMed]
- Nurgali, K.; Jagoe, R.T.; Abalo, R. Adverse effects of cancer chemotherapy: Anything new to improve tolerance and reduce sequelae? Front. Pharmacol. 2018, 9, 245. [Google Scholar] [CrossRef] [PubMed]
- Lyman, G.H. Impact of chemotherapy dose intensity on cancer patient outcomes. J. Natl. Compr. Cancer Netw. 2009, 7, 99–108. [Google Scholar] [CrossRef] [PubMed]
- Schirrmacher, V. From chemotherapy to biological therapy: A review of novel concepts to reduce the side effects of systemic cancer treatment. Int. J. Oncol. 2019, 54, 407–419. [Google Scholar] [CrossRef]
- Sanchez-Barcelo, E.J.; Mediavilla, M.D.; Alonso-Gonzalez, C.; Reiter, R.J. Melatonin uses in oncology: Breast cancer prevention and reduction of the side effects of chemotherapy and radiation. Expert. Opin. Investig. Drugs 2012, 21, 819–831. [Google Scholar] [CrossRef]
- Shi, J.; Kantoff, P.W.; Wooster, R.; Farokhzad, O.C. Cancer nanomedicine: Progress, challenges and opportunities. Nat. Rev. Cancer 2017, 17, 20–37. [Google Scholar] [CrossRef]
- Rai, P.; Mallidi, S.; Zheng, X.; Rahmanzadeh, R.; Mir, Y.; Elrington, S.; Khurshid, A.; Hasan, T. Development and Applications of Photo-Triggered Theranostic Agents. Adv. Drug Deliv. Rev. 2010, 62, 1094–1124. [Google Scholar] [CrossRef]
- Singh, M.; Harris-Birtill, D.C.C.; Markar, S.R.; Hanna, G.B.; Elson, D.S. Application of Gold Nanoparticles for Gastrointestinal Cancer Theranostics: A Systematic Review. Nanomed. Nanotechnol. Biol. Med. 2015, 11, 2083–2098. [Google Scholar] [CrossRef]
- Arvizo, R.; Bhattacharya, R.; Mukherjee, P. Gold Nanoparticles: Opportunities and Challenges in Nanomedicine. Expert Opin. Drug Deliv. 2010, 7, 753–763. [Google Scholar] [CrossRef]
- Biscaglia, F.; Ripani, G.; Rajendran, S.; Benna, C.; Mocellin, S.; Bocchinfuso, G.; Meneghetti, M.; Palleschi, A.; Gobbo, M. Gold Nanoparticle Aggregates Functionalized with Cyclic RGD Peptides for Targeting and Imaging of Colorectal Cancer Cells. ACS Appl. Nano Mater. 2019, 2, 6436–6444. [Google Scholar] [CrossRef]
- Mazzuca, C.; Di Napoli, B.; Biscaglia, F.; Ripani, G.; Rajendran, S.; Braga, A.; Benna, C.; Mocellin, S.; Gobbo, M.; Meneghetti, M.; et al. Understanding the Good and Poor Cell Targeting Activity of Gold Nanostructures Functionalized with Molecular Units for the Epidermal Growth Factor Receptor. Nanoscale Adv. 2019, 1, 1970–1979. [Google Scholar] [CrossRef] [PubMed]
- Biscaglia, F.; Rajendran, S.; Conflitti, P.; Benna, C.; Sommaggio, R.; Litti, L.; Mocellin, S.; Bocchinfuso, G.; Rosato, A.; Palleschi, A.; et al. Enhanced EGFR Targeting Activity of Plasmonic Nanostructures with Engineered GE11 Peptide. Adv. Healthc. Mater. 2017, 6, 1700596. [Google Scholar] [CrossRef] [PubMed]
- Gobbo, M.; Caligiuri, I.; Giannetti, M.; Litti, L.; Mazzuca, C.; Rizzolio, F.; Palleschi, A.; Meneghetti, M. SERS Nanostructures with Engineered Active Peptides against an Immune Checkpoint Protein. Nanoscale 2024, 16, 5206–5214. [Google Scholar] [CrossRef]
- Magiera-Mularz, K.; Skalniak, L.; Zak, K.M.; Musielak, B.; Rudzinska-Szostak, E.; Berlicki, Ł.; Kocik, J.; Grudnik, P.; Sala, D.; Zarganes-Tzitzikas, T.; et al. Bioactive Macrocyclic Inhibitors of the PD-1/PD-L1 Immune Checkpoint. Angew. Chem. Int. Ed. 2017, 56, 13732–13735. [Google Scholar] [CrossRef]
- Wang, T.; Wu, X.; Guo, C.; Zhang, K.; Xu, J.; Li, Z.; Jiang, S. Development of Inhibitors of the Programmed Cell Death-1/Programmed Cell Death-Ligand 1 Signaling Pathway. J. Med. Chem. 2019, 62, 1715–1730. [Google Scholar] [CrossRef]
- Zhan, M.M.; Hu, X.Q.; Liu, X.X.; Ruan, B.F.; Xu, J.; Liao, C. From Monoclonal Antibodies to Small Molecules: The Development of Inhibitors Targeting the PD-1/PD-L1 Pathway. Drug Discov. Today 2016, 21, 1027–1036. [Google Scholar] [CrossRef]
- Vadevoo, S.M.P.; Gurung, S.; Lee, H.S.; Gunassekaran, G.R.; Lee, S.M.; Yoon, J.W.; Lee, Y.K.; Lee, B. Peptides as multifunctional players in cancer therapy. Exp. Mol. Med. 2023, 55, 1099–1109. [Google Scholar] [CrossRef]
- Sui, X.; Niu, X.; Zhou, X.; Gao, Y. Peptide drugs: A new direction in cancer immunotherapy. Cancer Biol. Med. 2024, 21, 198–203. [Google Scholar] [CrossRef]
- Biscaglia, F.; Caligiuri, I.; Ripani, G.; Rizzolio, F.; Palleschi, A.; Meneghetti, M.; Gobbo, M. Protection against Proteolysis of a Cell Targeting Peptide on Gold Nanostructures. Nanoscale 2021, 13, 10544–10554. [Google Scholar] [CrossRef]
- Parvez, A.; Choudhary, F.; Mudgal, P.; Khan, R.; Qureshi, K.A.; Farooqi, H.; Aspatwar, A. PD-1 and PD-L1: Architects of immune symphony and immunotherapy breakthroughs in cancer treatment. Front. Immunol. 2023, 14, 1296341. [Google Scholar] [CrossRef]
- Mahoney, K.M.; Freeman, G.J.; McDermott, D.F. The next immune-checkpoint inhibitors: PD-1/PD-L1 blockade in melanoma. Clin. Ther. 2015, 37, 764–782. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Chen, Z.; Li, Y.; Zhao, W.; Wu, J.; Zhang, Z. PD-1/PD-L1 checkpoint inhibitors in tumor immunotherapy. Front. Pharmacol. 2021, 12, 731798. [Google Scholar] [CrossRef] [PubMed]
- Zak, K.M.; Kitel, R.; Przetocka, S.; Golik, P.; Guzik, K.; Musielak, B.; Dömling, A.; Dubin, G.; Holak, T.A. Structure of the Complex of Human Programmed Death 1, PD-1, and Its Ligand PD-L1. Structure 2015, 23, 2341–2348. [Google Scholar] [CrossRef]
- Magnez, R.; Thiroux, B.; Taront, S.; Segaoula, Z.; Quesnel, B.; Thuru, X. PD-1/PD-L1 Binding Studies Using Microscale Thermophoresis. Sci. Rep. 2017, 7, 17623. [Google Scholar] [CrossRef]
- Lin, X.; Lu, X.; Luo, G.; Xiang, H. Progress in PD-1/PD-L1 Pathway Inhibitors: From Biomacromolecules to Small Molecules. Eur. J. Med. Chem. 2020, 186, 111876. [Google Scholar] [CrossRef]
- Liu, W.; Jin, H.; Chen, T.; Zhang, G.; Lai, S.; Liu, G. Investigating the role of the N-terminal loop of PD-1 in binding process between PD-1 and Nivolumab via molecular dynamics simulation. Front. Mol. Biosci. 2020, 7, 574759. [Google Scholar] [CrossRef]
- Liu, K.; Tan, S.; Chai, Y.; Chen, D.; Song, H.; Zhang, C.W.H.; Shi, Y.; Liu, J.; Tan, W.; Lyu, J.; et al. Structural basis of anti-PD-L1 monoclonal antibody Avelumab for tumor therapy. Cell Res. 2017, 27, 151–153. [Google Scholar] [CrossRef]
- Boutros, C.; Tarhini, A.; Routier, E.; Lambotte, O.; Ladurie, F.L.; Carbonnel, F.; Izzeddine, H.; Marabelle, A.; Champiat, S.; Berdelou, A.; et al. Safety profiles of anti-CTLA-4 and anti-PD-1 antibodies alone and in combination. Nat. Rev. Clin. Oncol. 2016, 13, 473–486. [Google Scholar] [CrossRef]
- Jiang, Q.; Yao, F.; An, Y.; Lai, X.; Li, X.; Yu, Z.; Yang, X.D. Novel nanotherapeutics for cancer immunotherapy by albumin nanoparticles functionalized with PD-1 and PD-L1 aptamers. Cancer Nanotechnol. 2024, 15, 3. [Google Scholar] [CrossRef]
- Linde, C.; Chien, Y.T.; Chen, Z.; Mu, Q. Nanoparticle-enhanced PD-1/PD-L1 targeted combination therapy for triple negative breast cancer. Front. Oncol. 2024, 14, 1393492. [Google Scholar] [CrossRef]
- Song, S.; Shim, M.K.; Yang, S.; Lee, J.; Yun, W.S.; Cho, H.; Moon, Y.; Min, J.Y.; Han, E.H.; Yoon, H.Y.; et al. All-in-one glycol chitosan nanoparticles for co-delivery of doxorubicin and anti-PD-L1 peptide in cancer immunotherapy. Bioact. Mater. 2023, 28, 358–375. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Zhao, Z.; Zhang, L.; Li, Y.; Jain, A.; Barve, A.; Jin, W.; Liu, Y.; Fetse, J.; Cheng, K. Discovery of Low-Molecular Weight Anti-PD-L1 Peptides for Cancer Immunotherapy. J. Immunother. Cancer 2019, 7, 270. [Google Scholar] [CrossRef] [PubMed]
- Craik, D.J.; Fairlie, D.P.; Liras, S.; Price, D. The Future of Peptide-Based Drugs. Chem. Biol. Drug Des. 2013, 81, 136–147. [Google Scholar] [CrossRef] [PubMed]
- Moya, C.; Escudero, R.; Malaspina, D.C.; De La Mata, M.; Hernández-Saz, J.; Faraudo, J.; Roig, A. Insights into preformed human serum albumin corona on iron oxide nanoparticles: Structure, effect of particle size, impact on mri efficiency, and metabolization. ACS Appl. Bio Mater. 2019, 2, 3084–3094. [Google Scholar] [CrossRef]
- Kang-Pettinger, T.; Walker, K.; Brown, R.; Cowan, R.; Wright, H.; Baravalle, R.; Waters, L.C.; Muskett, F.W.; Bowler, M.W.; Sawmynaden, K.; et al. Identification, binding, and structural characterization of single domain anti-PD-L1 antibodies inhibitory of immune regulatory proteins PD-1 and CD80. J. Biol. Chem. 2023, 299, 102769. [Google Scholar] [CrossRef]
- Thomas, P.D.; Dill, K.A. An iterative method for extracting energy-like quantities from protein structures. Proc. Natl. Acad. Sci. USA 1996, 93, 11628–11633. [Google Scholar] [CrossRef]
- Dosztanyi, Z.; Csizmok, V.; Tompa, P.; Simon, I. The pairwise energy content estimated from amino acid composition discriminates between folded and intrinsically unstructured proteins. J. Mol. Biol. 2005, 347, 827–839. [Google Scholar] [CrossRef]
- Oostenbrink, C.; Villa, A.; Mark, A.E.; Van Gunsteren, W.F. A Biomolecular Force Field Based on the Free Enthalpy of Hydration and Solvation: The GROMOS Force-Field Parameter Sets 53A5 and 53A6. J. Comput. Chem. 2004, 25, 1656–1676. [Google Scholar] [CrossRef]
- Fuchs, P.F.J.; Hansen, H.S.; Hünenberger, P.H.; Horta, B.A.C. A GROMOS Parameter Set for Vicinal Diether Functions: Properties of Polyethyleneoxide and Polyethyleneglycol. J. Chem. Theory Comput. 2012, 8, 3943–3963. [Google Scholar] [CrossRef]
- Pu, Q.; Leng, Y.; Zhao, X.; Cummings, P.T. Molecular Simulations of Stretching Gold Nanowires in Solvents. Nanotechnol. 2007, 18, 424007. [Google Scholar] [CrossRef]
- Essmann, U.; Perera, L.; Berkowitz, M.L.; Darden, T.; Lee, H.; Pedersen, L.G. A Smooth Particle Mesh Ewald Method. J. Chem. Phys. 1995, 103, 8577–8593. [Google Scholar] [CrossRef]
- York, D.M.; Darden, T.A.; Pedersen, L.G. The Effect of Long-Range Electrostatic Interactions in Simulations of Macromolecular Crystals: A Comparison of the Ewald and Truncated List Methods. J. Chem. Phys. 1993, 99, 8345–8348. [Google Scholar] [CrossRef]
- Bussi, G.; Donadio, D.; Parrinello, M. Canonical Sampling through Velocity Rescaling. J. Chem. Phys. 2007, 126, 014101. [Google Scholar] [CrossRef]
- Berendsen, H.J.C.; Postma, J.P.M.; Van Gunsteren, W.F.; Dinola, A.; Haak, J.R. Molecular Dynamics with Coupling to an External Bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Messina, G.M.; Mazzuca, C.; Dettin, M.; Zamuner, A.; Di Napoli, B.; Ripani, G.; Marletta, G.; Palleschi, A. From nanoaggregates to mesoscale ribbons: The multistep self-organization of amphiphilic peptides. Nanoscale Adv. 2021, 3, 3605–3614. [Google Scholar]
- Kabsch, W.; Sander, C. Dictionary of Protein Secondary Structure: Pattern Recognition of Hydrogen-Bonded and Geometrical Features. Biopolymers 1983, 22, 2577–2637. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Peptide | Sequence 1 |
|---|---|
| CLP002 | Trp-His-Arg-Ser-Tyr-Tyr-Thr-Trp-Asn-Leu-Asn-Thr |
| SCLP002 | Thr-Arg-Trp-Ser-His-Tyr-Asn-Thr-Leu-Trp-Tyr-Asn |
| Linker | |
| C- | Ac-Cys-(O2oc)2- |
| P- | HS-PEG-Lys3-Gly2- |
| Linker and peptide | |
| C-CLP | Ac-Cys-(O2oc)2-CLP002-NH2 |
| P-CLP | HS-PEG-Lys3-Gly2-CLP002-NH2 |
| P-SCLP | HS-PEG-Lys3-Gly2-SCLP002-NH2 |
| System | Box (nm3) | Peptides | Water | Ions (Cl−) |
|---|---|---|---|---|
| NS@P-CLP | 10.32 × 9.99 × 27.55 | 45 | 79,050 | 180 |
| NS@P-SCLP | 10.30 × 9.97 × 27.50 | 45 | 78,349 | 180 |
| NS@C-CLP | 10.31 × 9.98 × 18.17 | 90 | 50,807 | 90 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Giannetti, M.; Gobbo, M.; Litti, L.; Caligiuri, I.; Rizzolio, F.; Meneghetti, M.; Mazzuca, C.; Palleschi, A. Unraveling the Specific Recognition Between PD-L1 and Engineered CLP002 Functionalized Gold Nanostructures: MD Simulation Studies. Molecules 2025, 30, 2045. https://doi.org/10.3390/molecules30092045
Giannetti M, Gobbo M, Litti L, Caligiuri I, Rizzolio F, Meneghetti M, Mazzuca C, Palleschi A. Unraveling the Specific Recognition Between PD-L1 and Engineered CLP002 Functionalized Gold Nanostructures: MD Simulation Studies. Molecules. 2025; 30(9):2045. https://doi.org/10.3390/molecules30092045
Chicago/Turabian StyleGiannetti, Micaela, Marina Gobbo, Lucio Litti, Isabella Caligiuri, Flavio Rizzolio, Moreno Meneghetti, Claudia Mazzuca, and Antonio Palleschi. 2025. "Unraveling the Specific Recognition Between PD-L1 and Engineered CLP002 Functionalized Gold Nanostructures: MD Simulation Studies" Molecules 30, no. 9: 2045. https://doi.org/10.3390/molecules30092045
APA StyleGiannetti, M., Gobbo, M., Litti, L., Caligiuri, I., Rizzolio, F., Meneghetti, M., Mazzuca, C., & Palleschi, A. (2025). Unraveling the Specific Recognition Between PD-L1 and Engineered CLP002 Functionalized Gold Nanostructures: MD Simulation Studies. Molecules, 30(9), 2045. https://doi.org/10.3390/molecules30092045