
Stereoselectivity of In Vivo Processes and Bioactivity of Farrerol Enantiomers


Abstract
:1. Introduction
2. Results and Discussion
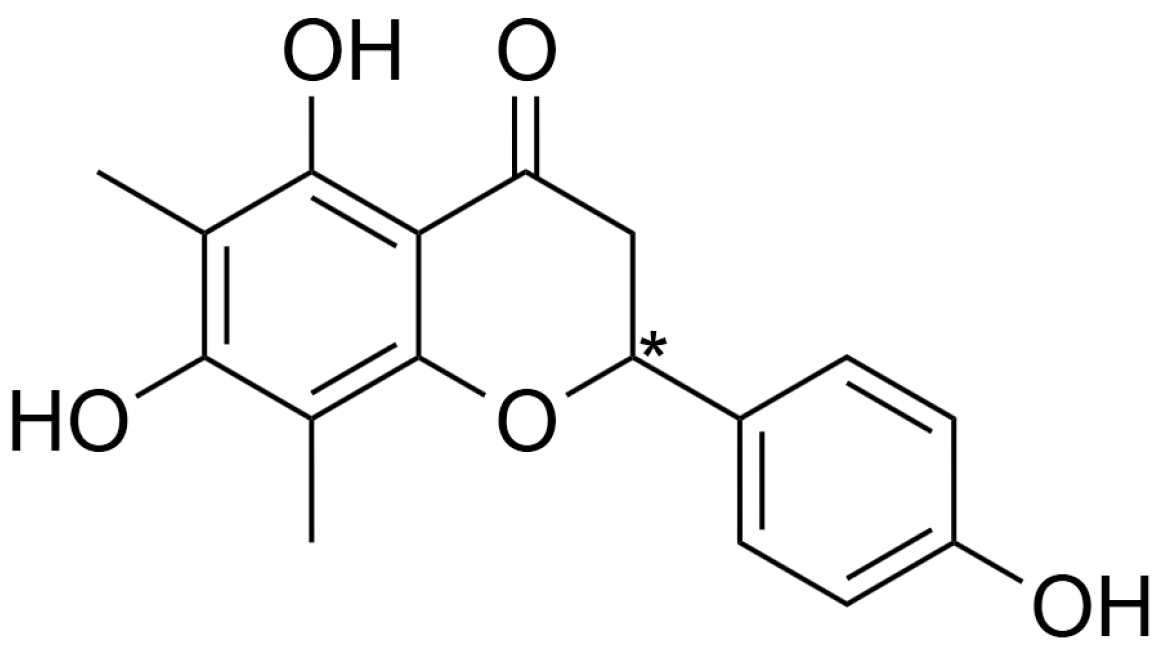
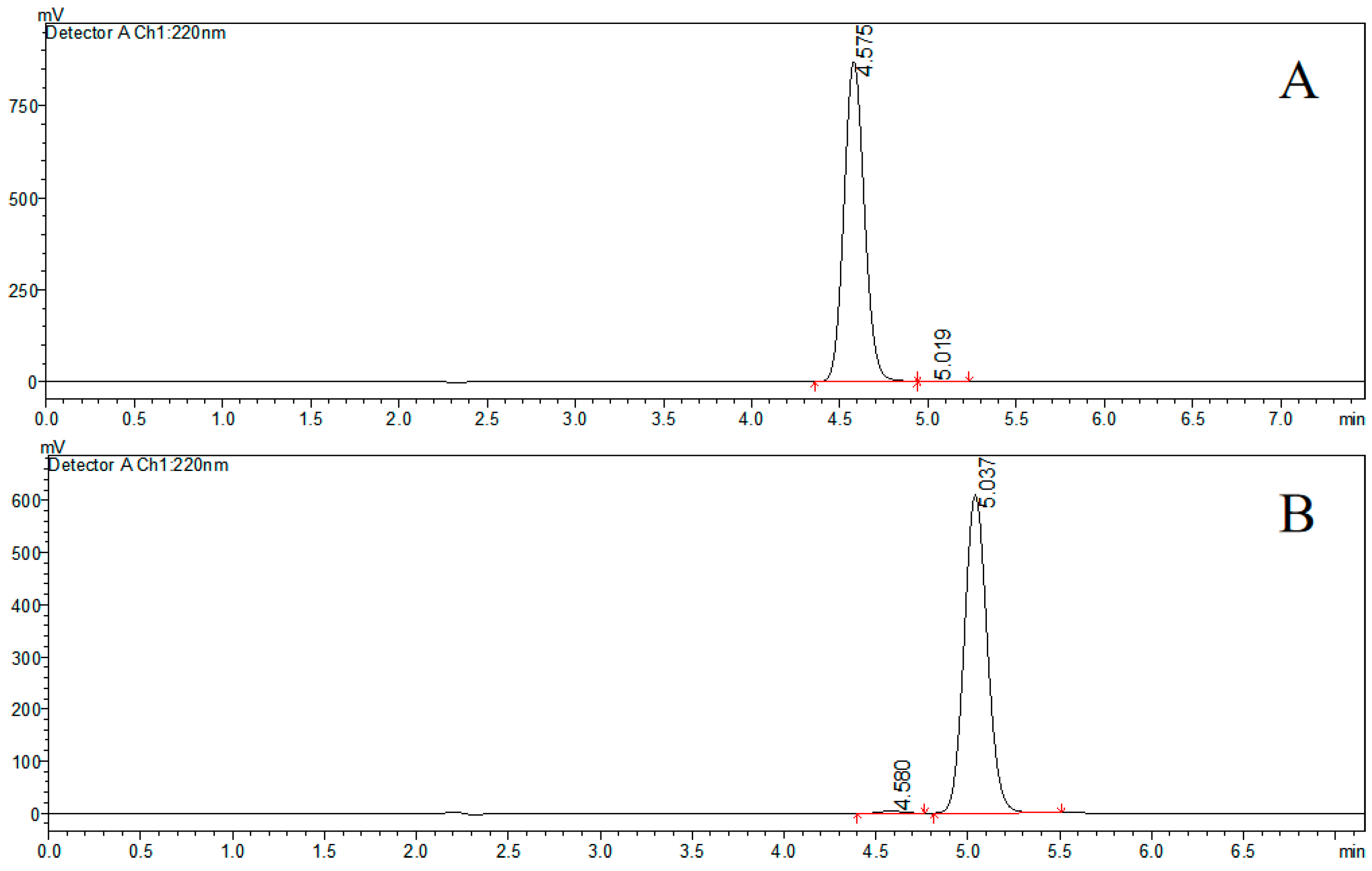
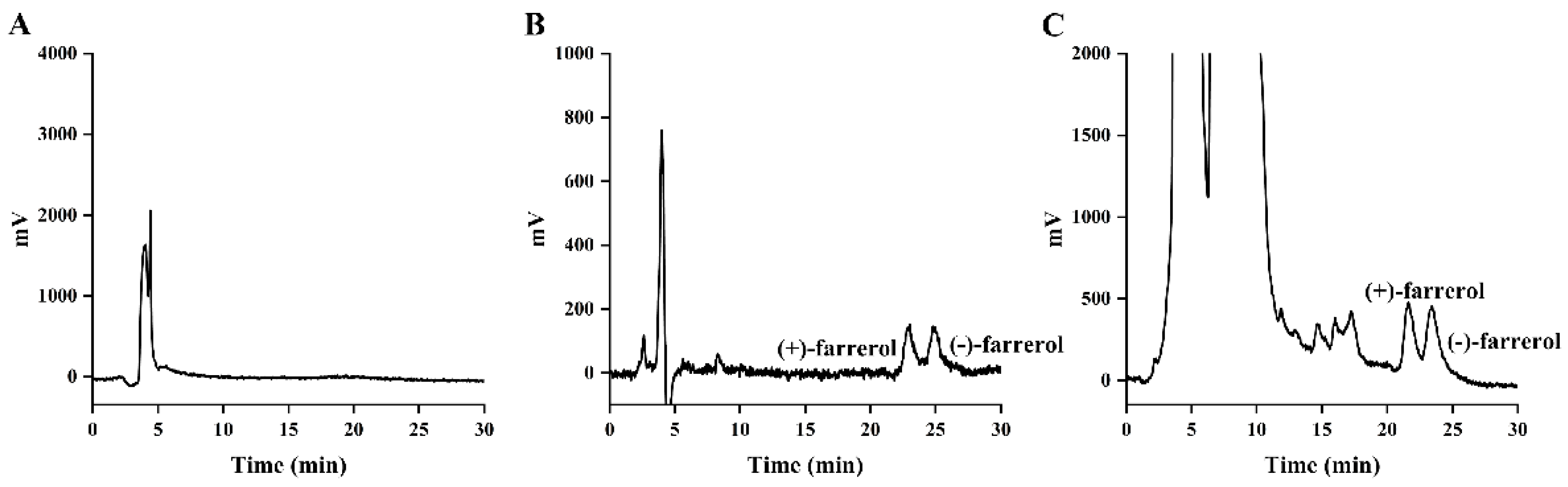
2.1. Preparation and Characterization of Farrerol Enantiomers
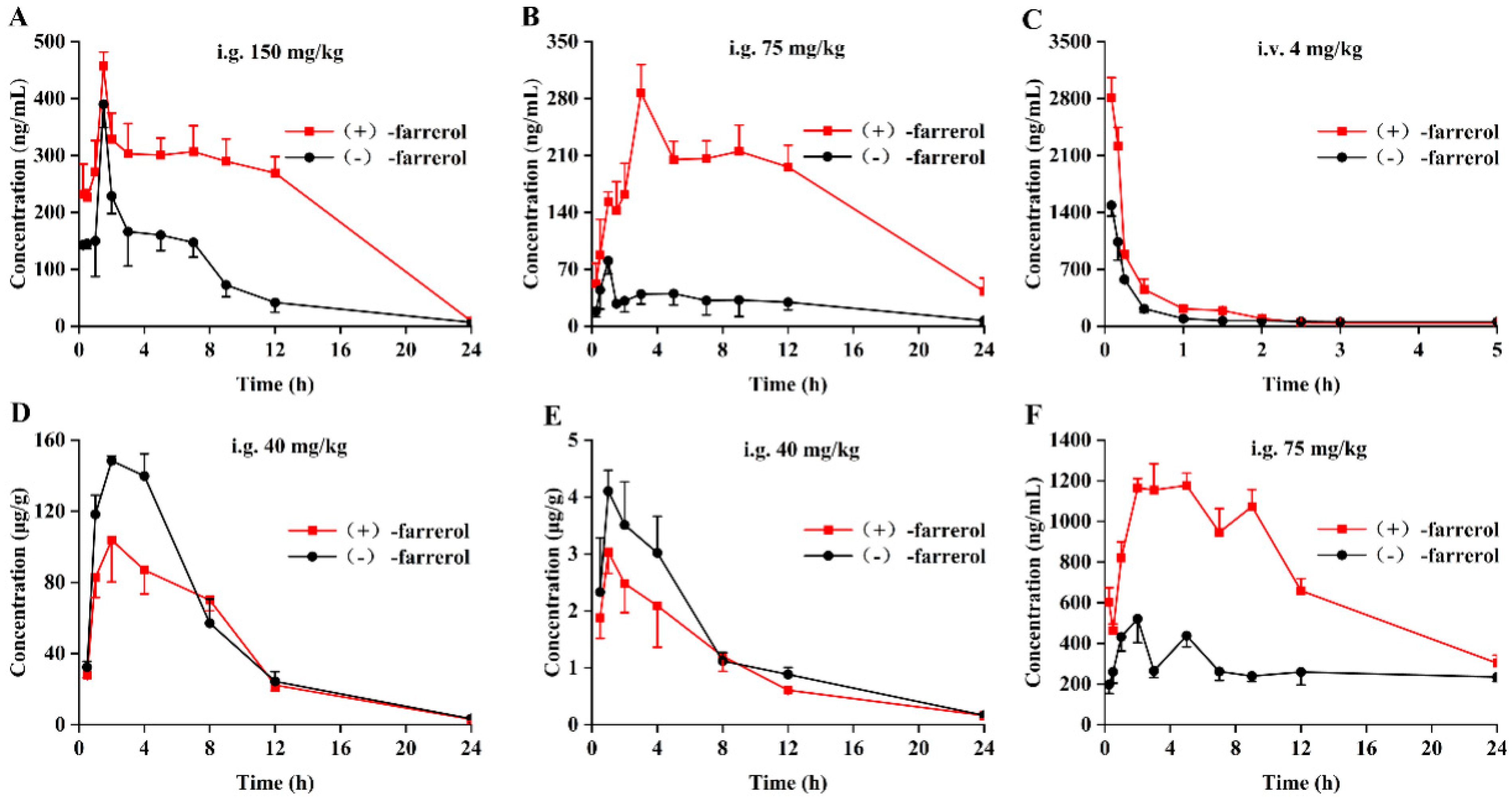
2.2. Stereoselective Pharmacokinetics Study in Rat Plasma
2.3. Stereoselective Pharmacokinetics Study in Rat Liver and Kidney
2.4. Stereoselective In Vitro Metabolism of Liver Microsomal
2.5. Stereoselectivity of Intestinal Absorption of Farrerol Enantiomers
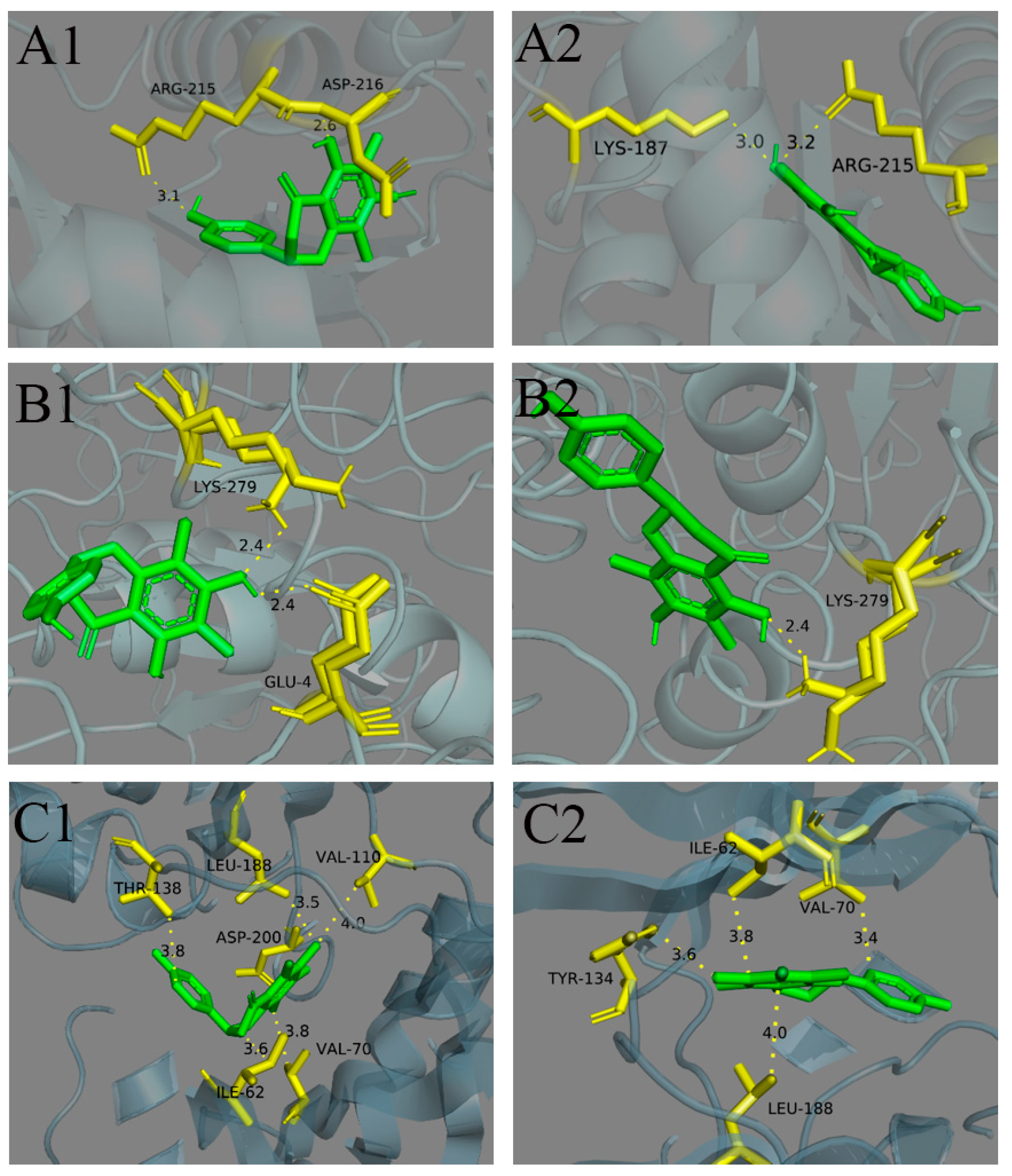
2.6. Stereoselective Binding of Farrerol Enantiomers to Antiproliferative Target
2.7. Stereoselective Growth Inhibition of HT-29 Cells by Farrerol Enantiomers
3. Materials and Methods
3.1. Chemicals and Reagents
3.2. Animals, Cells
3.3. Preparation and Characterization of Farrerol Enantiomers
3.4. Stereoselective Pharmacokinetics Studies
3.4.1. Pharmacokinetics Studies in Rat Plasma
3.4.2. Pharmacokinetics Studies in Rat Liver and Kidney Tissue
3.4.3. Chiral HPLC-MS/MS Conditions
3.4.4. Methods Validation
3.5. Stereoselective In Vitro Metabolism of Liver Microsomal
3.5.1. Liver Microsomal Preparation and Incubation
3.5.2. HPLC-MS/MS Conditions
3.6. Stereoselectivity of Intestinal Absorption
3.6.1. The Method of Rat Single-Pass Intestinal Perfusion
3.6.2. HPLC Method and Method Validation
3.7. Stereoselective Binding of Farrerol Enantiomers to UCHL3
3.8. Stereoselective Growth Inhibition of HT-29 Cells by Farrerol Enantiomers
3.9. Statistical Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Choi, S.E.; Park, K.H.; Han, B.H.; Jeong, M.S.; Seo, S.J.; Lee, D.I.; Joo, S.S.; Lee, M.W. Inhibition of Inducible Nitric Oxide Synthase and Cyclooxygenase-2 Expression by Phenolic Compounds from Roots of Rhododendron mucronulatum. Phytother. Res. 2011, 25, 1301–1305. [Google Scholar] [CrossRef]
- Ci, X.; Lv, H.; Wang, L.; Wang, X.; Peng, L.; Qin, F.X.-F.; Cheng, G. The antioxidative potential of farrerol occurs via the activation of Nrf2 mediated HO-1 signaling in RAW 264.7 cells. Chem.-Biol. Interact. 2015, 239, 192–199. [Google Scholar] [CrossRef]
- Fallon, P.G.; Ci, X.; Chu, X.; Wei, M.; Yang, X.; Cai, Q.; Deng, X. Different Effects of Farrerol on an OVA-Induced Allergic Asthma and LPS-induced Acute Lung Injury. PLoS ONE 2012, 7, e34634. [Google Scholar]
- Dai, F.; Gao, L.; Zhao, Y.; Wang, C.; Xie, S. Farrerol inhibited angiogenesis through Akt/mTOR, Erk and Jak2/Stat3 signal pathway. Phytomedicine 2016, 23, 686–693. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.-y.; Chen, L.; Zhu, Y.-h.; Zhang, M.; Wang, Y.-p.; Wang, M.-w. Involvement of estrogen receptor-β in farrerol inhibition of rat thoracic aorta vascular smooth muscle cell proliferation. Acta Pharmacol. Sin. 2011, 32, 433–440. [Google Scholar] [CrossRef] [PubMed]
- Li, J.-K.; Ge, R.; Tang, L.; Li, Q.-S. Protective effects of farrerol against hydrogen-peroxide-induced apoptosis in human endothelium-derived EA.hy926 cells. Can. J. Physiol. Pharmacol. 2013, 91, 733–740. [Google Scholar] [CrossRef]
- Volosov, A.; Yagen, B.; Bialer, M. Comparative Stereoselective Pharmacokinetic Analysis of 10-Hydroxycarbazepine After Oral Administration of its Individual Enantiomers and the Racemic Mixture to Dogs. Epilepsia 2000, 41, 1107–1111. [Google Scholar] [CrossRef]
- Ou, F.; Zhou, Y.; Lei, J.; Zeng, S.; Wu, F.; Zhang, N.; Yu, L. Development of a UHPLC-MS/MS method for the quantification of ilaprazole enantiomers in rat plasma and its pharmacokinetic application. J. Pharm. Anal. 2020, 10, 617–623. [Google Scholar] [CrossRef]
- Lun, J.; Zhang, W.; Zhao, Y.; Song, Y.; Guo, X. Enantiomeric Separation of Dioxopromethazine and its Stereoselective Pharmacokinetics in Rats by HPLC-MS/MS. J. Pharm. Sci. 2021, 110, 3082–3090. [Google Scholar] [CrossRef]
- Tekwani, B.L.; Avula, B.; Sahu, R.; Chaurasiya, N.D.; Khan, S.I.; Jain, S.; Fasinu, P.S.; Herath, H.M.T.B.; Stanford, D.; Nanayakkara, N.P.D.; et al. Enantioselective Pharmacokinetics of Primaquine in Healthy Human Volunteers. Drug Metab. Dispos. 2015, 43, 571–577. [Google Scholar] [CrossRef]
- Dubey, S.K.; Anand, A.; Saha, R.N. Enantioselective Tissue Distribution of Ketorolac and its Enantiomers in Rats. Drug Res. 2015, 65, 428–431. [Google Scholar] [CrossRef] [PubMed]
- Simões, R.A.; de Oliveira, A.R.M.; Bonato, P.S. Hollow fiber-based liquid-phase microextraction (HF-LPME) of isradipine and its main metabolite followed by chiral HPLC analysis: Application to an in vitro biotransformation study. Anal. Bioanal. Chem. 2011, 399, 2435–2443. [Google Scholar] [CrossRef] [PubMed]
- Saunders, D.; Vanachayangkul, P.; Imerbsin, R.; Khemawoot, P.; Siripokasupkul, R.; Tekwani, B.L.; Sampath, A.; Nanayakkara, N.P.D.; Ohrt, C.; Lanteri, C.; et al. Pharmacokinetics and Pharmacodynamics of (+)-Primaquine and (−)-Primaquine Enantiomers in Rhesus Macaques (Macaca mulatta). Antimicrob. Agents Chemother. 2014, 58, 7283–7291. [Google Scholar] [CrossRef]
- Zhang, X.; Gao, L.; Zhang, Z.; Tian, Y. Separation and determination of acetyl-glutamine enantiomers by HPLC–MS and its application in pharmacokinetic study. J. Pharm. Anal. 2017, 7, 303–308. [Google Scholar] [CrossRef] [PubMed]
- Cao, S.; Fu, Y.; Jiang, B.; Ge, X.; Wu, Q.; Tao, Y.; Zhao, P.; Wang, Z. Simultaneous stereoisomeric separation of loxoprofen sodium and its alcohol metabolites. Application to a stereoselective pharmacokinetic study. Arab. J. Chem. 2023, 16, 104942. [Google Scholar] [CrossRef]
- Ikuta, H.; Kawase, A.; Iwaki, M. Stereoselective Pharmacokinetics and Chiral Inversion of Ibuprofen in Adjuvant-induced Arthritic Rats. Drug Metab. Dispos. 2017, 45, 316–324. [Google Scholar] [CrossRef]
- Gao, L.B.; Wang, J.Z.; Yao, T.W.; Zeng, S. Study on the metabolic mechanism of chiral inversion of S-Mandelic acid in vitro. Chirality 2012, 24, 86–95. [Google Scholar] [CrossRef]
- Yu, L.; Pu, J.; Zuo, M.; Zhang, X.; Cao, Y.; Chen, S.; Lou, Y.; Zhou, Q.; Hu, H.; Jiang, H.; et al. Hepatic Glucuronidation of Isoneochamaejasmin A from the Traditional Chinese Medicine Stellera Chamaejasme L. Root. Drug Metab. Dispos. 2014, 42, 735–743. [Google Scholar] [CrossRef]
- Yang, Z.-z.; Li, L.; Wang, L.; Yuan, L.-m.; Xu, M.-c.; Gu, J.-k.; Jiang, H.-d.; Yu, L.-s.; Zeng, S. The regioselective glucuronidation of morphine by dimerized human UGT2B7, 1A1, 1A9 and their allelic variants. Acta Pharmacol. Sin. 2017, 38, 1184–1194. [Google Scholar] [CrossRef]
- Shen, Z.; Lv, C.; Zeng, S. Significance and challenges of stereoselectivity assessing methods in drug metabolism. J. Pharm. Anal. 2016, 6, 1–10. [Google Scholar] [CrossRef]
- Shen, Q.; Wang, L.; Zhou, H.; Jiang, H.-d.; Yu, L.-s.; Zeng, S. Stereoselective binding of chiral drugs to plasma proteins. Acta Pharmacol. Sin. 2013, 34, 998–1006. [Google Scholar] [CrossRef] [PubMed]
- Yan, T.; Lou, L.; Zhang, S.; Lishuang, Y.U.; Liu, Y. Separation of Farrerol Enantiomers and Its Content Determination in Chinese Materia Medica. Chin. J. Mod. Appl. Pharm. 2024, 41, 80–87. [Google Scholar]
- Wang, L.; Wei, W.; Xiao, Q.; Yang, H.; Ci, X. Farrerol Ameliorates APAP-induced Hepatotoxicity via Activation of Nrf2 and Autophagy. Int. J. Biol. Sci. 2019, 15, 788–799. [Google Scholar] [CrossRef] [PubMed]
- Zhang, W.; Wang, M.; Song, Z.; Fu, Q.; Chen, J.; Zhang, W.; Gao, S.; Sun, X.; Yang, G.; Zhang, Q.; et al. Farrerol directly activates the deubiqutinase UCHL3 to promote DNA repair and reprogramming when mediated by somatic cell nuclear transfer. Nat. Commun. 2023, 14, 1838. [Google Scholar] [CrossRef] [PubMed]
- Yan, C.; Zhang, X.; Miao, J.; Yuan, H.; Liu, E.; Liang, T.; Li, Q. Farrerol Directly Targets GSK-3β to Activate Nrf2-ARE Pathway and Protect EA.hy926 Cells against Oxidative Stress-Induced Injuries. Oxidative Med. Cell. Longev. 2020, 2020, 5967434. [Google Scholar] [CrossRef]
- Zhang, T.; Liu, H.; Liu, M.; Wang, C. Farrerol suppresses the progression of laryngeal squamous cell carcinoma via the mitochondria-mediated pathway. Eur. J. Pharmacol. 2021, 913, 174636. [Google Scholar] [CrossRef]
- Xiaojiang, Q.; Xiaomin, H.; Yiwei, S.; Jianke, Z.; Lin, S.; Xu, Z. The mechanism of farrerol improving middle cerebral arterial injury induced by nicotine in C57BL /6J mice through down-regulating Cx43. J. Toxicol. 2022, 36, 208–212. [Google Scholar]
- Shuangqiu, L.; Di, W.; Aimin, J.; Ziyi, L.; Yong, Z.; Guanghong, X. Protective effect of farrerol on liver injury induced by LPS/GalN in chicks. Chin. J. Vet. Sci. 2022, 42, 763–769. [Google Scholar]
- Stielow, M.; Witczyńska, A.; Kubryń, N.; Fijałkowski, Ł.; Nowaczyk, J.; Nowaczyk, A. The Bioavailability of Drugs—The Current State of Knowledge. Molecules 2023, 28, 8038. [Google Scholar] [CrossRef]
- Gao, J.; Cang, X.; Liu, L.; Lin, J.; Zhu, S.; Liu, L.; Liu, X.; Zhu, J.; Xu, C. Farrerol alleviates insulin resistance and hepatic steatosis of metabolic associated fatty liver disease by targeting PTPN1. J. Cell. Mol. Med. 2024, 28, e70096. [Google Scholar] [CrossRef]
- López-Yerena, A.; Vallverdú-Queralt, A.; Mols, R.; Augustijns, P.; Lamuela-Raventós, R.M.; Escribano-Ferrer, E. Absorption and Intestinal Metabolic Profile of Oleocanthal in Rats. Pharmaceutics 2020, 12, 134. [Google Scholar] [CrossRef] [PubMed]
- Gudjonsson, H.; Li, B.U.K.; Shug, A.L.; Olsen, W.A. In vivo studies of intestinal carnitine absorption in rats. Gastroenterology 1985, 88, 1880–1887. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | ee | [α]2⁰D (MeOH) |
|---|---|---|
| (+)-farrerol | 98% | +25.93° |
| (−)-farrerol | 98% | −26.13° |
| Cmax (ng/mL) | Tmax (h) | AUC(0–t) (ng/mL*h) | AUC(0–∞) (ng/mL*h) | MRT(0–t) (h) | t1/2 | F (%) | ||
|---|---|---|---|---|---|---|---|---|
| 1 | (+)-farrerol | 2662.7 226.4 | 0.08 | 1464.2 54.5 | 1552.9 56.0 | 2.70 0.10 | 10.7 0.10 | / |
| (−)-farrerol | 1695.0 224.6 ** | 0.08 | 1012.4 88.1 ** | 1195.7 49.7 ** | 3.48 0.07 * | 19.0 3.7 * | / | |
| 2 | (+)-farrerol | 457.2 24.6 | 1.50 | 3522.6 146.0 | 13,885.5 2550.4 | 5.98 0.14 | 22.9 2.6 | 6.4 0.04 |
| (−)-farrerol | 390.0 35.3 * | 1.50 | 1622.6 84.3 ** | 1837.8 226.9 ** | 4.65 0.06 | 3.5 0.4 ** | 4.3 0.7 * | |
| 3 | (+)-farrerol | 287.0 24.6 | 3.00 | 2399.1 102.3 | 6870.3 511.4 | 6.27 0.27 | 19.2 2.0 ** | 8.7 0.7 |
| (−)-farrerol | 80.5 7.5 ** | 1.00 * | 428.3 88.2 ** | 1341.8 337.6 ** | 5.61 0.56 | 8.5 1.5 ** | 2.3 0.05 ** | |
| 4 | (+)-farrerol | 1177.4 84.5 | 5.00 | 17,593.5 1273.3 | 22,122.4 1894.1 | 9.12 0.18 | 10.4 0.4 | / |
| 5 | (−)-farrerol | 520.9 91.7 # | 2.00 # | 6746.8 298.7 # | 8572.4 221.0 # | 10.84 0.47 | 12.0 0.5 | / |
| Liver | Kidney | |||
|---|---|---|---|---|
| (+)-Farrerol | (−)-farrerol | (+)-Farrerol | (−)-farrerol | |
| Cmax (µg/g) | 103.8 ± 18.05 | 148.4 ± 2.61 * | 3.02 ± 0.35 | 4.18 ± 0.48 * |
| Tmax (h) | 2.00 | 2.00 | 1.00 | 1.00 |
| AUC(0–t) (µg/g*h) | 976.25 ± 41.60 | 1200.96 ± 58.92 * | 23.93 ± 2.11 | 28.89 ± 2.53 * |
| AUC(0–∞) (µg/g*h) | 994.01 ± 40.84 | 1217.62 ± 65.03 * | 25.70 ± 2.09 | 30.31 ± 2.47 * |
| CYP Subtype | Marker Substrate | Positive Inhibitor | (+)-Farrerol | (−)-farrerol | |
|---|---|---|---|---|---|
| IC50/μmol·L−1 | IC50/μmol·L−1 | IC50/μmol·L−1 | |||
| 1A2 | phenacetin | β-naphthoflavone | 0.129 | 0.588 | 1.02 ** |
| 2C9 | diclofenac | sulfaphenazole | 0.219 | 1.97 | 1.57 * |
| 2C19 | S-mephenytoin | Benzenifanol | 0.106 | 1.61 | 2.07 * |
| 2D6 | dextromethorphan | quinindium | 0.0744 | >50 | >50 |
| 3A4/5(M) | midazolam | ketoconazole | 0.0104 | >50 | 20.9 ** |
| 2A6 | coumarin | Tranylcypromine | 0.383 | >50 | >50 |
| 2E1 | chlorzoxazone | 4-methylpyrazole | 1.46 | >50 | >50 |
| Concentration of Farrerol Racemate/μg·mL−1 | Peff (×10−1 cm·min−1) | Ka (×10−1 min) | |||
|---|---|---|---|---|---|
| (+)-Farrerol | (−)-farrerol | (+)-Farrerol | (−)-farrerol | ||
| Duodenum | 66.00 | 1.32 ± 0.03 | 1.33 ± 0.03 | 1.47 ± 0.02 | 1.47 ± 0.02 |
| 132.00 | 1.16 ± 0.11 | 1.17 ± 0.10 | 1.87 ± 0.43 | 1.87 ± 0.43 | |
| 180.00 | 1.30 ± 0.14 | 1.33 ± 0.11 | 2.04 ± 0.28 | 2.04 ± 0.28 | |
| 132.00+ verapamil | 1.42 ± 0.17 * | 1.43 ± 0.17 * | 1.79 ± 0.45 | 1.79 ± 0.45 | |
| Jejunum | 66.00 | 1.16 ± 0.23 | 1.17 ± 0.24 | 1.92 ± 0.10 | 1.92 ± 0.10 |
| 132.00 | 0.73 ± 0.03 | 0.74 ± 0.05 | 0.71 ± 0.17 | 0.71 ± 0.17 | |
| 180.00 | 1.03 ± 0.06 | 1.07 ± 0.10 | 1.14 ± 0.18 | 1.14 ± 0.18 | |
| 132.00+ verapamil | 1.12 ± 0.10 * | 1.12 ± 0.10 * | 1.16 ± 0.11 * | 1.16 ± 0.11 * | |
| Ileum | 66.00 | 1.12 ± 0.22 | 1.14 ± 0.22 | 2.05 ± 0.06 | 2.05 ± 0.06 |
| 132.00 | 0.87 ± 0.08 | 0.89 ± 0.10 | 0.84 ± 0.14 | 0.84 ± 0.14 | |
| 180.00 | 0.79 ± 0.04 | 0.78 ± 0.07 | 1.48 ± 0.39 | 1.48 ± 0.39 | |
| 132.00+ verapamil | 0.77 ± 0.11 | 0.77 ± 0.11 | 0.78 ± 0.28 | 0.78 ± 0.28 | |
| Colon | 66.00 | 1.09 ± 0.37 | 1.12 ± 0.41 | 1.73 ± 0.81 | 1.74 ± 0.81 |
| 132.00 | 0.93 ± 0.09 | 0.95 ± 0.10 | 0.83 ± 0.27 | 0.83 ± 0.27 | |
| 180.00 | 1.04 ± 0.12 | 1.12 ± 0.17 | 1.21 ± 0.16 | 1.21 ± 0.16 | |
| 132.00+ verapamil | 1.41 ± 0.23 * | 1.42 ± 0.23 * | 1.16 ± 0.36 * | 1.16 ± 0.36 * | |
| Antiproliferative Target | Receptor | Ligands | Residues Involved in Hydrogen Bond Formation | ΔG | |ΔΔG| |
|---|---|---|---|---|---|
| UCHL3 | 7yv4 | (+)-farrerol | ARG215, ASP216 | −3.63 | 7.57 |
| (−)-farrerol | LYS187, ARG215 | −11.20 | |||
| Stat3 | 1bg1 | (+)-farrerol | LEU706, ALA703 | −4.30 | 0.14 |
| (−)-farrerol | LEU706 | −4.44 | |||
| PTPN1 | 5qf5 | (+)-farrerol | GLU4, LYS279 | −18.57 | 1.30 |
| (−)-farrerol | LYS279 | −19.87 | |||
| GSK-3β | 4acc | (+)-farrerol | ASP200, GLN185 | −6.82 | 1.64 |
| (−)-farrerol | VAL135 | −8.46 | |||
| 4acd | (+)-farrerol | ILE62, VAL70, THR138, GLN185, LEU188 | −6.41 | 2.28 | |
| (−)-farrerol | ASP133, VAL135 | −8.69 | |||
| 4acg | (+)-farrerol | ILE62, VAL70, VAL110, THR138, LEU188 | −6.17 | 2.47 | |
| (−)-farrerol | ILE62, VAL70, TYP134, LEU188 | −8.64 | |||
| 4ach | (+)-farrerol | GLN185, ASP200 | −6.47 | 1.25 | |
| (−)-farrerol | ILE62, LEU132, TYP134, LEU188 | −7.72 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, L.; Yan, T.; Huang, D.; Xu, W.; Liu, Y.; Wang, X.; Li, H. Stereoselectivity of In Vivo Processes and Bioactivity of Farrerol Enantiomers. Molecules 2025, 30, 2038. https://doi.org/10.3390/molecules30092038
Chen L, Yan T, Huang D, Xu W, Liu Y, Wang X, Li H. Stereoselectivity of In Vivo Processes and Bioactivity of Farrerol Enantiomers. Molecules. 2025; 30(9):2038. https://doi.org/10.3390/molecules30092038
Chicago/Turabian StyleChen, Lirong, Tang Yan, Dongting Huang, Wei Xu, Yongjing Liu, Xiaoying Wang, and Hua Li. 2025. "Stereoselectivity of In Vivo Processes and Bioactivity of Farrerol Enantiomers" Molecules 30, no. 9: 2038. https://doi.org/10.3390/molecules30092038
APA StyleChen, L., Yan, T., Huang, D., Xu, W., Liu, Y., Wang, X., & Li, H. (2025). Stereoselectivity of In Vivo Processes and Bioactivity of Farrerol Enantiomers. Molecules, 30(9), 2038. https://doi.org/10.3390/molecules30092038