
Novel Crown Ether-Functionalized Fusidic Acid Butyl Ester: Synthesis, Biological Evaluation, In Silico ADMET, and Molecular Docking Studies

Abstract
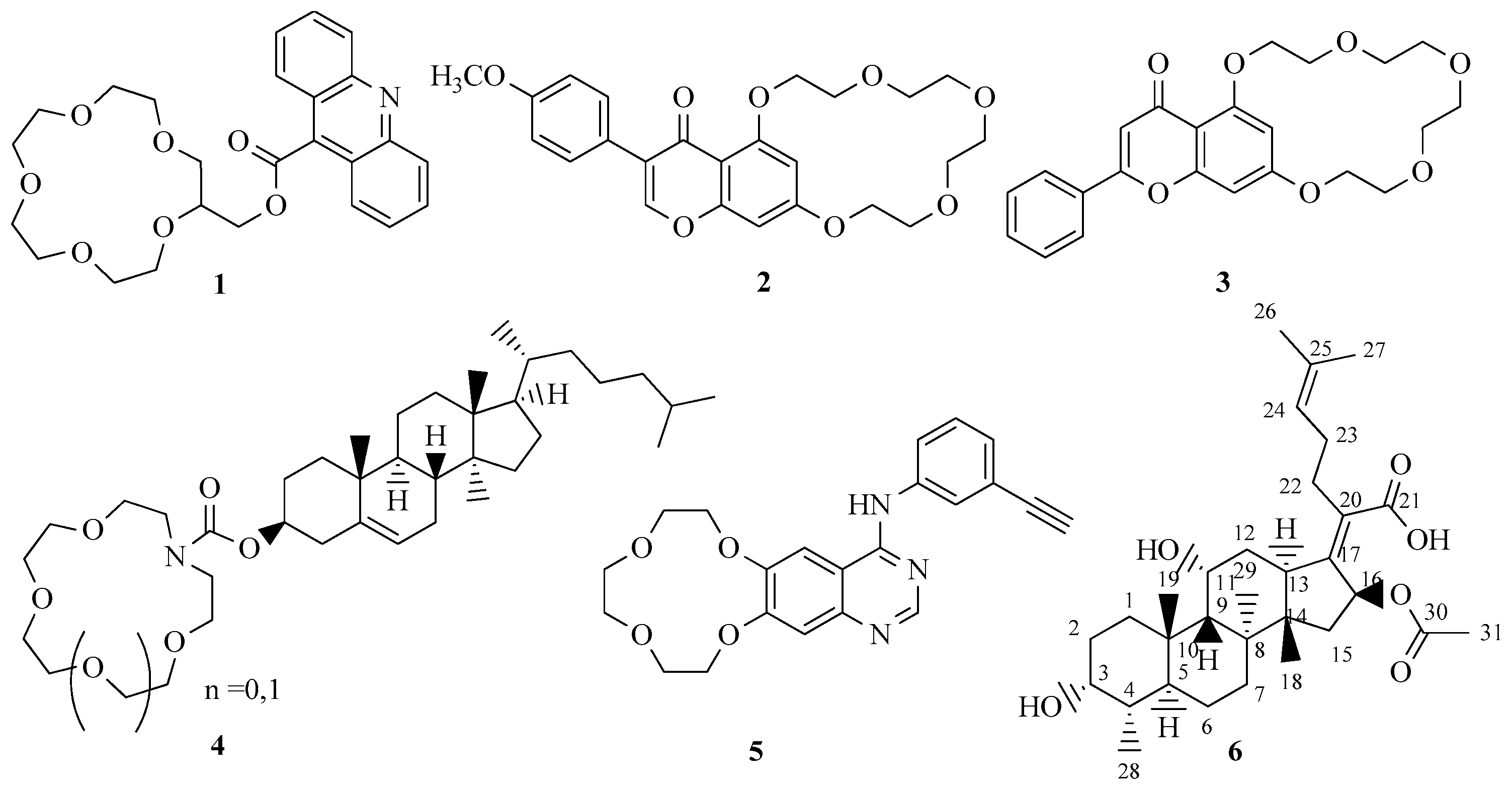
1. Introduction
2. Results and Discussion
2.1. Chemistry
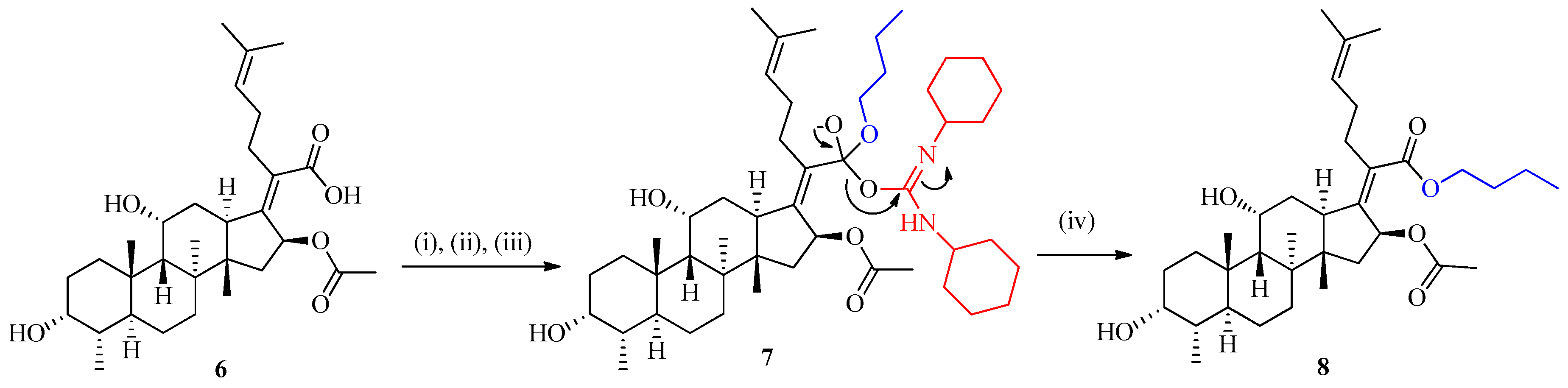
2.1.1. Preparation of Fusidic Acid C-21 Butyl Ester (FABE) 8
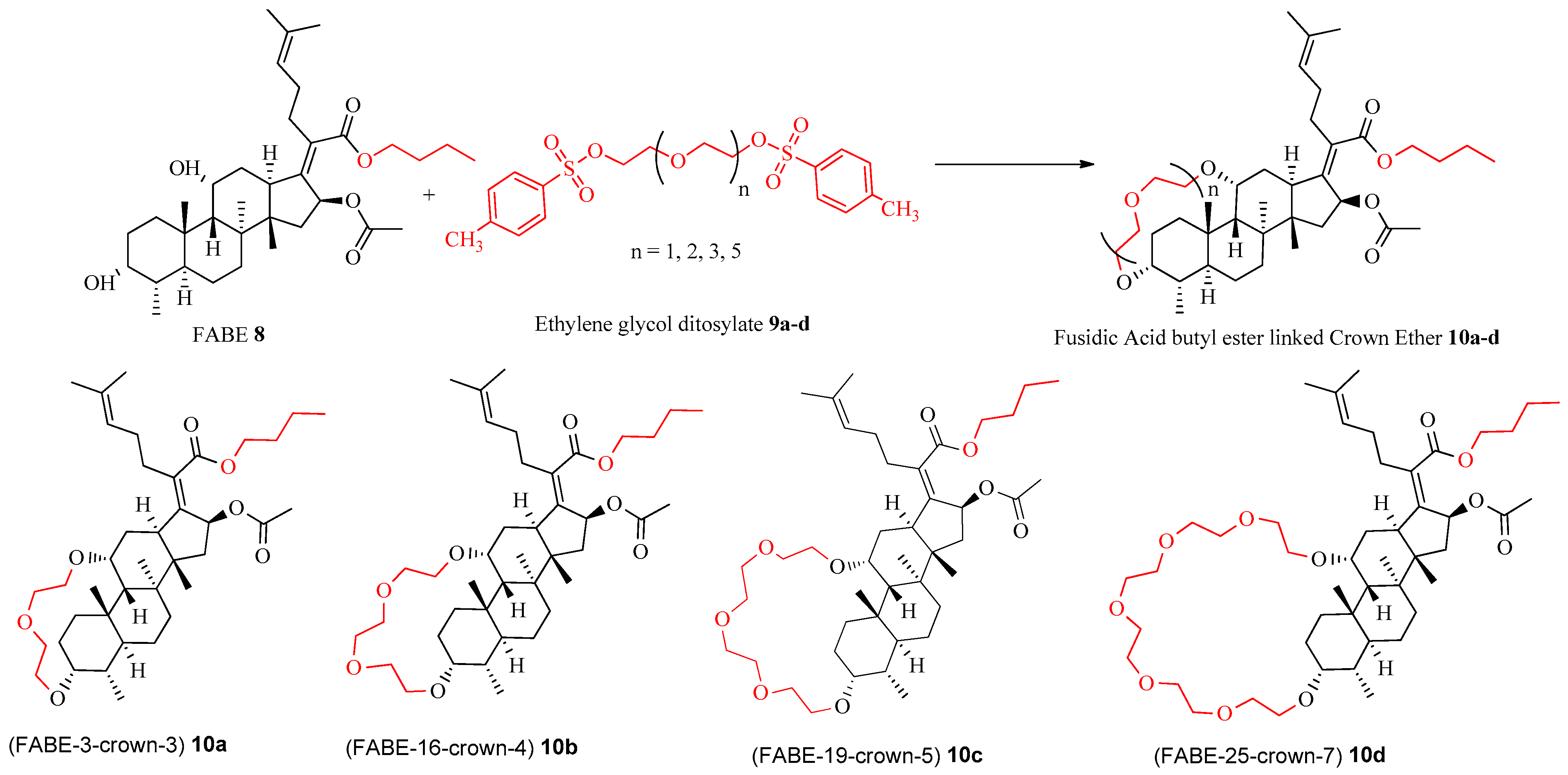
2.1.2. Preparation of Fusidic Acid C-21 Butyl Ester-Linked Crown Ethers 10a–d
2.1.3. An Alternate Approach for the Synthesis of Fusidic Acid Butyl Ester-Linked Crown Ether 10a–d
2.2. Spectral Characterization
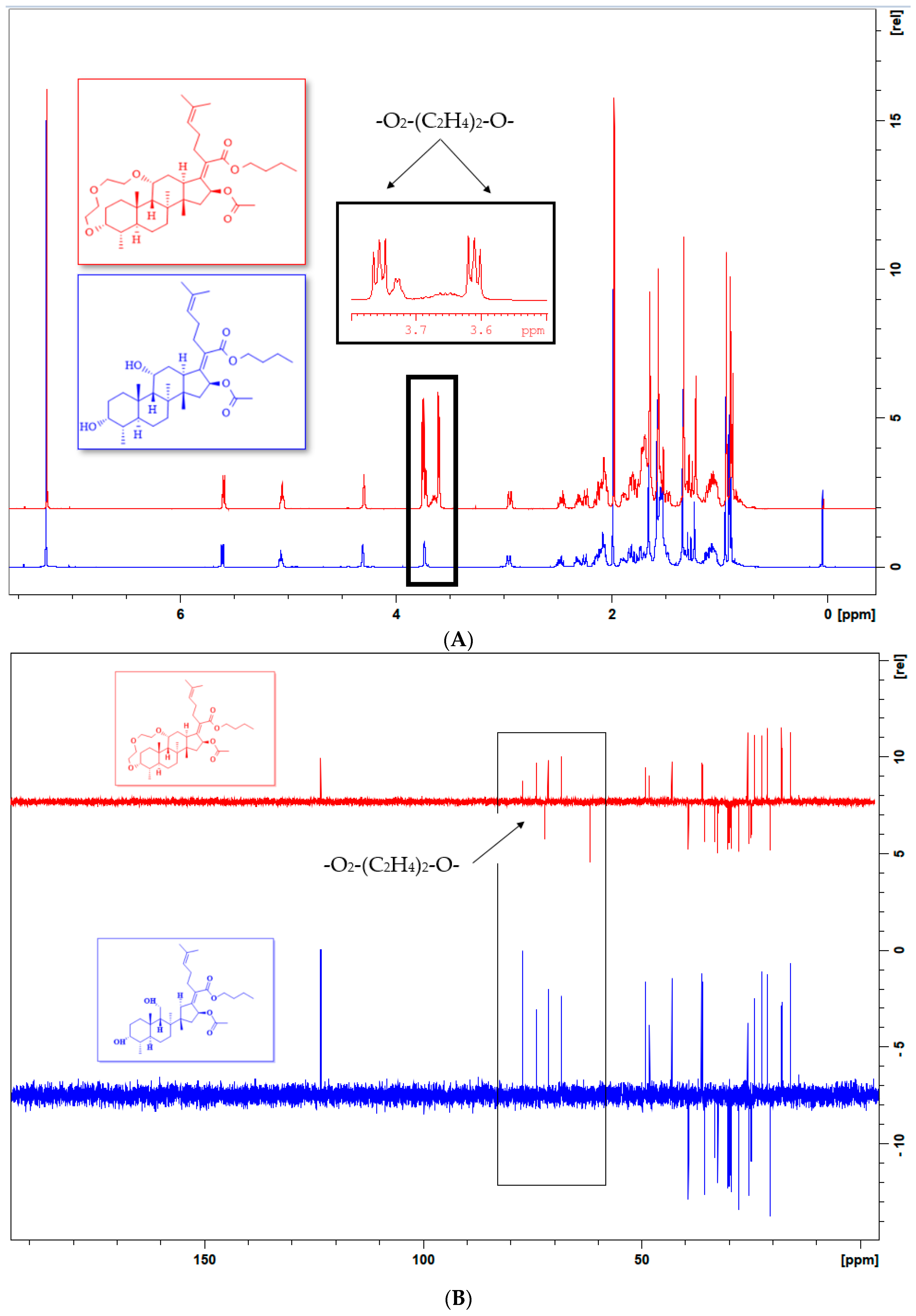
2.2.1. 1HNMR Spectral Analysis
2.2.2. 13C NMR Spectral Analysis
2.2.3. FT-IR Spectral Analysis
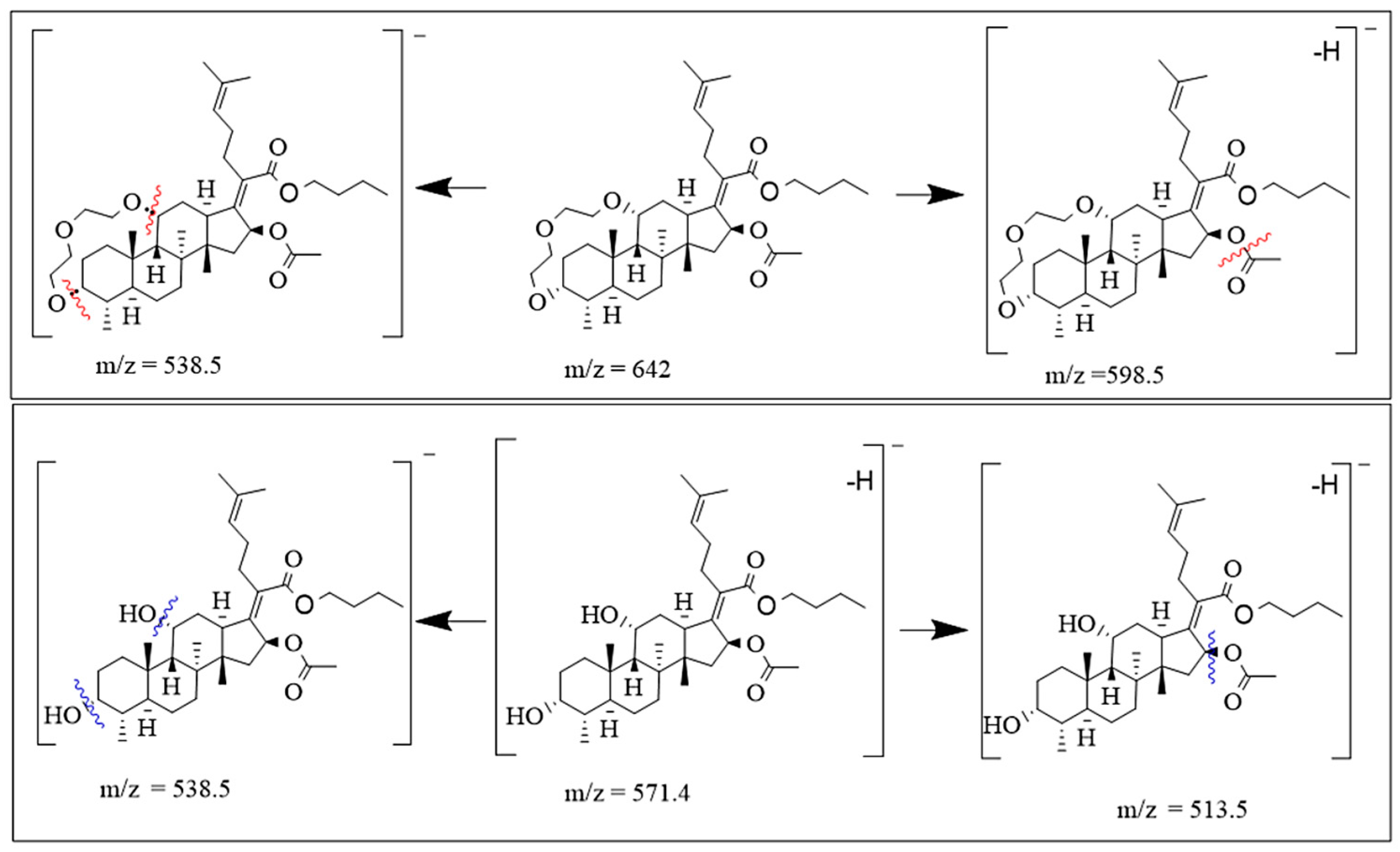
2.2.4. ESIMS Analysis
2.3. Biological Studies
2.3.1. Antioxidant Activity Investigations
2.3.2. α-Glucosidase Inhibition Investigations
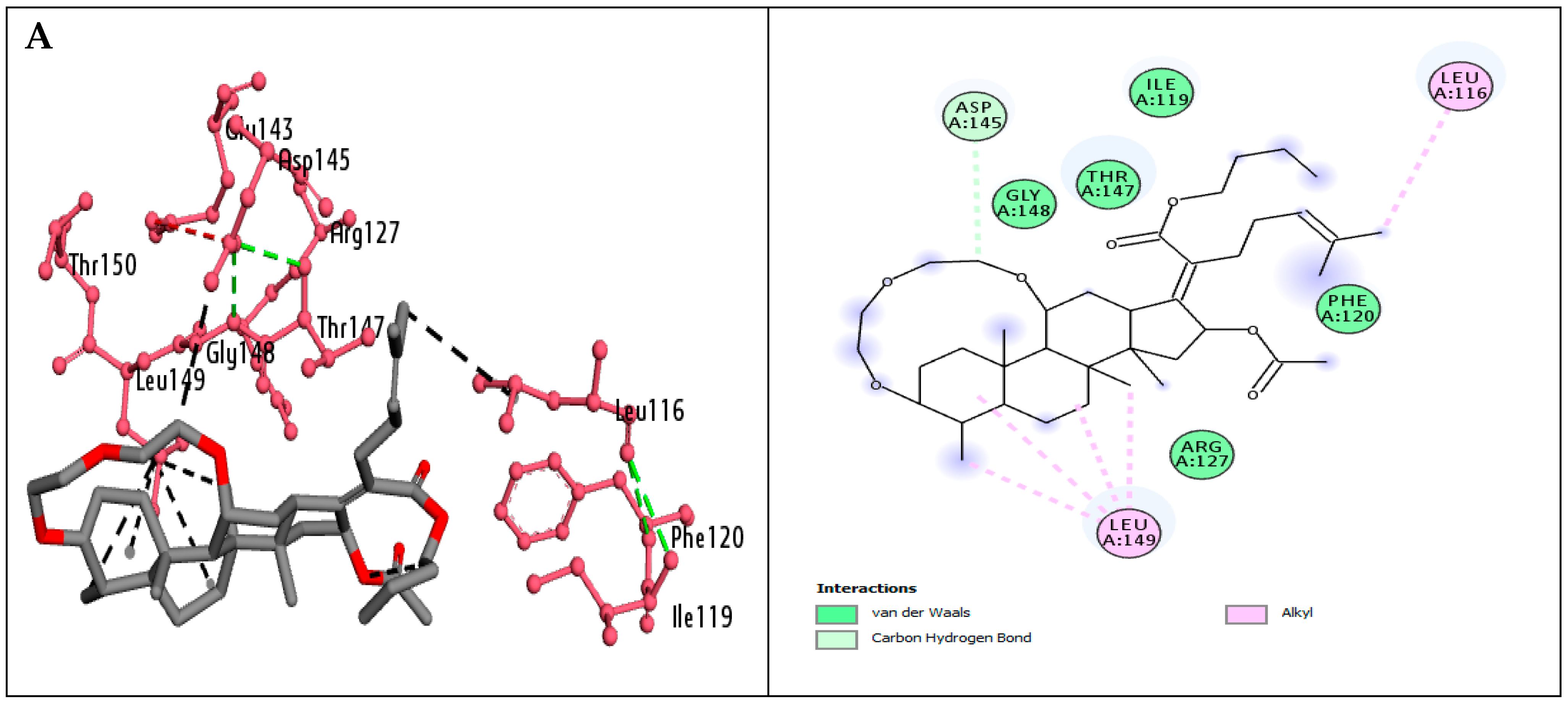
2.4. Molecular Docking Investigations
2.5. ADME-T Result Interpretation
3. Materials and Methods
3.1. General
3.2. Synthetic Approach
3.2.1. Synthetic Procedure for the Synthesis of Fusidic Acid Butyl Ester 8
Butyl(2Z)-2-[(4a,8a,14b,16b)-16-(acetyloxy)-3,11-dihydroxy-4,8,10,14-tetramethylgonan-17-ylidene]-6-methylhept-5-enoate (8)
3.2.2. Synthetic Procedure for the Synthesis of Fusidic Acid Butyl Ester-Linked Crown Ethers
Butyl(2Z)-2-[(3aS,16S,17aS,17bS,18S)-16-(acetyloxy)-3a,17a,17b,18-tetramethyloctahydro-3,6 methanocyclopenta[3,4]naptho[1,8hi][1,4,7]trioxyacylotridecin-15(1H)-ylidene]-6-methylhept-5-enoate (FABE-13-crown 3) 10a
Butyl(2Z)-2-[(3aS,19S,20aS,20bS,21S)-19-(acetyloxy)-3a,20a,20b,21-tetramethylicosahydro-3,6-methanocyclopenta[3,4]naphtho[1,8-kl][1,4,7,10]tetraoxacyclohexadecin-18(1H)-ylidene]-6-methylhept-5-enoate (FABE-16-crown-4) 10b
Butyl(2Z)-2-[(3aS,22S,23aS,23bS,24S)-22-(acetyloxy)-3a,23a,23b,24-tetramethyldocosahydro-3,6-methanocyclopenta[3,4]naphtho[1,8-no][1,4,7,10,13]pentaoxacyclononadecin-21(1H)-ylidene]-6- methylhept-5-enoate (FABE 19-crown-5) 10c
Butyl(2Z)-2-[(3aS,27S,28aS,28bS,29S)-27-(acetyloxy)-3a,28a,28b,29-tetramethyltetracosahydro-3,6-methanocyclopenta[3,4] naphtho[1,8-st][1,3,6,9,12,15,18]heptaoxacyclotetracosin-26(1H)-ylidene]-6- methylhept-5-enoate (FABE-25-crown-7) 10d
3.3. Biological Evaluation
3.3.1. Antioxidant Assay
3.3.2. α-Glucosidase Inhibition Assay
3.4. Methodology of Molecular Docking
3.5. In Silico ADMET Investigations
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| FABE | Fusidic acid butyl ester |
| BHA | Butyl hydroxyanisole |
| ADMET | Absorption, Digestion, Metabolism, Excretion, and Toxicity |
| DPPH | 2,2-diphenyl-1-picrylhydrazyl |
References
- Pham, H.T.; Julian, R.R. Mass Shifting and Radical Delivery with Crown Ether Attachment for Separation and Analysis of Phosphatidylethanolamine Lipids. Anal. Chem. 2014, 86, 3020–3027. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Calle, S.; Miller, A.J.M. Tunable and Switchable Catalysis Enabled by Cation-Controlled Gating with Crown Ether Ligands. Acc. Chem. Res. 2023, 56, 971–981. [Google Scholar] [CrossRef]
- Berkecz, R.; Németi, G.; Péter, A.; Ilisz, I. Liquid Chromatographic Enantioseparations Utilizing Chiral Stationary Phases Based on Crown Ethers and Cyclofructans. Molecules 2021, 26, 4648. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Awate, S.S.; Fullerton-Shirey, S.K. Bistable Electrical Switching Using a Crown Ether-Based Monolayer Electrolyte on WSe2 Field-Effect Transistors with Various Salts. ACS Appl. Eng. Mater. 2025, 3, 494–501. [Google Scholar] [CrossRef]
- Fong, C.W. Physiology of Ionophore Transport of Potassium and Sodium Ions across Cell Membranes: Valinomycin and 18-Crown-6 Ether. Int. J. Comput. Biol. Drug Des. 2016, 9, 228. [Google Scholar] [CrossRef]
- Ullah, F.; Khan, T.A.; Iltaf, J.; Anwar, S.; Khan, M.F.A.; Khan, M.R.; Ullah, S.; Rehman, M.F.U.; Mustaqeem, M.; Kotwica-Mojzych, K.; et al. Heterocyclic Crown Ethers with Potential Biological and Pharmacological Properties: From Synthesis to Applications. Appl. Sci. 2022, 12, 1102. [Google Scholar] [CrossRef]
- Chehardoli, G.; Bahmani, A. The Role of Crown Ethers in Drug Delivery. Supramol. Chem. 2019, 31, 221–238. [Google Scholar] [CrossRef]
- Ahmad, M.S.; Hawaiz, F.E. Novel Chalcone-Based Crown Ethers: Synthesis, Characterization, Antioxidant Activity, Biological Evaluations, and Wastewater Remediation. RSC Adv. 2024, 14, 2369–2379. [Google Scholar] [CrossRef]
- Hamzah, A.E.; Mahdi, I.K.; Radhi, A.J.; Mohsin, D.H.; Mohammed, L.J. Synthesis, Characterization of Thia-Crown Ether Sugar Derivatives and Evaluation of Their Anticancer Activity with Computational Insight. J. Wildl. Biodivers. 2023, 7, 526–546. [Google Scholar] [CrossRef]
- Chávez-Riveros, A.; Hernández-Vázquez, E.; Nieto-Camacho, A.; Ramírez-Apan, T.; Miranda, L.D. Synthesis of Diphenylamine Macrocycles and Their Anti-Inflammatory Effects. Org. Biomol. Chem. 2019, 17, 1423–1435. [Google Scholar] [CrossRef]
- Nhung, D.T.; Tram, N.N.; Tam, P.T.T.; Alekseeva, K.A.; Khrustalev, V.N.; Hoang, D.T.; Duan, L.T.; Anh, L.T. Synthesis, α-Glucosidase Inhibitory Activity, and Molecular Docking Studies of Tri(Benzo)-5-Azacrownophanes with a γ-Piperidone Moiety. ChemistrySelect 2023, 8, e202300860. [Google Scholar] [CrossRef]
- Wang, Q.; Huang, W.; Sun, Q.; Le, M.; Cai, L.; Jia, Y.-G. Facially Amphiphilic Skeleton-Derived Antibacterial Crown Ether/Silver Ion Complexes. Soft Matter 2025, 21, 2152–2159. [Google Scholar] [CrossRef]
- Surur, A.S.; Sun, D. Macrocycle-Antibiotic Hybrids: A Path to Clinical Candidates. Front. Chem. 2021, 9, 659845. [Google Scholar] [CrossRef]
- Majhi, S.; Das, D. Chemical Derivatization of Natural Products: Semisynthesis and Pharmacological Aspects- A Decade Update. Tetrahedron 2021, 78, 131801. [Google Scholar] [CrossRef]
- Maier, M.E. Design and Synthesis of Analogues of Natural Products. Org. Biomol. Chem. 2015, 13, 5302–5343. [Google Scholar] [CrossRef] [PubMed]
- Kralj, M.; Tušek-Božić, L.; Frkanec, L. Biomedical Potentials of Crown Ethers: Prospective Antitumor Agents. ChemMedChem 2008, 3, 1478–1492. [Google Scholar] [CrossRef] [PubMed]
- Sabah, K.; Heidelberg, T.; Hashim, R. Novel Crown Ethers on Glucose Based Glycolipids. Carbohydr. Res. 2011, 346, 891–896. [Google Scholar] [CrossRef] [PubMed]
- Golcs, Á.; Vezse, P.; Ádám, B.Á.; Huszthy, P.; Tóth, T. Comparison in Practical Applications of Crown Ether Sensor Molecules Containing an Acridone or an Acridine Unit—A Study on Protonation and Complex Formation. J. Incl. Phenom. Macrocycl. Chem. 2021, 101, 63–75. [Google Scholar] [CrossRef]
- Iqbal, T.; Arshad, N.; Hashim, J.; Ali, S.A.; Zehra, B.; Ahmad, M.S.; Hassan, N.; Ullah, A.; Hamid, S.Z.; Isaac, I.O. Natural Products Based Crown Ethers: Synthesis and Their Anticancer Potential. J. Asian Nat. Prod. Res. 2022, 24, 268–277. [Google Scholar] [CrossRef]
- Arshad, N.; Hameed, A.; Iqbal, T. Natural Products Embedded Crown Ethers as Potent Insulin Secretory Agents. Artic. Pak. J. Pharm. Sci. 2021, 34, 2003–2008. [Google Scholar] [CrossRef]
- Sewbalas, A.; Islam, R.U.; Van Otterlo, W.A.L.; De Koning, C.B.; Singh, M.; Arbuthnot, P.; Ariatti, M. Enhancement of Transfection Activity in HEK293 Cells by Lipoplexes Containing Cholesteryl Nitrogen-Pivoted Aza-Crown Ethers. Med. Chem. Res. 2013, 22, 2561–2569. [Google Scholar] [CrossRef]
- Hu, S.; Xie, G.; Zhang, D.X.; Davis, C.; Long, W.; Hu, Y.; Wang, F.; Kang, X.; Tan, F.; Ding, L.; et al. Synthesis and Biological Evaluation of Crown Ether Fused Quinazoline Analogues as Potent EGFR Inhibitors. Bioorg Med. Chem. Lett. 2012, 22, 6301–6305. [Google Scholar] [CrossRef]
- Long, J.; Ji, W.; Zhang, D.; Zhu, Y.; Bi, Y. Bioactivities and Structure–Activity Relationships of Fusidic Acid Derivatives: A Review. Front. Pharmacol. 2021, 12, 759220. [Google Scholar] [CrossRef] [PubMed]
- Curbete, M.M.; Salgado, H.R.N. A Critical Review of the Properties of Fusidic Acid and Analytical Methods for Its Determination. Crit. Rev. Anal. Chem. 2016, 46, 352–360. [Google Scholar] [CrossRef]
- Jackson, W.B.; Low, D.E.; Dattani, D.; Whitsitt, P.F.; Leeder, R.G.; Macdougall, R. Treatment of Acute Bacterial Conjunctivitis: 1% Fusidic Acid Viscous Drops vs. 0.3% Tobramycin Drops. Can. J. Ophthalmol. 2002, 37, 228–237. [Google Scholar] [CrossRef]
- Klein, S.; Nurjadi, D.; Eigenbrod, T.; Bode, K.A. Evaluation of Antibiotic Resistance to Orally Administrable Antibiotics in Staphylococcal Bone and Joint Infections in One of the Largest University Hospitals in Germany: Is There a Role for Fusidic Acid? Int. J. Antimicrob. Agents 2016, 47, 155–157. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.L.; Tang, H.J.; Hsieh, P.H.; Chiu, F.Y.; Chen, Y.H.; Chang, M.C.; Huang, C.T.; Liu, C.P.; Lau, Y.J.; Hwang, K.P.; et al. Fusidic Acid for the Treatment of Bone and Joint Infections Caused by Meticillin-Resistant Staphylococcus Aureus. Int. J. Antimicrob. Agents 2012, 40, 103–107. [Google Scholar] [CrossRef]
- DAVIS, J.S. Management of Bone and Joint Infections Due to Staphylococcus aureus. Intern. Med. J. 2005, 35, S79–S96. [Google Scholar] [CrossRef]
- Bortolin, M.; Bidossi, A.; De Vecchi, E.; Avveniente, M.; Drago, L. In Vitro Antimicrobial Activity of Chlorquinaldol against Microorganisms Responsible for Skin and Soft Tissue Infections: Comparative Evaluation with Gentamicin and Fusidic Acid. Front. Microbiol. 2017, 8, 1039. [Google Scholar] [CrossRef]
- Long, B. Chapter 2. Fusidic Acid in Skin and Soft-Tissue Infections. Acta Derm. Venereol. 2008, 88, 14–20. [Google Scholar] [CrossRef]
- Alm, R.A.; Lahiri, S.D. Narrow-Spectrum Antibacterial Agents—Benefits and Challenges. Antibiotics 2020, 9, 418. [Google Scholar] [CrossRef] [PubMed]
- Huang, X.; Shen, Q.K.; Guo, H.Y.; Quan, Z.S.; Li, X. Research, Development and Pharmacological Activity of Fusidic Acid and Its Derivatives. J. Mol. Struct. 2023, 1291, 135942. [Google Scholar] [CrossRef]
- Espinoza-Moraga, M.; Singh, K.; Njoroge, M.; Kaur, G.; Okombo, J.; De Kock, C.; Smith, P.J.; Wittlin, S.; Chibale, K. Synthesis and Biological Characterisation of Ester and Amide Derivatives of Fusidic Acid as Antiplasmodial Agents. Bioorg Med. Chem. Lett. 2017, 27, 658–661. [Google Scholar] [CrossRef] [PubMed]
- Tu, B.; Cao, N.; Zhang, B.; Zheng, W.; Li, J.; Tang, X.; Su, K.; Li, J.; Zhang, Z.; Yan, Z.; et al. Synthesis and Biological Evaluation of Novel Fusidic Acid Derivatives as Two-in-One Agent with Potent Antibacterial and Anti-Inflammatory Activity. Antibiotics 2022, 11, 1026. [Google Scholar] [CrossRef]
- Munawar, S.; Zahoor, A.F.; Hussain, S.M.; Ahmad, S.; Mansha, A.; Parveen, B.; Ali, K.G.; Irfan, A. Steglich Esterification: A Versatile Synthetic Approach toward the Synthesis of Natural Products, Their Analogues/Derivatives. Heliyon 2024, 10, e23416. [Google Scholar] [CrossRef]
- Neises, B.; Steglich, W. Simple Method for the Esterification of Carboxylic Acids. Angew. Chem. Int. Ed. Engl. 1978, 17, 522–524. [Google Scholar] [CrossRef]
- Lutjen, A.B.; Quirk, M.A.; Barbera, A.M.; Kolonko, E.M. Synthesis of (E)-Cinnamyl Ester Derivatives via a Greener Steglich Esterification. Bioorg. Med. Chem. 2018, 26, 5291–5298. [Google Scholar] [CrossRef]
- Munir, R.; Zahoor, A.F.; Anjum, M.N.; Mansha, A.; Irfan, A.; Chaudhry, A.R.; Irfan, A.; Kotwica-Mojzych, K.; Glowacka, M.; Mojzych, M. Yamaguchi Esterification: A Key Step toward the Synthesis of Natural Products and Their Analogs—A Review. Front. Chem. 2024, 12, 1477764. [Google Scholar] [CrossRef]
- Cavallaro, P.A.; De Santo, M.; Greco, M.; Marinaro, R.; Belsito, E.L.; Liguori, A.; Leggio, A. Titanium Tetrachloride-Assisted Direct Esterification of Carboxylic Acids. Molecules 2024, 29, 777. [Google Scholar] [CrossRef]
- Iwasawa, T.; Wash, P.; Gibson, C.; Rebek, J. Reaction of an Introverted Carboxylic Acid with Carbodiimide. Tetrahedron 2007, 63, 6506–6511. [Google Scholar] [CrossRef]
- Uray, G.; Kelterer, A.M.; Hashim, J.; Glasnov, T.N.; Oliver Kappe, C.; Fabian, W.M.F. Bisquinolones as Chiral Fluorophores—A Combined Experimental and Computational Study of Absorption and Emission Characteristics. J. Mol. Struct. 2009, 929, 85–96. [Google Scholar] [CrossRef]
- Barman, S.; Roy, M.N. Hollow Circular Compound-Based Inclusion Complexes of an Ionic Liquid. RSC Adv. 2016, 6, 76381–76389. [Google Scholar] [CrossRef]
- Kruk, J.; Aboul-Enein, H.Y. Reactive Oxygen and Nitrogen Species in Carcinogenesis: Implications of Oxidative Stress on the Progression and Development of Several Cancer Types. Mini-Rev. Med. Chem. 2017, 17, 904–919. [Google Scholar] [CrossRef] [PubMed]
- Teleanu, D.M.; Niculescu, A.G.; Lungu, I.I.; Radu, C.I.; Vladâcenco, O.; Roza, E.; Costăchescu, B.; Grumezescu, A.M.; Teleanu, R.I. An Overview of Oxidative Stress, Neuroinflammation and Neurodegenerative Diseases. Int. J. Mol. Sci. 2022, 23, 5938. [Google Scholar] [CrossRef]
- Francenia Santos-Sánchez, N.; Salas-Coronado, R.; Villanueva-Cañongo, C.; Hernández-Carlos, B. Antioxidant Compounds and Their Antioxidant Mechanism. In Antioxidants; IntechOpen: London, UK, 2019. [Google Scholar] [CrossRef]
- Lefack Bongmo, L.V.; Nouga, A.B.; Happi, G.M.; Tabekoueng, G.B.; Lateef, M.; Kamdem Waffo, A.F.; Ali, M.S.; Choudhary, M.I.; Wansi, J.D. Phytochemical Compounds of Guibourtia Ehie and Their Antioxidant, Urease and α-Glucosidase Inhibitory Activities. Nat. Resour. Hum. Health 2022, 2, 306–312. [Google Scholar] [CrossRef]
- Asmat, U.; Abad, K.; Ismail, K. Diabetes Mellitus and Oxidative Stress—A Concise Review. Saudi Pharm. J. 2016, 24, 547–553. [Google Scholar] [CrossRef]
- Sarian, M.N.; Ahmed, Q.U.; Mat So’Ad, S.Z.; Alhassan, A.M.; Murugesu, S.; Perumal, V.; Syed Mohamad, S.N.A.; Khatib, A.; Latip, J. Antioxidant and Antidiabetic Effects of Flavonoids: A Structure-Activity Relationship Based Study. Biomed. Res. Int. 2017, 2017, 8386065. [Google Scholar] [CrossRef]
- Van de Laar, F.A.; Lucassen, P.L.B.J.; Akkermans, R.P.; Van de Lisdonk, E.H.; Rutten, G.E.H.M.; Van Weel, C. Alpha-Glucosidase Inhibitors for Type 2 Diabetes Mellitus. Cochrane Database Syst. Rev. 2005, 2009, CD003639. [Google Scholar] [CrossRef] [PubMed]
- Derosa, G.; Maffioli, P. α-Glucosidase Inhibitors and Their Use in Clinical Practice. Arch. Med. Sci. 2012, 8, 899–906. [Google Scholar] [CrossRef]
- Akhter, P.; Ashiq, U.; Jamal, R.A.; Shaikh, Z.; Mahroof-Tahir, M.; Lateef, M.; Badar, R. Chemistry, Alpha-Glucosidase and Radical Scavenging Properties of Uranyl(VI) Hydrazide Complexes. Med. Chem. 2019, 15, 923–936. [Google Scholar] [CrossRef]
- Adardour, M.; Ait Lahcen, M.; Oubahmane, M.; Ettahiri, W.; Hdoufane, I.; Bouamama, H.; Alanazi, M.M.; Cherqaoui, D.; Taleb, M.; Garcia, E.Z.; et al. Design, Synthesis, Molecular Modeling and Biological Evaluation of Novel Pyrazole Benzimidazolone Derivatives as Potent Antioxidants. Pharmaceuticals 2023, 16, 1648. [Google Scholar] [CrossRef] [PubMed]
- Aliye, M.; Dekebo, A.; Tesso, H.; Abdo, T.; Eswaramoorthy, R.; Melaku, Y. Molecular Docking Analysis and Evaluation of the Antibacterial and Antioxidant Activities of the Constituents of Ocimum Cufodontii. Sci. Rep. 2021, 11, 10101. [Google Scholar] [CrossRef] [PubMed]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A Free Web Tool to Evaluate Pharmacokinetics, Drug-Likeness and Medicinal Chemistry Friendliness of Small Molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [PubMed]
- Pires, D.E.V.; Blundell, T.L.; Ascher, D.B. PkCSM: Predicting Small-Molecule Pharmacokinetic and Toxicity Properties Using Graph-Based Signatures. J. Med. Chem. 2015, 58, 4066–4072. [Google Scholar] [CrossRef]
- Guan, L.; Yang, H.; Cai, Y.; Sun, L.; Di, P.; Li, W.; Liu, G.; Tang, Y. ADMET-Score-a Comprehensive Scoring Function for Evaluation of Chemical Drug-Likeness. Medchemcomm 2019, 10, 148–157. [Google Scholar] [CrossRef]
- Wang, S.; Li, Y.; Wang, J.; Chen, L.; Zhang, L.; Yu, H.; Hou, T. ADMET Evaluation in Drug Discovery. 12. Development of Binary Classification Models for Prediction of HERG Potassium Channel Blockage. Mol. Pharm. 2012, 9, 996–1010. [Google Scholar] [CrossRef]
- Lipinski, C.A. Lead- and Drug-like Compounds: The Rule-of-Five Revolution. Drug Discov. Today Technol. 2004, 1, 337–341. [Google Scholar] [CrossRef]
- Caron, G.; Kihlberg, J.; Goetz, G.; Ratkova, E.; Poongavanam, V.; Ermondi, G. Steering New Drug Discovery Campaigns: Permeability, Solubility, and Physicochemical Properties in the BRo5 Chemical Space. ACS Med. Chem. Lett. 2021, 12, 13–23. [Google Scholar] [CrossRef]
- Guezane-Lakoud, S.; Ferrah, M.; Merabet-Khelassi, M.; Touil, N.; Toffano, M.; Aribi-Zouioueche, L. 2-Hydroxymethyl-18-Crown-6 as an Efficient Organocatalyst for α-Aminophosphonates Synthesized under Eco-Friendly Conditions, DFT, Molecular Docking and ADME/T Studies. J. Biomol. Struct. Dyn. 2024, 42, 3332–3348. [Google Scholar] [CrossRef]
- Caron, G.; Ermondi, G. Updating Molecular Properties during Early Drug Discovery. Drug Discov. Today 2017, 22, 835–840. [Google Scholar] [CrossRef]
- Ren, S.; Lien, E.J. Caco-2 Cell Permeability vs. Human Gastro-Intestinal Absorption: QSPR Analysisl. In Progress in Drug Research; Birkhäuser: Basel, Switzerland, 2000. [Google Scholar]
- Azman, M.; Sabri, A.H.; Anjani, Q.K.; Mustaffa, M.F.; Hamid, K.A. Intestinal Absorption Study: Challenges and Absorption Enhancement Strategies in Improving Oral Drug Delivery. Pharmaceuticals 2022, 15, 975. [Google Scholar] [CrossRef] [PubMed]
- Amin, M.L. P-Glycoprotein Inhibition for Optimal Drug Delivery. Drug Target Insights 2013, 2013, 27–34. [Google Scholar] [CrossRef]
- Callaghan, R.; Luk, F.; Bebawy, M. Inhibition of the Multidrug Resistance P-Glycoprotein: Time for a Change of Strategy? Drug Metab. Dispos. 2014, 42, 623–631. [Google Scholar] [CrossRef]
- Dewanjee, S.; Dua, T.K.; Bhattacharjee, N.; Das, A.; Gangopadhyay, M.; Khanra, R.; Joardar, S.; Riaz, M.; De Feo, V.; Zia-Ul-Haq, M. Natural Products as Alternative Choices for P-Glycoprotein (P-Gp) Inhibition. Molecules 2017, 22, 871. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 Enzymes in Drug Metabolism: Regulation of Gene Expression, Enzyme Activities, and Impact of Genetic Variation. Pharmacol. Ther. 2013, 138, 103–141. [Google Scholar] [CrossRef] [PubMed]
- Arshad, N.; Jawaid, S.; Hashim, J.; Ullah, I.; Gul, S.; Aziz, A.; Wadood, A.; Khan, A. Highly Potent Anti-Inflammatory, Analgesic and Antioxidant Activities of 3,5-Disubstituted Tetrahydro-2H-1,3,5-Thiadiazine Thiones. Bioorg Med. Chem. Lett. 2023, 79, 129068. [Google Scholar] [CrossRef]
- Abbas, M.; Arshad, N. Synthesis, Highly Potent α-Glucosidase Inhibition, Antioxidant and Molecular Docking of Various Novel Dihydropyrimidine Derivatives to Treat Diabetes Mellitus. Bioorg. Med. Chem. Lett. 2025, 115, 130016. [Google Scholar] [CrossRef] [PubMed]
- Declercq, J.P.; Evrard, C.; Clippe, A.; Vander Stricht, D.; Bernard, A.; Knoops, B. Crystal Structure of Human Peroxiredoxin 5, a Novel Type of Mammalian Peroxiredoxin at 1.5 Å Resolution. J. Mol. Biol. 2001, 311, 751–759. [Google Scholar] [CrossRef]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef]
- Biovia Discovery Studio. Discovery Studio Visualizer; Biovia Discovery Studio: San Diego, CA, USA, 2017; Volume 936, pp. 240–249. [Google Scholar]
- O’Boyle, N.M.; Banck, M.; James, C.A.; Morley, C.; Vandermeersch, T.; Hutchison, G.R. Open Babel: An Open Chemical Toolbox. J. Cheminform. 2011, 3, 33. [Google Scholar] [CrossRef]
- Morris, G.M.; Ruth, H.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. Software News and Updates AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound Name | Antioxidant IC50 Value (μM) | * Binding Affinities (Kcal/mol) | α-Glucosidase Inhibition IC50 Value (μM) |
|---|---|---|---|
| Intermediate 7 | 32.9 ± 0.4 | - | 75.4 ± 0.7 |
| 8 | 54.6 ± 0.2 | −6.1 | 43.2 ± 0.8 |
| 10a | 65.5 ± 0.3 | −5.8 | 67.5 ± 0.8 |
| 10b | 32.1 ± 0.3 | −6.7 | 76.5 ± 0.1 |
| 10c | 22.5 ± 0.2 | −6.5 | 23.5 ± 0.2 |
| 10d | 45.4 ± 0.9 | −7.1 | 54.3 ± 0.1 |
| Butylated hydroxyanisole | 44.2 ± 0.3 | −5.3 | - |
| Acarbose | - | - | 5.2 ± 0.8 |
| Compound | MW | LogPo/w | log Sw | HBA | HBD | TPSA | RotB | Vb(RO5) | Vveber |
|---|---|---|---|---|---|---|---|---|---|
| Fusidic Acid 6 | 516.71 | 3.94 | −4.294 | 6 | 3 | 104.06 | 6 | 0 | 0 |
| Intermediate 7 | 796.60 | 6.45 | −3.721 | 9 | 4 | 126.35 | 16 | 0 | 0 |
| 8 | 572.82 | 5.69 | −4.44 | 6 | 2 | 93.06 | 10 | 0 | 0 |
| 10a | 642.91 | 6.07 | −3.79 | 7 | 0 | 80.29 | 10 | 0 | 0 |
| 10b | 686.96 | 5.96 | −3.774 | 8 | 0 | 89.52 | 10 | 0 | 0 |
| 10c | 731.01 | 6.66 | −3.729 | 9 | 0 | 98.75 | 10 | 0 | 0 |
| 10d | 805.09 | 7.16 | −3.573 | 11 | 0 | 117.21 | 10 | 0 | 0 |
| Compound | CaCO-2 cm/s | HIA (%) | Log Kp | BBB (log BB) | P-gp Substrate | CYP3A4 Inhibitor | AMES Toxicity | Hepato Toxicity |
|---|---|---|---|---|---|---|---|---|
| Fusidic Acid 6 | 0.632 | 74.652 | −2.735 | −0.952 | Yes | Yes | No | No |
| Intermediate 7 | 0.796 | 100 | −2.723 | −1.393 | Yes | No | No | Yes |
| 8 | 0.958 | 100 | −2.543 | 0.07 | Yes | Yes | No | No |
| 10a | 1.18 | 99.216 | −2.726 | −0.736 | No | Yes | No | No |
| 10b | 1.241 | 99.735 | −2.729 | −0.921 | No | Yes | No | No |
| 10c | 1.036 | 100 | −2.73 | −1.106 | No | Yes | No | No |
| 10d | 1.03 | 100 | −2.733 | −1.49 | No | Yes | No | No |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sultan, H.; Arshad, N.; Lateef, M. Novel Crown Ether-Functionalized Fusidic Acid Butyl Ester: Synthesis, Biological Evaluation, In Silico ADMET, and Molecular Docking Studies. Molecules 2025, 30, 2033. https://doi.org/10.3390/molecules30092033
Sultan H, Arshad N, Lateef M. Novel Crown Ether-Functionalized Fusidic Acid Butyl Ester: Synthesis, Biological Evaluation, In Silico ADMET, and Molecular Docking Studies. Molecules. 2025; 30(9):2033. https://doi.org/10.3390/molecules30092033
Chicago/Turabian StyleSultan, Hira, Nuzhat Arshad, and Mehreen Lateef. 2025. "Novel Crown Ether-Functionalized Fusidic Acid Butyl Ester: Synthesis, Biological Evaluation, In Silico ADMET, and Molecular Docking Studies" Molecules 30, no. 9: 2033. https://doi.org/10.3390/molecules30092033
APA StyleSultan, H., Arshad, N., & Lateef, M. (2025). Novel Crown Ether-Functionalized Fusidic Acid Butyl Ester: Synthesis, Biological Evaluation, In Silico ADMET, and Molecular Docking Studies. Molecules, 30(9), 2033. https://doi.org/10.3390/molecules30092033