
Development and Characterization of pFluor50, a Fluorogenic-Based Kinetic Assay System for High-Throughput Inhibition Screening and Characterization of Time-Dependent Inhibition and Inhibition Type for Six Human CYPs
 , , ,
, , ,
Abstract
1. Introduction
2. Results
2.1. Calibration Curves for Different Products
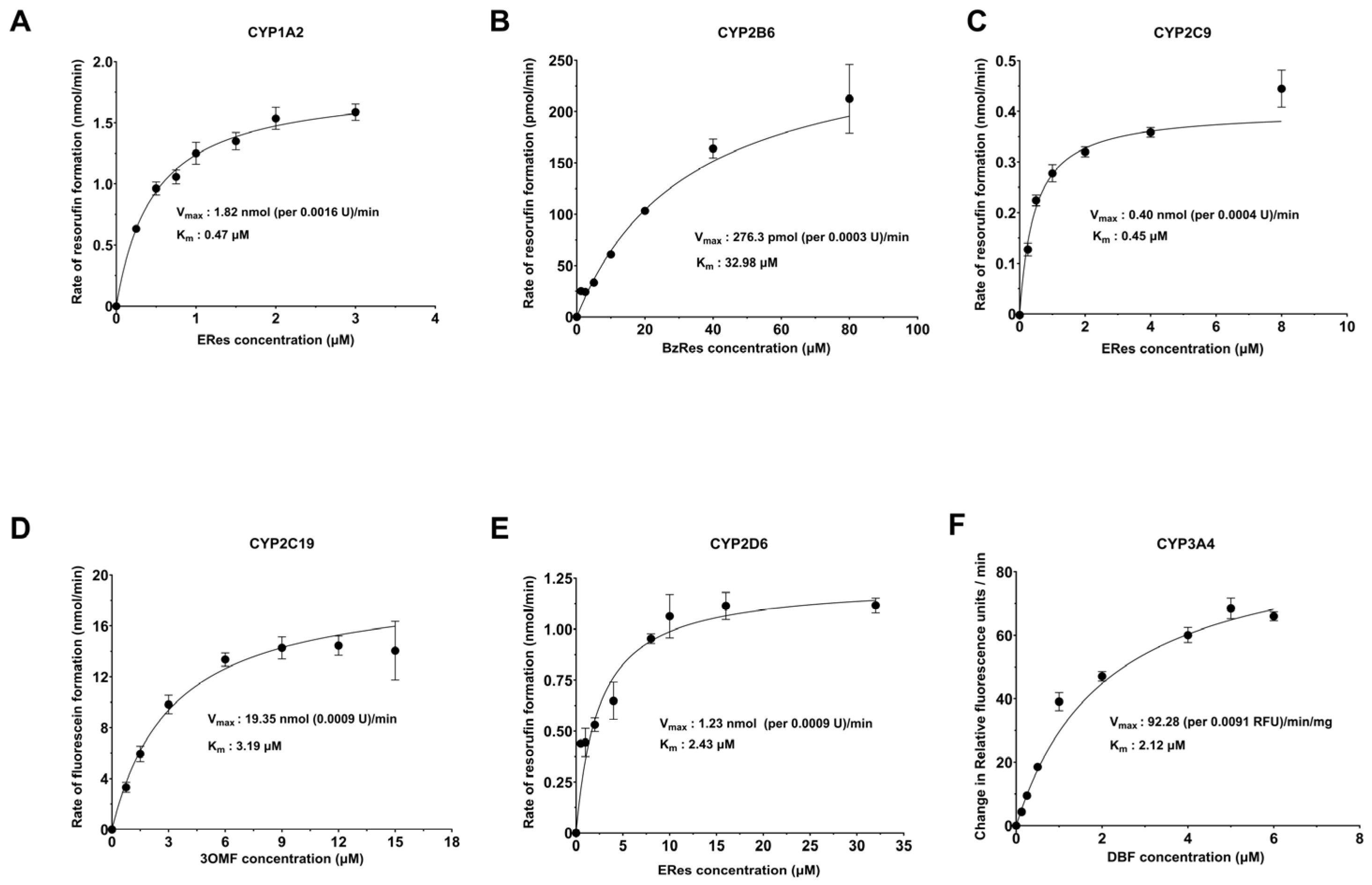
2.2. Michaelis–Menten Analysis for Determination of Kinetic Parameters of CYP–Substrate Pairs
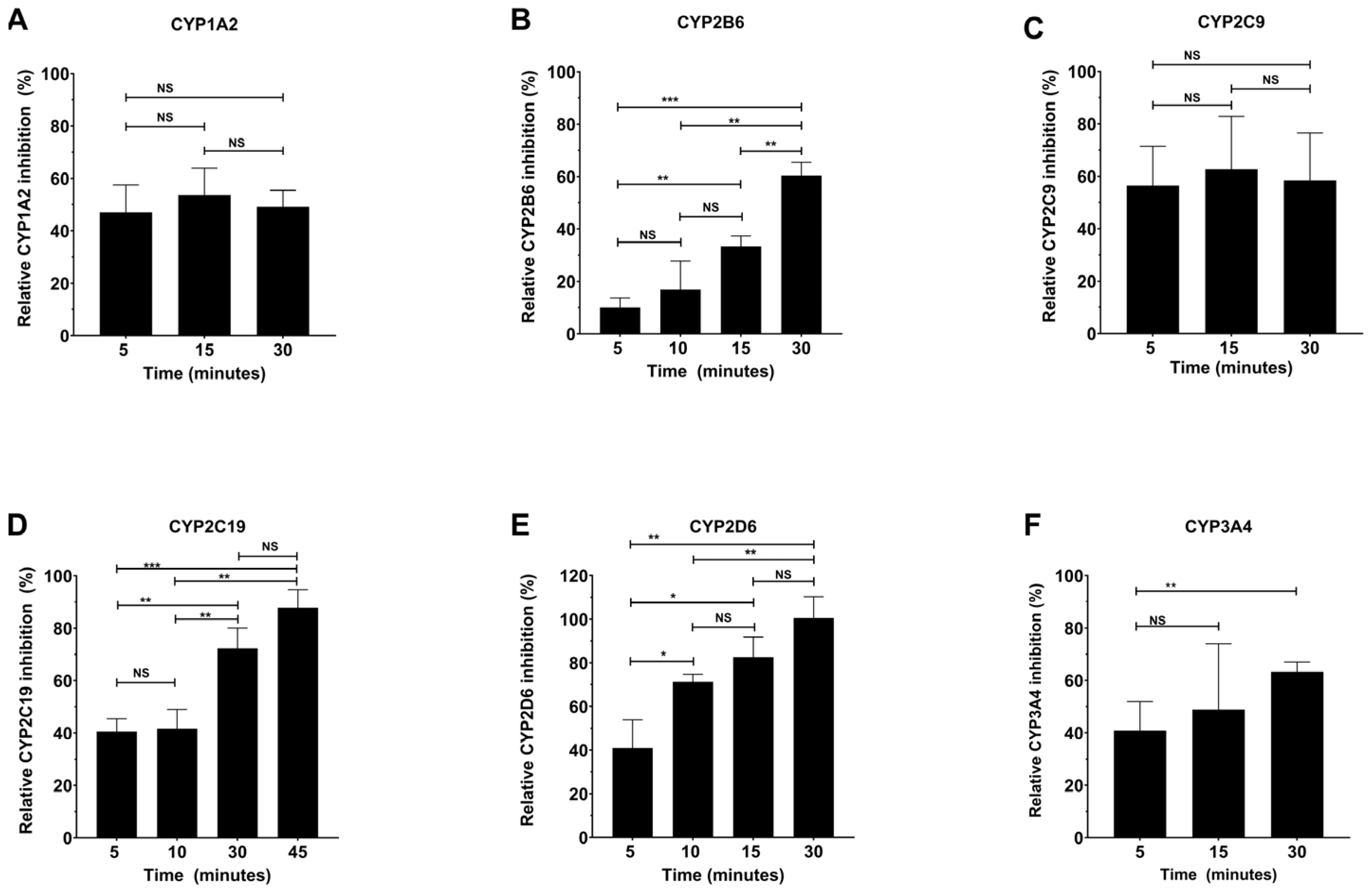
2.3. Determination of the Time-Dependence of CYP Inhibition by Specific Inhibitors
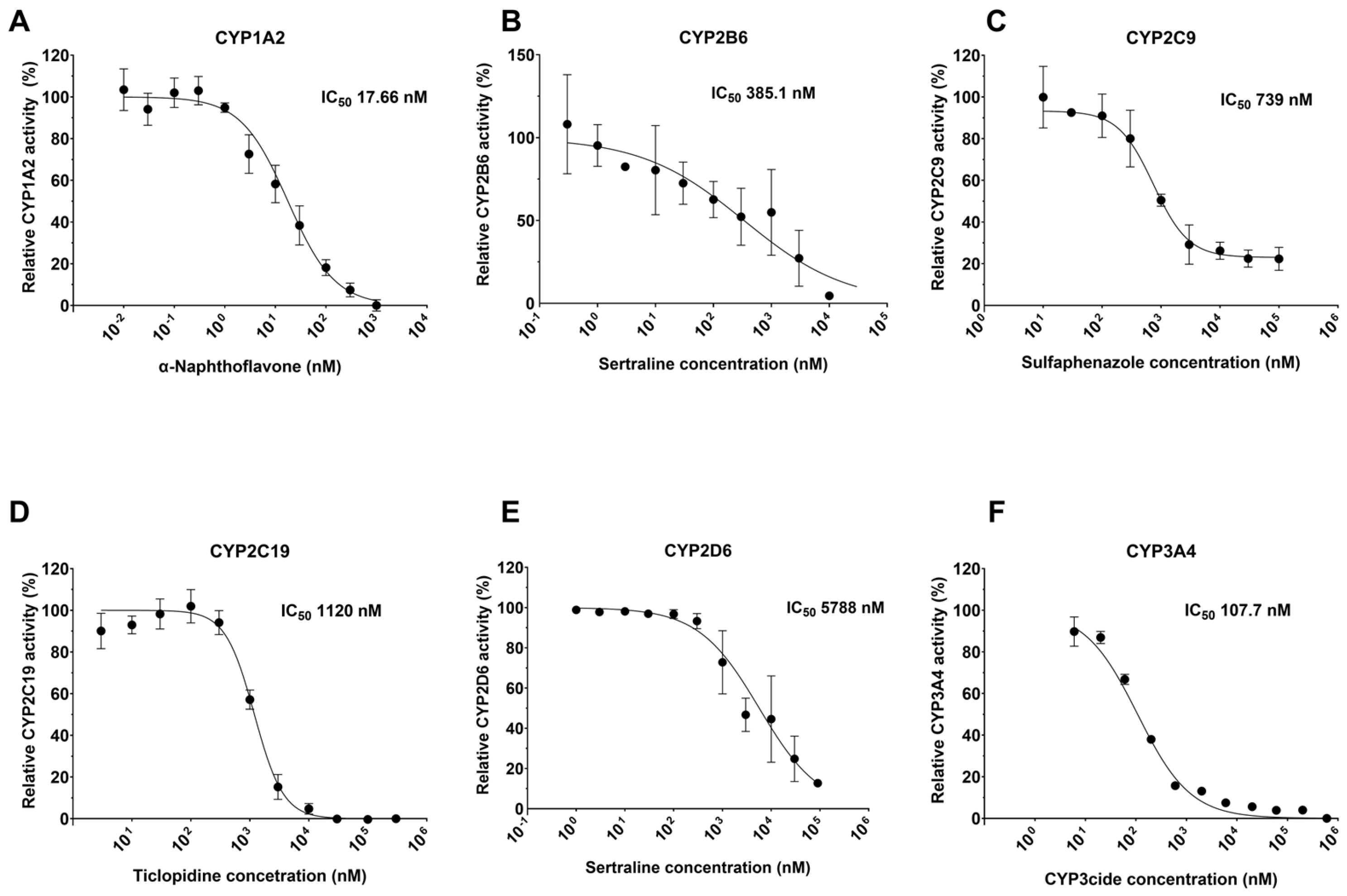
2.4. Determination of IC50 for Known Inhibitors
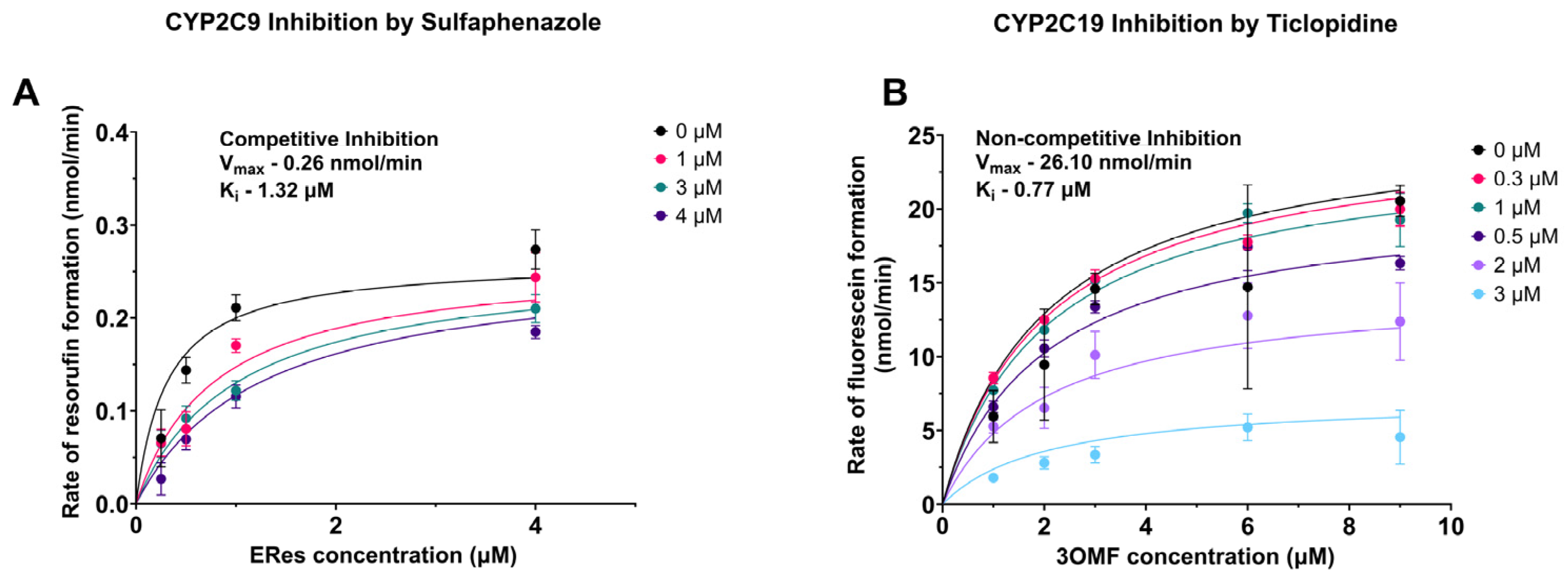
2.5. Determination of the Type of Inhibition for Select CYP–Inhibitor Pairs
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. pFluor50 Assay System Development for CYP Activity Using Fluorogenic Substrates
4.3. Michaelis–Menten Kinetics for Each CYP–Substrate Pair
4.4. Determination of IC50 Values for Each CYP-Specific Inhibitor
4.5. Determination of Product Formation Rate
4.6. Data Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| CYP450 | Cytochrome 450 enzymes |
| CYPs | Cytochrome P450 |
| CYP1A2 | Cytochrome P450-1A2 |
| CYP2B6 | Cytochrome P450-2B6 |
| CYP2C9 | Cytochrome P450-2C9 |
| CYP2C19 | Cytochrome P450-2C19 |
| CYP2D6 | Cytochrome P450-2D6 |
| CYP3A4 | Cytochrome P450-3A4 |
| Eres | Resorufin ethoxy ether |
| Bzres | Resorufin benzyl ether |
| 3OMF | 3-O-methyl fluorescein |
| DBF | Dibenzyl fluorescein |
References
- McKinnon, R.A.; Sorich, M.J.; Ward, M.B. Cytochrome P450 part 1: Multiplicity and function. J. Pharm. Pract. Res. 2008, 38, 55–57. [Google Scholar] [CrossRef]
- Lamb, D.C.; Lei, L.; Warrilow, A.G.; Lepesheva, G.I.; Mullins, J.G.; Waterman, M.R.; Kelly, S.L. The first virally encoded cytochrome p450. J. Virol. 2009, 83, 8266–8269. [Google Scholar] [CrossRef] [PubMed]
- Attar, M.; Shen, J.; Ling, K.H.J.; Tang-Liu, D. Ophthalmic drug delivery considerations at the cellular level: Drug-metabolising enzymes and transporters. Expert Opin. Drug Deliv. 2005, 2, 891–908. [Google Scholar] [CrossRef]
- Sadler, N.C.; Nandhikonda, P.; Webb-Robertson, B.J.; Ansong, C.; Anderson, L.N.; Smith, J.N.; Corley, R.A.; Wright, A.T. Hepatic cytochrome P450 activity, abundance, and expression throughout human development. Drug Metab. Dispos. 2016, 44, 984–991. [Google Scholar] [CrossRef]
- Ogu, C.C.; Maxa, J.L. Drug interactions due to cytochrome P450. In Baylor University Medical Center Proceedings; Taylor & Francis: New York, NY, USA, 2000; Volume 143, pp. 421–423. [Google Scholar]
- Hasler, J.A.; Estabrook, R.; Murray, M.; Pikuleva, I.; Waterman, M.; Capdevila, J.; Holla, V.; Helvig, C.; Falck, J.R.; Farrell, G.; et al. Human cytochromes P450. Mol. Asp. Med. 1999, 20, 1–137. [Google Scholar] [CrossRef]
- Yan, Z.; Caldwell, G.W. Cytochrome P450: In Vitro Methods and Protocols; Springer: New York, NY, USA, 2021; pp. 1–25. [Google Scholar]
- Galetin, A.; Gertz, M.; Houston, J.B. Contribution of intestinal cytochrome p450-mediated metabolism to drug-drug inhibition and induction interactions. Drug. Metab. Pharmacokinet. 2010, 25, 28–47. [Google Scholar] [CrossRef]
- Zanger, U.M.; Schwab, M. Cytochrome P450 enzymes in drug metabolism: Regulation of gene expression, enzyme activities, and impact of genetic variation. Pharmacol. Ther. 2010, 138, 103–141. [Google Scholar] [CrossRef]
- Donato, M.T.; Jimenez, N.; Castell, J.V.; Gómez-Lechón, M.J. Fluorescence-based assays for screening nine cytochrome P450 (P450) activities in intact cells expressing individual human P450 enzymes. Drug. Metab. Dispos. 2004, 32, 699–706. [Google Scholar] [CrossRef]
- Miller, V.P.; Stresser, D.M.; Blanchard, A.P.; Turner, S.; Crespi, C.L. Fluorometric high-throughput screening for inhibitors of cytochrome P450. Ann. N. Y. Acad. Sci. 2000, 919, 26–32. [Google Scholar] [CrossRef]
- Zhao, M.; Ma, J.; Li, M.; Zhang, Y.; Jiang, B.; Zhao, X.; Huai, C.; Shen, L.; Zhang, N.; He, L.; et al. Cytochrome P450 enzymes and drug metabolism in humans. Int. J. Mol. Sci. 2021, 22, 12808. [Google Scholar] [CrossRef]
- Laine, K.; Tybring, G.; Härtter, S.; Andersson RN, K.; Svensson, J.O.; Widén, J.; Bertilsson, L. Inhibition of cytochrome P4502D6 activity with paroxetine normalizes the ultrarapid metabolizer phenotype as measured by nortriptyline pharmacokinetics and the debrisoquin test. Clin. Pharmacol. Ther. 2001, 70, 327–335. [Google Scholar] [CrossRef] [PubMed]
- Okudaira, T.; Kotegawa, T.; Imai, H.; Tsutsumi, K.; Nakano, S.; Ohashi, K. Effect of the treatment period with erythromycin on cytochrome P450 3A activity in humans. J. Clin. Pharm. 2007, 47, 871–876. [Google Scholar] [CrossRef] [PubMed]
- Deodhar, M.; Al Rihani, S.B.; Arwood, M.J.; Darakjian, L.; Dow, P.; Turgeon, J.; Michaud, V. Mechanisms of CYP450 inhibition: Understanding drug-drug interactions due to mechanism-based inhibition in clinical practice. Pharmaceutics 2020, 12, 846. [Google Scholar] [CrossRef] [PubMed]
- Stresser, D.M. High-Throughput Screening of Human Cytochrome P450 Inhibitors Using Fluorometric Substrates: Methodology for 25 Enzyme/Substrate Pairs. In Optimization in Drug Discovery: In Vitro Methods; Humana Totowa: Totowa, NJ, USA, 2004; pp. 215–230. [Google Scholar]
- Hinkson, I.V.; Madej, B.; Stahlberg, E.A. Accelerating therapeutics for opportunities in medicine: A paradigm shift in drug discovery. Front. Pharmacol. 2020, 11, 770. [Google Scholar] [CrossRef]
- Sun, D.; Gao, W.; Hu, H.; Zhou, S. Why 90% of clinical drug development fails and how to improve it? Acta Phar. Sin. B 2022, 12, 3049–3062. [Google Scholar] [CrossRef]
- Ortiz de Montellano, P.R. Cytochrome P450-activated prodrugs. Future Med. Chem. 2013, 5, 213–228. [Google Scholar] [CrossRef]
- Ayrton, J.; Plumb, R.; Leavens, W.J.; Mallett, D.; Dickins, M.; Dear, G.J. Application of a generic fast gradient liquid chromatography tandem mass spectrometry method for the analysis of cytochrome P450 probe substrates. Rapid Commun. Mass Spectrom. 1998, 12, 217–224. [Google Scholar] [CrossRef]
- Nayadu, S.; Behera, D.; Sharma, M.; Kaur, G.; Gudi, G. Fluorescent probe based CYP inhibition assay: A high throughput tool for early drug discovery screening. Int. J. Pharm. Pharm. Sci. 2013, 5, 303–307. [Google Scholar]
- Sukumaran, S.M.; Potsaid, B.; Lee, M.Y.; Clark, D.S.; Dordick, J.S. Development of a fluorescence-based, ultra high-throughput screening platform for nanoliter-scale cytochrome P450 microarrays. J. Biomol. Screen. 2009, 14, 668–678. [Google Scholar] [CrossRef]
- Ung, Y.T.; Ong, C.E.; Pan, Y. Current high-throughput approaches of screening modulatory effects of xenobiotics on cytochrome P450 (CYP) enzymes. High-Throughput 2018, 7, 29. [Google Scholar] [CrossRef]
- Kabir, M.L.; Wang, F.; Clayton, A.H. Intrinsically fluorescent anti-cancer drugs. Biology 2022, 11, 1135. [Google Scholar] [CrossRef] [PubMed]
- Le, S.B.; Holmuhamedov, E.L.; Narayanan, V.L.; Sausville, E.A.; Kaufmann, S.H. Adaphostin and other anticancer drugs quench the fluorescence of mitochondrial potential probes. Cell Death Differ. 2006, 13, 151–159. [Google Scholar] [CrossRef] [PubMed]
- Bell, L.; Bickford, S.; Nguyen, P.H.; Wang, J.; He, T.; Zhang, B.; Friche, Y.; Zimmerlin, A.; Urban, L.; Bojanic, D. Evaluation of Fluorescence-and Mass Spectrometry–Based CYP Inhibition Assays for Use in Drug Discovery. SLAS Discov. 2008, 13, 343–353. [Google Scholar] [CrossRef] [PubMed]
- Paradise, E.; Chaturvedi, P.; Ter-Ovanesyan, E. Cytochrome P450 inhibition assays using traditional and fluorescent substrates. Curr. Protoc. Pharmacol. 2007, 39, 7–11. [Google Scholar] [CrossRef]
- Cohen, L.H.; Remley, M.J.; Raunig, D.; Vaz, A.D. In vitro drug interactions of cytochrome p450: An evaluation of fluorogenic to conventional substrates. Drug Metab. Dispos. 2003, 31, 1005–1015. [Google Scholar] [CrossRef]
- Raunio, H.; Pentikäinen, O.; Juvonen, R.O. Coumarin-based profluorescent and fluorescent substrates for determining xenobiotic-metabolizing enzyme activities in vitro. Int. J. Mol. Sci. 2020, 21, 4708. [Google Scholar] [CrossRef]
- Murayama, N.; Soyama, A.; Saito, Y.; Nakajima, Y.; Komamura, K.; Ueno, K.; Kamakura, S.; Kitakaze, M.; Kimura, H.; Goto, Y.I.; et al. Six novel nonsynonymous CYP1A2 gene polymorphisms: Catalytic activities of the naturally occurring variant enzymes. J. Pharmacol. Exp. Ther. 2004, 308, 300–306. [Google Scholar] [CrossRef]
- Chandrashekar, D.V.; Mehvar, R. UPLC-MS/MS analysis of CYP1A-mediated ethoxyresorufin-O-deethylation activity in the rat kidney microsomes. J. Chromatogr. B 2020, 1153, 122272. [Google Scholar] [CrossRef]
- Juvonen, R.O.; Ahinko, M.; Jokinen, E.M.; Huuskonen, J.; Raunio, H.; Pentikainen, O.T. Substrate selectivity of coumarin derivatives by human CYP1 enzymes: In vitro enzyme kinetics and in silico modeling. ACS Omega 2021, 6, 11286–11296. [Google Scholar] [CrossRef]
- Niwa, T.; Inoue-Yamamoto, S.; Shiraga, T.; Takagi, A. Effect of antifungal drugs on cytochrome P450 (CYP) 1A2, CYP2D6, and CYP2E1 activities in human liver microsomes. Biol. Pharm. Bull. 2005, 28, 1813–1816. [Google Scholar] [CrossRef]
- Lautraite, S.; Musonda, A.C.; Doehmer, J.; Edwards, G.O.; Chipman, J.K. Flavonoids inhibit genetic toxicity produced by carcinogens in cells expressing CYP1A2 and CYP1A1. Mutagenesis 2002, 17, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Appiah-Opong, R.; Commandeur, J.N.; van Vugt-Lussenburg, B.; Vermeulen, N.P. Inhibition of human recombinant cytochrome P450s by curcumin and curcumin decomposition products. Toxicology 2007, 235, 83–91. [Google Scholar] [CrossRef] [PubMed]
- Ekins, S.; Vandenbranden, M.; Ring, B.J.; Gillespie, J.S.; Yang, T.J.; Gelboin, H.V.; Wrighton, S.A. Further characterization of the expression in liver and catalytic activity of CYP2B6. J. Pharmacol. Exp. Ther. 1998, 286, 1253–1259. [Google Scholar] [CrossRef] [PubMed]
- Sudsakorn, S.; Skell, J.; Williams, D.A.; O’Shea, T.J.; Liu, H. Evaluation of 3-O-methylfluorescein as a selective fluorometric substrate for CYP2C19 in human liver microsomes. Drug Metab. Dispos. 2007, 35, 841–847. [Google Scholar] [CrossRef]
- Foti, R.S.; Rock, D.A.; Han, X.; Flowers, R.A.; Wienkers, L.C.; Wahlstrom, J.L. Ligand-based design of a potent and selective inhibitor of cytochrome P450 2C19. J. Med. Chem. 2012, 55, 1205–1214. [Google Scholar] [CrossRef]
- Jiang, R.; Yamaori, S.; Okamoto, Y.; Yamamoto, I.; Watanabe, K. Cannabidiol is a potent inhibitor of the catalytic activity of cytochrome P450 2C19. Drug Metab. Pharmacokinet. 2013, 28, 332–338. [Google Scholar] [CrossRef]
- Stresser, D.M.; Blanchard, A.P.; Turner, S.D.; Erve, J.C.; Dandeneau, A.A.; Miller, V.P.; Crespi, C.L. Substrate-dependent modulation of CYP3A4 catalytic activity: Analysis of 27 test compounds with four fluorometric substrates. Drug Metab. Dispos. 2000, 28, 1440–1448. [Google Scholar] [CrossRef]
- Moutinho, D.; Marohnic, C.C.; Panda, S.P.; Rueff, J.; Masters, B.S.; Kranendonk, M. Altered human CYP3A4 activity caused by Antley-Bixler syndrome-related variants of NADPH-cytochrome P450 oxidoreductase measured in a robust in vitro system. Drug Metab. Dispos. 2012, 40, 754–760. [Google Scholar] [CrossRef]
- Thomford, N.E.; Dzobo, K.; Chopera, D.; Wonkam, A.; Maroyi, A.; Blackhurst, D.; Dandara, C. In vitro reversible and time-dependent CYP450 inhibition profiles of medicinal herbal plant extracts Newbouldia laevis and Cassia abbreviata: Implications for herb-drug interactions. Molecules 2016, 21, 891. [Google Scholar] [CrossRef]
- Obach, R.S.; Cox, L.M.; Tremaine, L.M. Sertraline is metabolized by multiple cytochrome P450 enzymes, monoamine oxidases, and glucuronyl transferases in human: An in vitro study. Drug Metab. Dispos. 2005, 33, 262–270. [Google Scholar] [CrossRef]
- Pihlaja, T.; Kiiski, I.; Sikanen, T. HLM chip—A microfluidic approach to study the mechanistic basis of cytochrome P450 inhibition using immobilized human liver microsomes. Eur. J. Pharm. Sci. 2024, 197, 106773. [Google Scholar] [CrossRef] [PubMed]
- Nishiya, Y.; Hagihara, K.; Kurihara, A.; Okudaira, N.; Farid, N.A.; Okazaki, O.; Ikeda, T. Comparison of mechanism-based inhibition of human cytochrome P450 2C19 by ticlopidine, clopidogrel, and prasugrel. Xenobiotica 2009, 39, 836–843. [Google Scholar] [CrossRef] [PubMed]
- Walsky, R.L.; Obach, R.S.; Hyland, R.; Kang, P.; Zhou, S.; West, M.; Geoghegan, K.F.; Helal, C.J.; Walker, G.S.; Goosen, T.C.; et al. Selective mechanism-based inactivation of CYP3A4 by CYP3cide (PF-04981517) and its utility as an in vitro tool for delineating the relative roles of CYP3A4 versus CYP3A5 in the metabolism of drugs. Drug Metab. Dispos. 2012, 40, 1686–1697. [Google Scholar] [CrossRef]
- Park, E.J.; Park, K.; Durai, P.; Kim, K.Y.; Park, S.Y.; Kwon, J.; Lee, H.J.; Pan, C.H.; Liu, K.H. Potent and selective inhibition of CYP1A2 enzyme by obtusifolin and its chemopreventive effects. Pharmaceutics 2022, 14, 2683. [Google Scholar] [CrossRef]
- Juvonen, R.O.; Jokinen, E.M.; Javaid, A.; Lehtonen, M.; Raunio, H.; Pentikäinen, O.T. Inhibition of human CYP1 enzymes by a classical inhibitor α-naphthoflavone and a novel inhibitor N-(3, 5-dichlorophenyl) cyclopropanecarboxamide: An in vitro and in silico study. Chem. Biol. Drug Des. 2020, 95, 520–533. [Google Scholar] [CrossRef]
- Shimada, T.; Yamazaki, H.; Foroozesh, M.; Hopkins, N.E.; Alworth, W.L.; Guengerich, F.P. Selectivity of polycyclic inhibitors for human cytochrome P450s 1A1, 1A2, and 1B1. Chem. Res. Toxicol. 1998, 11, 1048–1056. [Google Scholar] [CrossRef]
- Ramachandran, S.D.; Vivarès, A.; Klieber, S.; Hewitt, N.J.; Muenst, B.; Heinz, S.; Walles, H.; Braspenning, J. Applicability of second-generation upcyte® human hepatocytes for use in CYP inhibition and induction studies. Pharmacol. Res. Perspect. 2015, 3, e00161. [Google Scholar] [CrossRef]
- Perloff, E.S.; Mason, A.K.; Dehal, S.S.; Blanchard, A.P.; Morgan, L.; Ho, T.; Dandeneau, A.; Crocker, R.M.; Chandler, C.M.; Boily, N.; et al. Validation of cytochrome p450 time-dependent inhibition assays: A two-time point ic50 shift approach facilitates k inact assay design. Xenobiotica 2009, 39, 99–112. [Google Scholar] [CrossRef]
- Otton, S.V.; Ball, S.E.; Cheung, S.W.; Inaba, T.; Rudolph, R.L.; Sellers, E.M. Venlafaxine oxidation in vitro is catalysed by CYP2D6. Br. J. Clin. Pharmacol. 1996, 41, 149–156. [Google Scholar] [CrossRef]
- Hesse, L.M.; Venkatakrishnan, K.; Court, M.H.; von Moltke, L.L.; Duan, S.X.; Shader, R.I.; Greenblatt, D.J. CYP2B6 mediates the in vitro hydroxylation of bupropion: Potential drug interactions with other antidepressants. Drug Metab. Dispos. 2000, 28, 1176–1183. [Google Scholar] [CrossRef]
- Kahma, H.; Paludetto, M.N.; Neuvonen, M.; Kurkela, M.; Filppula, A.M.; Niemi, M.; Backman, J.T. Screening of 16 major drug glucuronides for time-dependent inhibition of nine drug-metabolizing CYP enzymes–detailed studies on CYP3A inhibitors. Eur. J. Pharm. Sci. 2024, 198, 106735. [Google Scholar] [CrossRef] [PubMed]
- Ha-Duong, N.T.; Marques-Soares, C.; Dijols, S.; Sari, M.A.; Dansette, P.M.; Mansuy, D. Interaction of new sulfaphenazole derivatives with human liver cytochrome p450 2Cs: Structural determinants required for selective recognition by CYP 2C9 and for inhibition of human CYP 2Cs. Arch. Biochem. Biophys. 2001, 394, 189–200. [Google Scholar] [CrossRef] [PubMed]
- Giancarlo, G.M.; Venkatakrishnan, K.; Granda, B.W.; von Moltke, L.L.; Greenblatt, D.J. Relative contributions of CYP2C9 and 2C19 to phenytoin 4-hydroxylation in vitro: Inhibition by sulfaphenazole, omeprazole, and ticlopidine. Eur. J. Clin. Pharmacol. 2001, 57, 31–36. [Google Scholar] [CrossRef] [PubMed]
- McGinnity, D.F.; Tucker, J.; Trigg, S.; Riley, R.J. Prediction of CYP2C9-mediated drug-drug interactions: A comparison using data from recombinant enzymes and human hepatocytes. Drug Metab. Dispos. 2005, 33, 1700–1707. [Google Scholar] [CrossRef]
- Zi, J.; Liu, D.; Ma, P.; Huang, H.; Zhu, J.; Wei, D.; Yang, J.; Chen, C. Effects of CYP2C9* 3 and CYP2C9* 13 on diclofenac metabolism and inhibition-based drug-drug interactions. Drug Metab. Pharmacokinet. 2010, 25, 343–350. [Google Scholar] [CrossRef]
- Donahue, S.R.; Flockhart, D.A.; Abernethy, D.R.; Ko, J.W. Ticlopidine inhibition of phenytoin metabolism mediated by potent inhibition of CYP2C19. Clin. Pharmacol. Ther. 1997, 62, 572–577. [Google Scholar] [CrossRef]
- Hagihara, K.; Nishiya, Y.; Kurihara, A.; Kazui, M.; Farid, N.A.; Ikeda, T. Comparison of human cytochrome P450 inhibition by the thienopyridines prasugrel, clopidogrel, and ticlopidine. Drug Metab. Pharmacokinet. 2008, 23, 412–420. [Google Scholar] [CrossRef]
- Ko, J.W.; Desta, Z.; Soukhova, N.V.; Tracy, T.; Flockhart, D.A. In vitro inhibition of the cytochrome P450 (CYP450) system by the antiplatelet drug ticlopidine: Potent effect on CYP2C19 and CYP2D6. Br. J. Clin. Pharmacol. 2010, 49, 343–351. [Google Scholar] [CrossRef]
- Kahma, H.; Aurinsalo, L.; Neuvonen, M.; Katajamäki, J.; Paludetto, M.N.; Viinamäki, J.; Launiainen, T.; Filppula, A.M.; Tornio, A.; Niemi, M.; et al. An automated cocktail method for in vitro assessment of direct and time-dependent inhibition of nine major cytochrome P450 enzymes–application to establishing CYP2C8 inhibitor selectivity. Eur. J. Pharm. Sci. 2021, 162, 105810. [Google Scholar] [CrossRef]
- Kozakai, K.; Yamada, Y.; Oshikata, M.; Kawase, T.; Suzuki, E.; Haramaki, Y.; Taniguchi, H. Cocktail-substrate approach-based high-throughput assay for evaluation of direct and time-dependent inhibition of multiple cytochrome P450 isoforms. Drug Metab. Pharmacokinet. 2014, 29, 198–207. [Google Scholar] [CrossRef]
- Jin, X.; Potter, B.; Luong, T.L.; Nelson, J.; Vuong, C.; Potter, C.; Xie, L.; Zhang, J.; Zhang, P.; Sousa, J.; et al. Pre-clinical evaluation of CYP 2D6 dependent drug–drug interactions between primaquine and SSRI/SNRI antidepressants. Malar J. 2016, 15, 280. [Google Scholar] [CrossRef] [PubMed]
- McGinnity, D.F.; Waters, N.J.; Tucker, J.; Riley, R.J. Integrated in vitro analysis for the in vivo prediction of cytochrome P450-mediated drug-drug interactions. Drug Metab. Dispos. 2008, 36, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Work, H.M.; Kandel, S.E.; Lampe, J.N. Comparison of the CYP3A Selective Inhibitors CYP3cide, Clobetasol, and Azamulin for Their Potential to Distinguish CYP3A7 Activity in the Presence of CYP3A4/5. Drug Metab. Dispos. 2024, 52, 1224–1233. [Google Scholar] [CrossRef] [PubMed]
- Leonard, E.; Koffas, M.A. Engineering of artificial plant cytochrome P450 enzymes for synthesis of isoflavones by Escherichia coli. Appl. Environ. Microbiol. 2007, 73, 7246–7251. [Google Scholar] [CrossRef]
- Nelson, D.R. The cytochrome p450 homepage. Hum. Genom. 2009, 4, 59–65. [Google Scholar] [CrossRef]
- Quehl, P.; Hollender, J.; Schüürmann, J.; Brossette, T.; Maas, R.; Jose, J. Co-expression of active human cytochrome P450 1A2 and cytochrome P450 reductase on the cell surface of Escherichia coli. Microb. Cell Factories 2016, 15, 26. [Google Scholar] [CrossRef]
- Hochachka, P.W.; Lewis, J.K. Interacting effects of pH and temperature on the Km values for fish tissue lactate dehydrogenases. Comp. Biochem. Physiol. Part B Comp. Biochem. 1971, 39, 925–933. [Google Scholar] [CrossRef]
- Burton, A.C. Human calorimetry: II. The average temperature of the tissues of the body: Three figures. J. Nutr. 1935, 9, 261–280. [Google Scholar] [CrossRef]
- Proksch, E. pH in nature, humans and skin. J. Dermatol. 2018, 45, 1044–1052. [Google Scholar] [CrossRef]
- Nelson, D.L.; Lehninger, A.L.; Cox, M.M. Lehninger Principles of Biochemistry; Macmillan: New York, NY, USA, 2008. [Google Scholar]
- Busby, W.F., Jr.; Ackermann, J.M.; Crespi, C.L. Effect of methanol, ethanol, dimethyl sulfoxide, and acetonitrile on in vitro activities of cDNA-expressed human cytochromes P-450. Drug Metab. Dispos. 1999, 27, 246–249. [Google Scholar] [CrossRef]
- Li, D.; Han, Y.; Meng, X.; Sun, X.; Yu, Q.; Li, Y.; Wan, L.; Huo, Y.; Guo, C. Effect of regular organic solvents on cytochrome P450-mediated metabolic activities in rat liver microsomes. Drug Metab. Dispos. 2010, 38, 1922–1925. [Google Scholar] [CrossRef] [PubMed]
- Greenblatt, D.J.; von Moltke, L.L.; Harmatz, J.S.; Shader, R.I. Human cytochromes mediating sertraline biotransformation: Seeking attribution. J. Clin. Psychopharmacol. 2010, 19, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, K.; Ishizuka, T.; Shimada, N.; Yoshimura, Y.; Kamijima, K.; Chiba, K. Sertraline N-demethylation is catalyzed by multiple isoforms of human cytochrome P-450 in vitro. Drug Metab. Dispos. 1999, 27, 763–766. [Google Scholar] [CrossRef] [PubMed]
- Palacharla, R.C.; Nirogi, R.; Uthukam, V.; Manoharan, A.; Ponnamaneni, R.K.; Kalaikadhiban, I. Quantitative in vitro phenotyping and prediction of drug interaction potential of CYP2B6 substrates as victims. Xenobiotica 2018, 48, 663–675. [Google Scholar] [CrossRef]
- Xu, Z.H.; Wang, W.; Zhao, X.J.; Huang, S.L.; Zhu, B.; He, N.; Shu, Y.; Liu, Z.Q.; Zhou, H.H. Evidence for involvement of polymorphic CYP2C19 and 2C9 in the N-demethylation of sertraline in human liver microsomes. Br. J. Clin. Pharmacol. 1999, 48, 416–423. [Google Scholar] [CrossRef]
- Thelingwani, R.S.; Dhansay, K.; Smith, P.; Chibale, K.; Masimirembwa, C.M. Potent inhibition of CYP1A2 by Frutinone A, an active ingredient of the broad spectrum antimicrobial herbal extract from P. fruticosa. Xenobiotica 2012, 42, 989–1000. [Google Scholar] [CrossRef]
- Walsky, R.L.; Astuccio, A.V.; Obach, R.S. Evaluation of 227 drugs for in vitro inhibition of cytochrome P450 2B6. J. Clin. Pharmacol. 2006, 46, 1426–1438. [Google Scholar] [CrossRef]
- Khan, M.; Mohan, I.K.; Kutala, V.K.; Kumbala, D.; Kuppusamy, P. Cardioprotection by sulfaphenazole, a cytochrome p450 inhibitor: Mitigation of ischemia-reperfusion injury by scavenging of reactive oxygen species. J. Pharmacol. Exp. Ther. 2007, 323, 813–821. [Google Scholar] [CrossRef]
- Sproule, B.A.; Otton, S.V.; Cheung, S.W.; Zhong, X.H.; Romach, M.K.; Sellers, E.M. CYP2D6 inhibition in patients treated with sertraline. J. Clin. Psychopharmacol. 1997, 17, 102–106. [Google Scholar] [CrossRef]
- Atkins, W.M. Non-Michaelis-Menten kinetics in cytochrome P450-catalyzed reactions. Annu. Rev. Pharmacol. Toxicol. 2005, 45, 291–310. [Google Scholar] [CrossRef]
- Beam, A.; Motsinger-Reif, A. Beyond IC50s: Towards robust statistical methods for in vitro association studies. J. Pharmacogenom. Pharmacoproteom. 2014, 5, 1000121. [Google Scholar] [PubMed]
- Reynolds, K.; Yang, X.; Peters, S.A.; Sinha, V.; Heymann, H.; Borges, L.N.; Einolf, H.J.; Fu, S.; Hoshino, M.; Li, L.; et al. ICH M12 Drug Interaction Studies: Summary of the Efforts to Achieve Global Convergence. Clin. Pharmacol. Ther. 2025. online ahead of print. [Google Scholar] [CrossRef] [PubMed]
- VandenBrink, B.M.; Foti, R.S.; Rock, D.A.; Wienkers, L.C.; Wahlstrom, J.L. Evaluation of CYP2C8 inhibition in vitro: Utility of montelukast as a selective CYP2C8 probe substrate. Drug Metab. Dispos. 2011, 3, 1546–1554. [Google Scholar] [CrossRef] [PubMed]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| CYPexpress (Amount) | CYPexpress Activity [Units/(mg/mL)] 1 | Substrate | Ex:Em (nm) | Read Time (min) | Read Time Interval (sec) | Master Mix | Substrate Concentration Range (µM) |
|---|---|---|---|---|---|---|---|
| CYP1A2 (1 mg/mL) | CYP1A2 (0.0016 U) | Eres | 560:590 | 10 | 60 | NADPH-0.52 mM MgCl2-2.64 mM | 0.25–3 |
| CYP2B6 (1 mg/mL) | CYP2B6 (0.0003 U) | Bzres | 535:590 | 60 | 90 | NADPH-0.33 mM MgCl2-3.3 mM NADP+-1.3 mM G6P-3.3 mM G6PDH-0.4 unit/mL | 1.25–80 |
| CYP2C9 (1 mg/mL) | CYP2C9 (0.0004 U) | Eres | 560:590 | 40 | 120 | NADPH-0.65 mM MgCl2-3.3 mM | 0.25–8 |
| CYP2C19 (2 mg/mL) | CYP2C19 (0.0009 U) | 3OMF | 485:525 | 20 | 30 | NADPH-0.33 mM MgCl2-3.3 mM NADP+-1.3 mM G6P-3.3 mM G6PDH-0.4 unit/mL | 1–12 |
| CYP2D6 (1 mg/mL) | CYP2D6 (0.0009 U) | Eres | 560:590 | 75 | 90 | NADPH-2.6 mM MgCl2-3.3 mM | 1–32 |
| CYP3A4 (1 mg/mL) | CYP3A4 (91.2 RFU/min/ (mg/mL) 2) | DBF | 485:535 | 20 | 30 | NADPH-0.33 mM MgCl2-3.3 mM NADP+-1.3 mM G6P-3.3 mM G6PDH-0.4 unit/mL | 0.25–6 |
| CYPexpress | Substrate (Concentration) | Inhibitor | Solvent (Final Concentration) | Pre-Incubation Time (min) | Inhibitor Concentrations (µM) |
|---|---|---|---|---|---|
| CYP1A2 (1 mg/mL) | Eres (2 µM) | α-Naphthoflavone | DMSO (1%) | 10 | 0.00001–1 |
| CYP2B6 (1 mg/mL) | Bzres (10 µM) | Sertraline | Methanol (0.5%) | 30 | 0.0003–30 |
| CYP2C9 (1 mg/mL) | Eres (2 µM) | Sulfaphenazole | DMSO (1%) | 10 | 0.003–300 |
| CYP2C19 (2 mg/mL) | 3OMF (2 µM) | Ticlopidine | Acetonitrile (1.67%) | 5 | 0.0003–300 |
| CYP2D6 (1 mg/mL) | Eres (2 µM) | Sertraline | Methanol (0.5%) | 30 | 0.001–100 |
| CYP3A4 (1 mg/mL) | DBF (4 µM) | CYP3cide | DMSO (0.5%) | 5 | 0.001–100 |
| CYP (Substrate) | Average Vmax (nmol/min) | Average KM (µM) | Literature KM (µM) | Ref |
|---|---|---|---|---|
| CYP1A2 (Eres) | 1.61 ± 0.2 | 0.35 ± 0.1 | 0.62 ± 0.14 1.72 ± 0.24 0.56 ± 0.13 0.22 0.26 ± 0.06 | [30,31,32,33,34] |
| CYP2B6 (Bzres) | 0.33 ± 0.1 | 40.61 ± 19.6 | 34.0 ± 10.4 1.3 ± 0.1 (human Bl cell) | [35,36] |
| CYP2C9 (Eres) | 0.43 ± 0.05 | 0.45 ± 0.0 | NA | NA |
| CYP2C19 (3OMF) | 18.2 ± 2.0 | 2.31 ± 0.8 | 1.1 ± 0.9 1.18 ± 0.06 2.32 | [37,38,39] |
| CYP2D6 (Eres) | 0.93 ± 0.2 | 1.63 ± 0.7 | NA | NA |
| CYP3A4 (DBF) | 91.20 ± 13.9 (RFU/min) | 1.77 ± 0.3 | 0.87 ± 0.12 1.37 ± 0.22 | [40,41] |
| CYP (Inhibitor) | Average IC50 (nM) | Literature IC50 nM [Reference] | Literature Substrate/Method |
|---|---|---|---|
| CYP1A2 (α-Naphthoflavone) | 12.2 ± 4.7 | 6.1 ± 0.8 [47] 8.8 ± 2.5 [48] 6 [49] 0.15 ± 0.04 [50] 9.1 [51] | Phenacetin/LC-MS/MS Fluoregenic (Eres) Fluoregenic (Eres) Acetominaphen/LC-MS/MS Phenacetin/LC-MS/MS |
| CYP2B6 (Sertraline) | 751.9 ± 557.9 | 200 ± 24 [52] 3200 ± 900 HLM [53] 1430 (1070–1900) [54] | Bupropion/LC-MS/MS Bupropion/LC-MS/MS Bupropion/LC-MS/MS |
| CYP2C9 (Sulfaphenazole) | 724.5 ± 89.8 | 600 [55] 480 ± 50 [56] 400 [57] 700 ± 80 [58] | Teinilic acid/LC-MS/MS Phenytoin/HPLC Diclofenac/LC-MS/MS Diclofenac/LC-MS/MS |
| CYP2C19 (Ticlopidine) | 781.9 ± 413.1 | 370 ± 70 [59] 203 ± 124 [60] Ki 1200 ± 500 [61] 640 [62] 141 [63] | (S)-Mephenytoin/LC-MS/MS (S)-Mephenytoin/LC-MS/MS (S)-Mephenytoin/LC-MS/MS (S)-Mephenytoin/LC-MS/MS (S)-Mephenytoin/LC-MS/MS |
| CYP2D6 (Sertraline) | 4417 ± 1292 | 4350 ± 500 [64] 2000 [65] | Primaquine/LC-MS/MS Bufuralol/LC-MS/MS |
| CYP3A4 (CYP3cide) | 126.1 ± 66.2 | 300 ± 20 [46] 690 ± 22 [46] 273 (160–443) [66] 119 (78–177) [66] | Midazolam/LC-MS/MS Testosterone/LC-MS/MS DBF/fluoregenic Midazolam/LC-MS/MS |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shriwas, P.; Revnew, A.; Roo, S.; Bender, A.; Miller, K.; Hadad, C.M.; Lane, T.R.; Ekins, S.; McElroy, C.A. Development and Characterization of pFluor50, a Fluorogenic-Based Kinetic Assay System for High-Throughput Inhibition Screening and Characterization of Time-Dependent Inhibition and Inhibition Type for Six Human CYPs. Molecules 2025, 30, 2032. https://doi.org/10.3390/molecules30092032
Shriwas P, Revnew A, Roo S, Bender A, Miller K, Hadad CM, Lane TR, Ekins S, McElroy CA. Development and Characterization of pFluor50, a Fluorogenic-Based Kinetic Assay System for High-Throughput Inhibition Screening and Characterization of Time-Dependent Inhibition and Inhibition Type for Six Human CYPs. Molecules. 2025; 30(9):2032. https://doi.org/10.3390/molecules30092032
Chicago/Turabian StyleShriwas, Pratik, Andre Revnew, Sarah Roo, Alex Bender, Kevin Miller, Christopher M. Hadad, Thomas R. Lane, Sean Ekins, and Craig A. McElroy. 2025. "Development and Characterization of pFluor50, a Fluorogenic-Based Kinetic Assay System for High-Throughput Inhibition Screening and Characterization of Time-Dependent Inhibition and Inhibition Type for Six Human CYPs" Molecules 30, no. 9: 2032. https://doi.org/10.3390/molecules30092032
APA StyleShriwas, P., Revnew, A., Roo, S., Bender, A., Miller, K., Hadad, C. M., Lane, T. R., Ekins, S., & McElroy, C. A. (2025). Development and Characterization of pFluor50, a Fluorogenic-Based Kinetic Assay System for High-Throughput Inhibition Screening and Characterization of Time-Dependent Inhibition and Inhibition Type for Six Human CYPs. Molecules, 30(9), 2032. https://doi.org/10.3390/molecules30092032