
Triethylamine-Capped Calcium Phosphate Oligomers/Polyacrylamide Synergistically Reinforced α-Hemihydrate Gypsum Composites: A Mechanistic Study on Mechanical Strengthening via Organic/Inorganic Interpenetrating Networks

 , , , , ,
, , , , ,
Abstract
1. Introduction
2. Results and Discussion
2.1. Influence of CPO/PAM Precursor Addition on the Properties of CPO/PAM/-HHG Composites
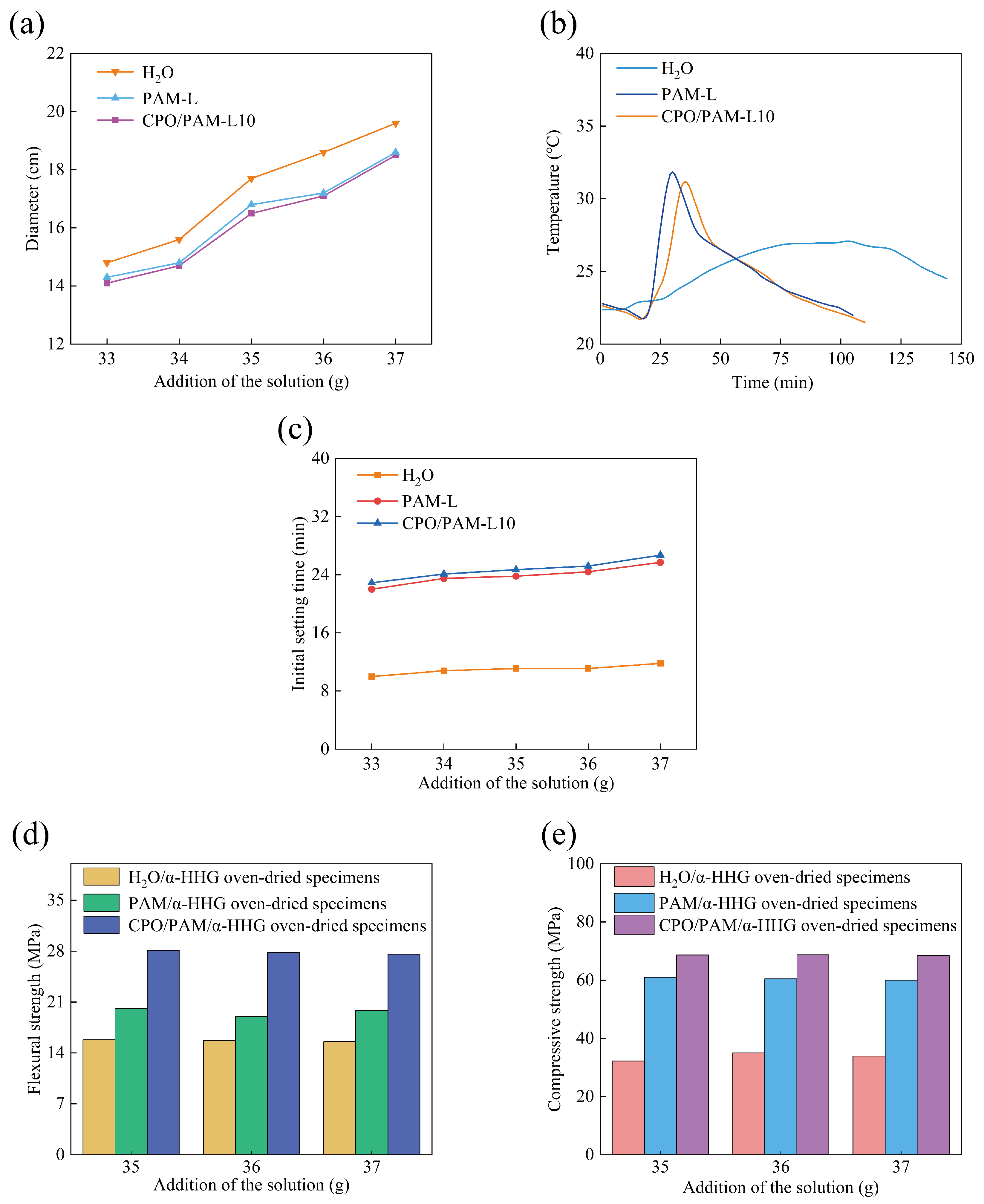
2.1.1. Influence on the Flowability of CPO/PAM/-HHG Composite Slurries
2.1.2. Influence on the Hydration–Hardening Temperature of CPO/PAM/-HHG Composites
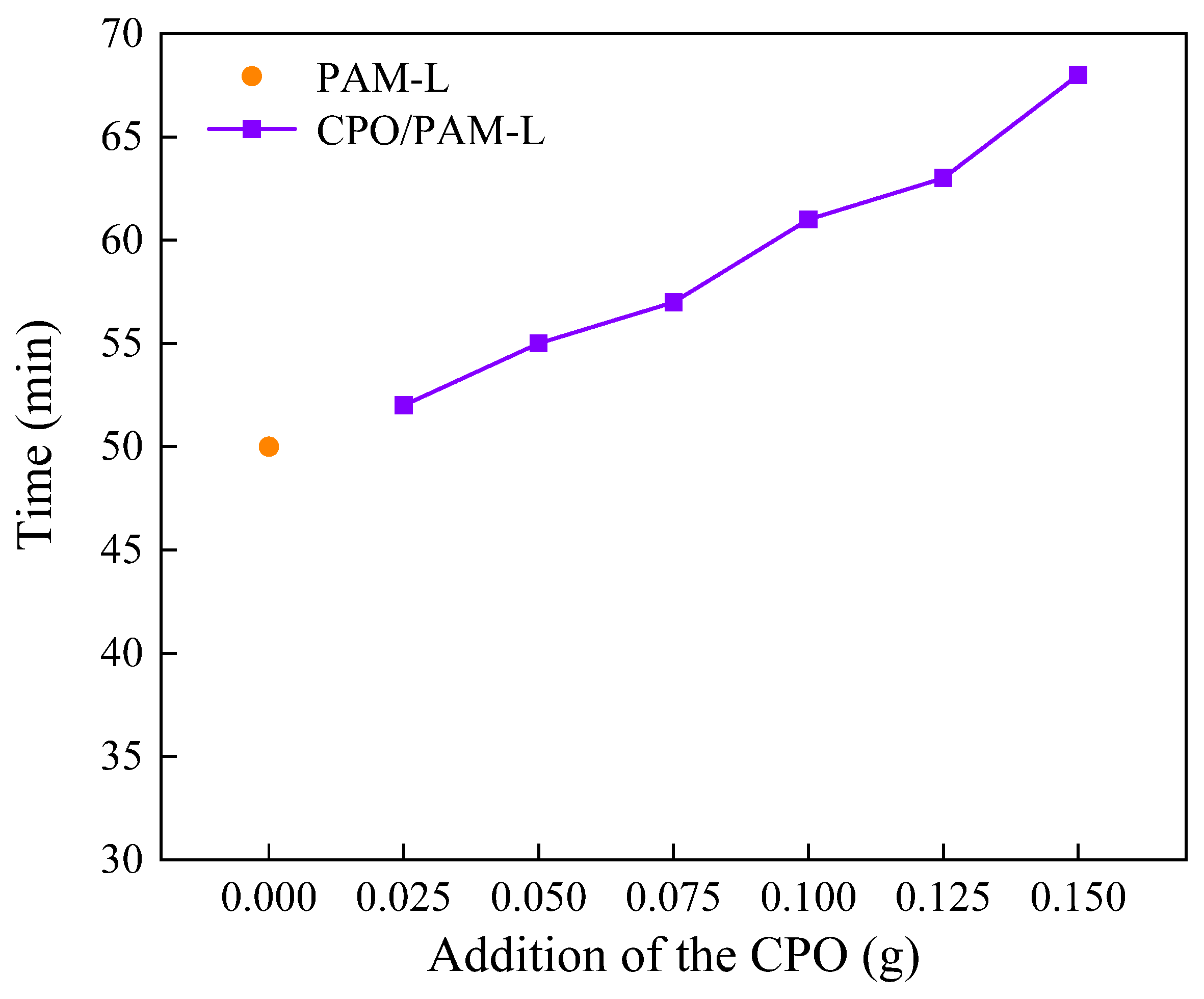
2.1.3. Influence on the Initial Setting Time of CPO/PAM/-HHG Composites
2.1.4. Influence on the Mechanical Properties of CPO/PAM/-HHG Composites
2.2. Influence of CPO’s Addition in Precursor Solution on the Properties of CPO/PAM/-HHG Composites
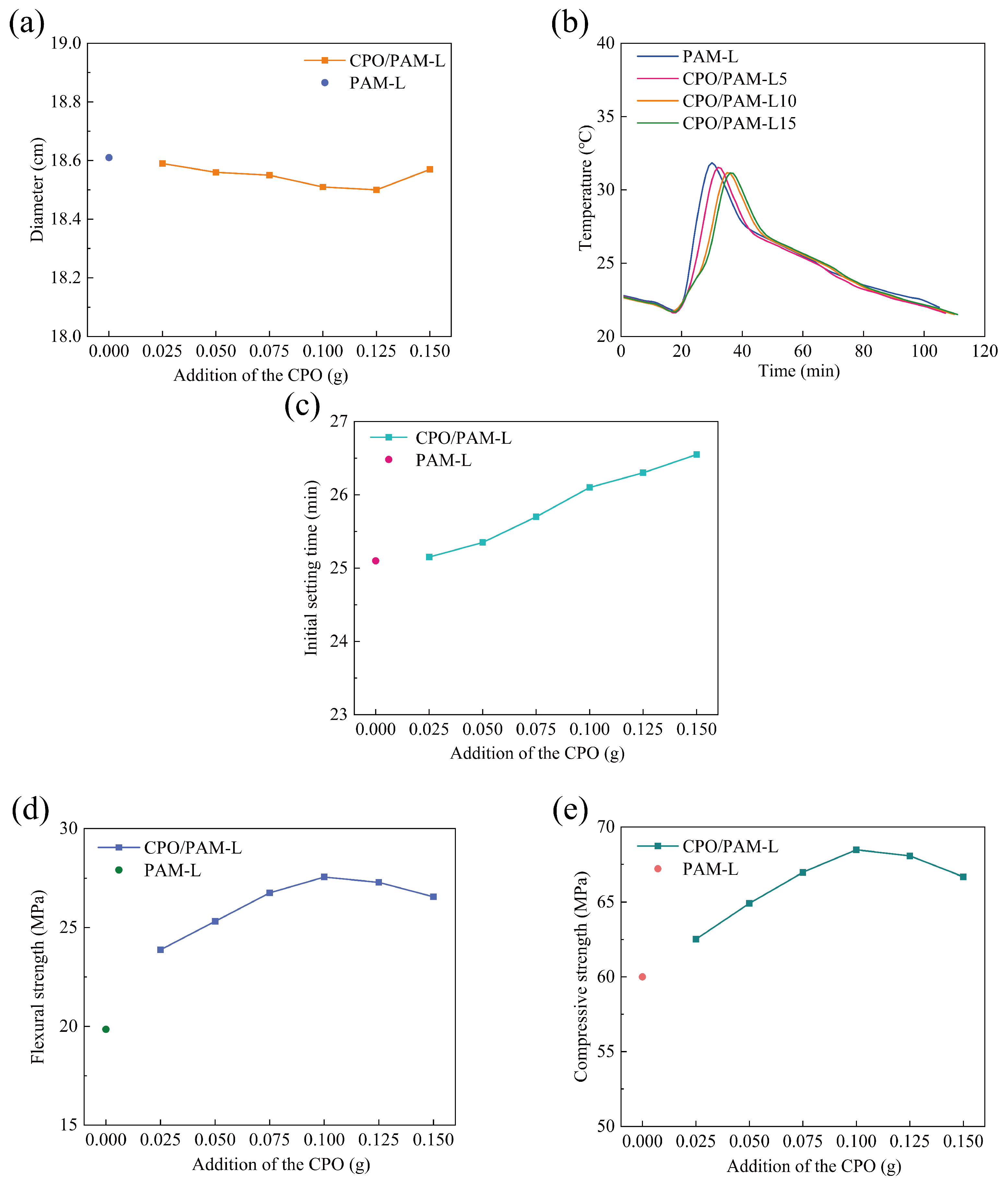
2.2.1. Influence on the Flowability of CPO/PAM/-HHG Composite Slurries
2.2.2. Influence on the Hydration–Hardening Temperature of CPO/PAM/-HHG Composites
2.2.3. Influence on the Initial Setting Time of CPO/PAM/-HHG Composites
2.2.4. Influence on the Mechanical Properties of CPO/PAM/-HHG Composites
2.3. Phase Analysis of CPO/PAM/-HHG Composite Materials
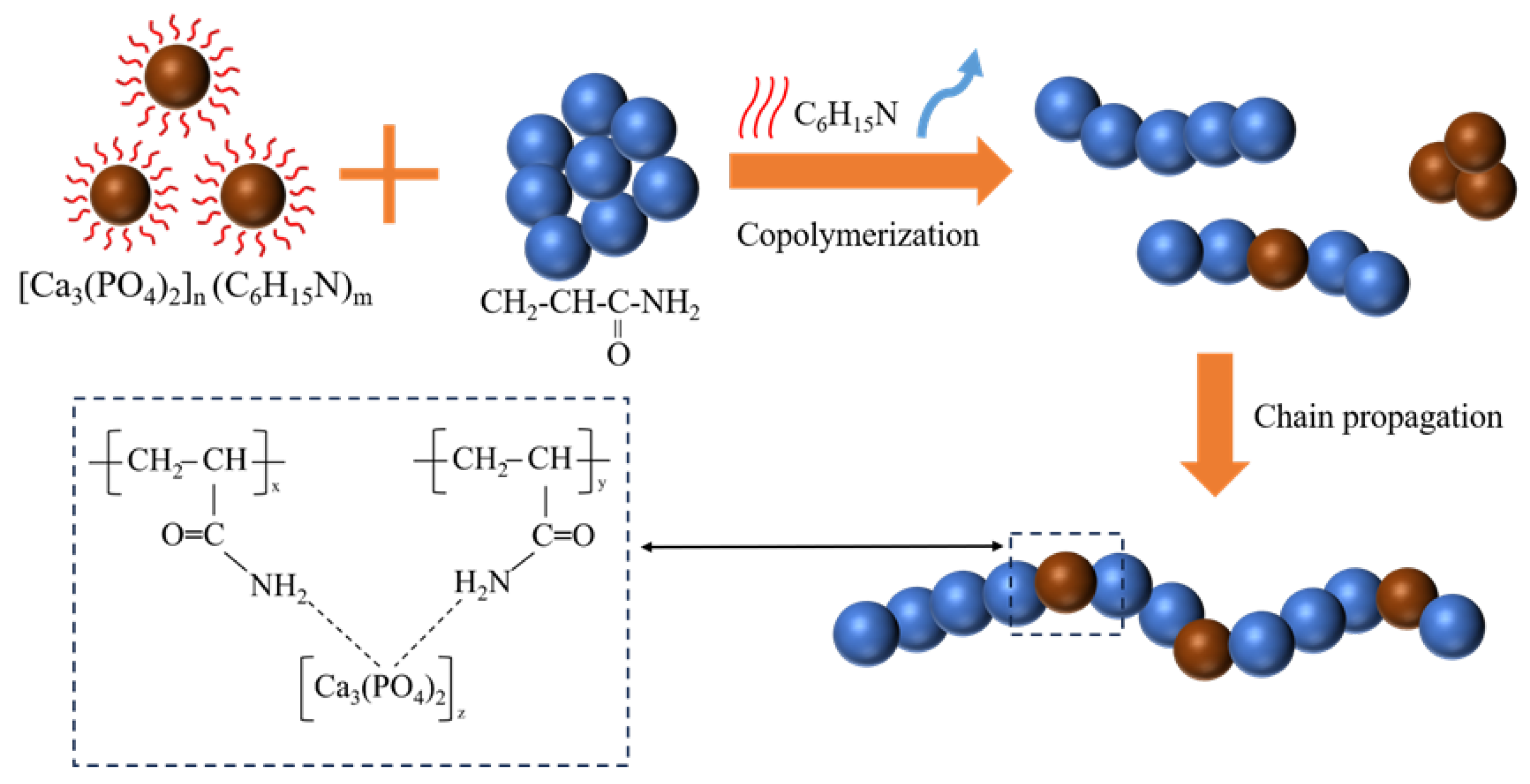
2.4. Fracture Analysis of CPO/PAM/-HHG Composite Materials and Microstructural Analysis of TEA-Capped CPO
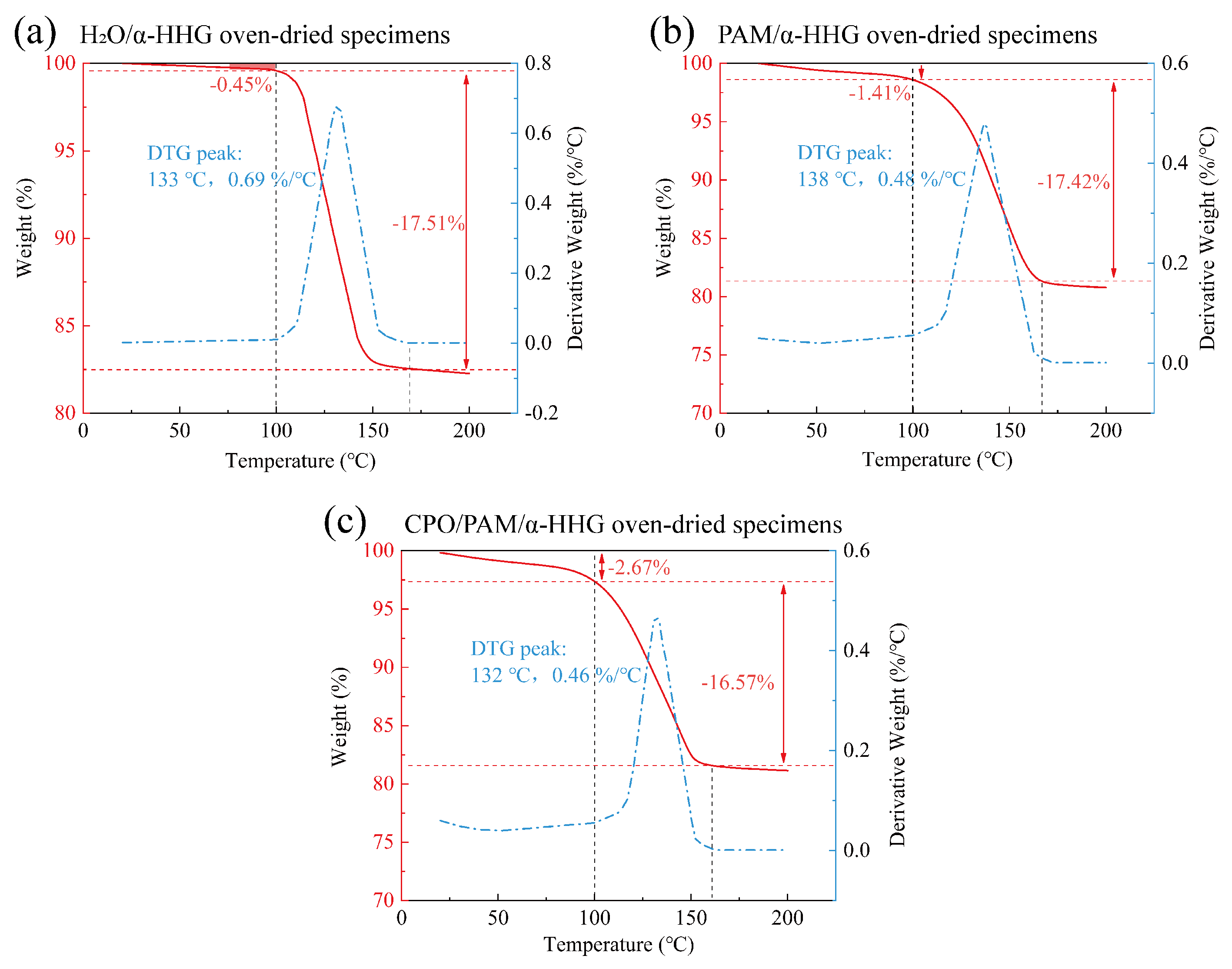
2.5. Thermogravimetric Analysis of CPO/PAM/-HHG Composite Materials
2.6. Water Resistance Analysis
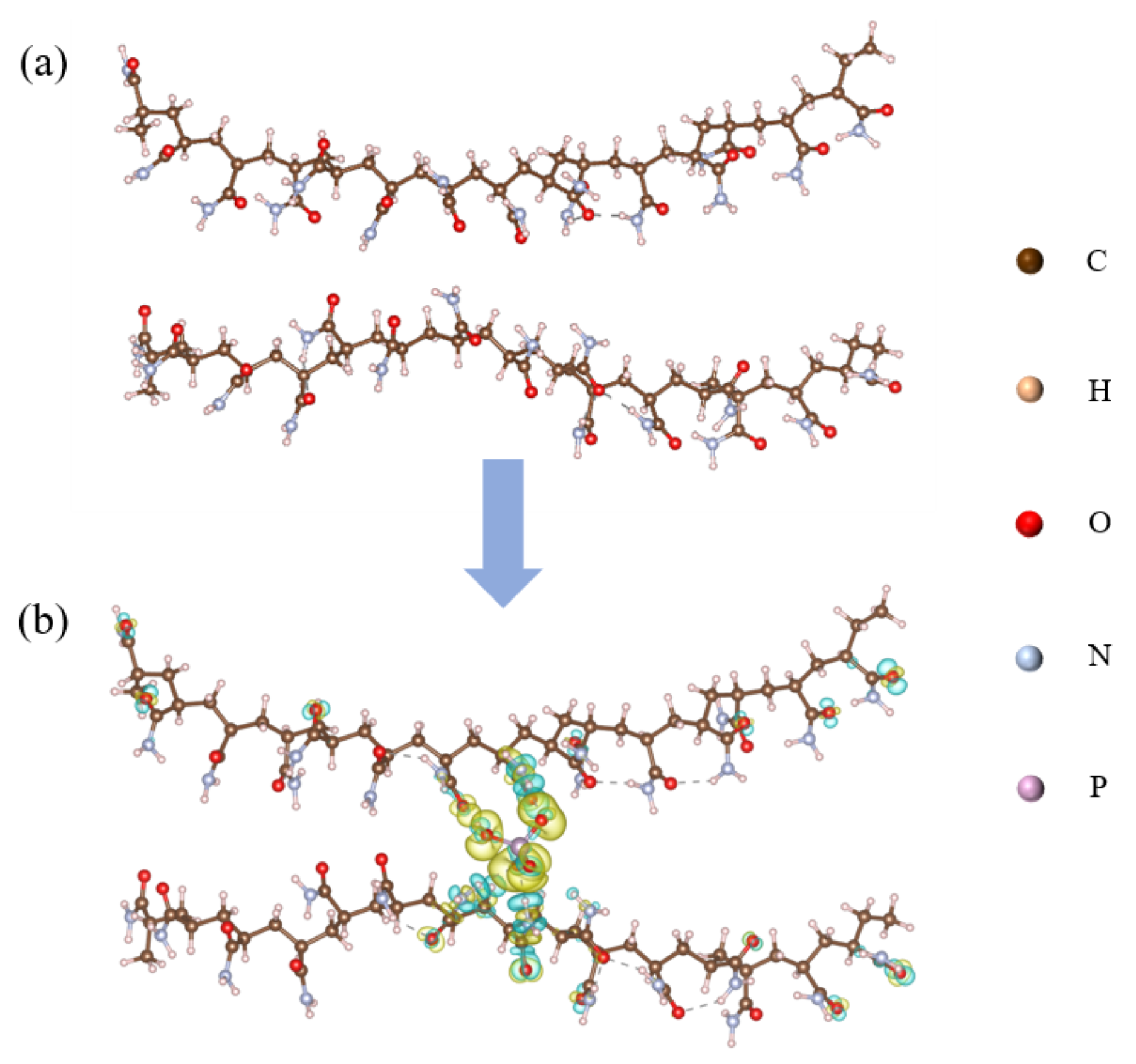
2.7. Theoretical Investigation of Adsorption Behaviour Between PAM and CPO
3. Materials and Methods
3.1. Materials
3.2. Preparation of TEA-Capped CPO
3.3. Preparation of CPO/PAM Precursor Solutions
3.4. Evaluation of the Drawable Time of CPO/PAM Composite Hydrogels
3.5. Preparation and Mechanical Property Testing of CPO/PAM/-HHG Composites
3.6. Evaluation of Physical Properties of CPO/PAM/-HHG Composite Slurry
3.7. Structural Analysis of CPO/PAM/-HHG Composites
3.8. Thermogravimetric Analysis
3.9. Water Resistance Analysis
3.10. Computational Details
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Ye, Y.; Huang, Q.; Li, X. Effect of Fiber Loading on Mechanical and Flame-Retardant Properties of Poplar-Fiber-Reinforced Gypsum Composites. Molecules 2024, 29, 2674. [Google Scholar] [CrossRef] [PubMed]
- Guo, G.; Gao, G.; Jiang, W.; Wang, X.; Pei, M.; Wang, L. Synthesis of Triblock Polycarboxylate Superplasticizers with Well-Defined Structure and Its Dispersing Performance in β-Hemihydrate Gypsum. Molecules 2023, 28, 513. [Google Scholar] [CrossRef] [PubMed]
- Xie, S.; Ji, Z.; Yang, Y.; Hou, G.; Wang, J. Electromagnetic wave absorption enhancement of carbon black/gypsum based composites filled with expanded perlite. Compos. Part B Eng. 2016, 106, 10–19. [Google Scholar] [CrossRef]
- Xie, Z.; Liu, X.; Zhang, Z.; Wei, C.; Gu, J. Application of the Industrial Byproduct Gypsum in Building Materials: A Review. Materials 2024, 17, 1837. [Google Scholar] [CrossRef]
- Ravenhill, E.R.; Kirkman, P.M.; Unwin, P.R. Microscopic Studies of Calcium Sulfate Crystallization and Transformation at Aqueous-Organic Interfaces. Cryst. Growth Des. 2016, 16, 5887–5895. [Google Scholar] [CrossRef]
- Vidales-Barriguete, A.; Santa-Cruz-Astorqui, J.; Pina-Ramirez, C.; Kosior-Kazberuk, M.; Kalinowska-Wichrowska, K.; Atanes-Sanchez, E. Study of the Mechanical and Physical Behavior of Gypsum Boards with Plastic Cable Waste Aggregates and Their Application to Construction Panels. Materials 2021, 14, 2255. [Google Scholar] [CrossRef]
- Tang, Y.; Gao, J.; Liu, C.; Chen, X.; Zhao, Y. Dehydration Pathways of Gypsum and the Rehydration Mechanism of Soluble Anhydrite γ-CaSO4. ACS Omega 2019, 4, 7636–7642. [Google Scholar] [CrossRef]
- Xie, Y.; Yang, L.; Wang, C.; Feng, X.; Tang, Z.; Liu, Z. Production and properties of the polyvinyl alcohol modified macro-defect-free alpha-hemihydrate gypsum composite. Constr. Build. Mater. 2023, 375, 130721. [Google Scholar] [CrossRef]
- Triantafyllou, E.; Karydis-Messinis, A.; Moschovas, D.; Kyriakaki, C.; Vasilopoulos, K.C.; Giannakas, A.E.; Karakassides, M.A.; Avgeropoulos, A.; Zafeiropoulos, N.E.; Salmas, C.E. Microwave-Assisted Extraction of Cellulose from Aloe Vera Plant Residue and Preparation of Cellulose Nanocrystal-Poly(vinyl alcohol) Hydrogels. Molecules 2024, 29, 6012. [Google Scholar] [CrossRef]
- Schexnailder, P.; Schmidt, G. Nanocomposite polymer hydrogels. Colloid Polym. Sci. 2009, 287, 1–11. [Google Scholar]
- El Sayed, M.M. Production of Polymer Hydrogel Composites and Their Applications. J. Polym. Environ. 2023, 31, 2855–2879. [Google Scholar] [CrossRef]
- Song, X.; Lu, G.; Wang, J.; Zheng, J.; Sui, S.; Li, Q.; Zhang, Y. Molecular Dynamics-Assisted Design of High Temperature-Resistant Polyacrylamide/Poloxamer Interpenetrating Network Hydrogels. Molecules 2022, 27, 5326. [Google Scholar] [CrossRef] [PubMed]
- Ho, T.C.; Chang, C.C.; Chan, H.P.; Chung, T.W.; Shu, C.W.; Chuang, K.P.; Duh, T.H.; Yang, M.H.; Tyan, Y.C. Hydrogels: Properties and Applications in Biomedicine. Molecules 2022, 27, 2902. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.K.; Lai, C.W.; Johan, M.R.; Gan, S.S.; Chua, W.W. Recent advances of modified polyacrylamide in drilling technology. J. Pet. Sci. Eng. 2022, 215, 110566. [Google Scholar] [CrossRef]
- Zhao, Y.; Zhou, J.; Xu, X.; Liu, W.; Zhang, J.; Fan, M.; Wang, J. Synthesis and characterization of a series of modified polyacrylamide. Colloid Polym. Sci. 2009, 287, 237–241. [Google Scholar]
- Ma, T.; Wu, Y.; Liu, N.; Wu, Y. Hydrolyzed polyacrylamide modified diatomite waste as a novel adsorbent for organic dye removal: Adsorption performance and mechanism studies. Polyhedron 2020, 175, 114227. [Google Scholar] [CrossRef]
- Kaşgöz, H.; Özgümüş, S.; Orbay, M. Modified polyacrylamide hydrogels and their application in removal of heavy metal ions. Polymer 2003, 44, 1785–1793. [Google Scholar]
- Zheng, X.; Zheng, H.; Xiong, Z.; Zhao, R.; Liu, Y.; Zhao, C.; Zheng, C. Novel anionic polyacrylamide-modify-chitosan magnetic composite nanoparticles with excellent adsorption capacity for cationic dyes and pH-independent adsorption capability for metal ions. Chem. Eng. J. 2020, 392, 123706. [Google Scholar] [CrossRef]
- Yu, Y.; Mu, Z.; Jin, B.; Liu, Z.; Tang, R. Organic-Inorganic Copolymerization for a Homogenous Composite without an Interphase Boundary. Angew. Chem. Int. Ed. 2020, 59, 2071–2075. [Google Scholar] [CrossRef]
- Sophia, M.; Sakthieswaran, N. Synergistic effect of mineral admixture and bio-carbonate fillers on the physico-mechanical properties of gypsum plaster. Constr. Build. Mater. 2019, 204, 419–439. [Google Scholar] [CrossRef]
- Dima, C.; Badanoiu, A.; Cirstea, S.; Nicoara, A.I.; Stoleriu, S. Lightweight Gypsum Materials with Potential Use for Thermal Insulations. Materials 2020, 13, 5454. [Google Scholar] [CrossRef] [PubMed]
- Zha, F.; Qiao, B.; Kang, B.; Xu, L.; Chu, C.; Yang, C. Engineering properties of expansive soil stabilized by physically amended titanium gypsum. Constr. Build. Mater. 2021, 303, 124456. [Google Scholar] [CrossRef]
- Ding, X.; Wang, S.; Dai, R.; Chen, H.; Shan, Z. Hydrogel beads derived from chrome leather scraps for the preparation of lightweight gypsum. Environ. Technol. Innov. 2022, 25, 102224. [Google Scholar] [CrossRef]
- Wu, Q.; Zhu, Z.; Li, S.; Wang, S.; Chen, B. Effect of polyacrylic ester emulsion on mechanical properties of macro-defect free desulphurization gypsum plaster. Constr. Build. Mater. 2017, 153, 656–662. [Google Scholar] [CrossRef]
- Jia, C.; Wu, L.; Chen, Q.; Ke, P.; De Yoreo, J.J.; Guan, B. Structural evolution of amorphous calcium sulfate nanoparticles into crystalline gypsum phase. CrystEngComm 2020, 22, 6805–6810. [Google Scholar] [CrossRef]
- Charai, M.; Mghazli, M.O.; Channouf, S.; El Hammouti, A.; Jagadesh, P.; Moga, L.; Mezrhab, A. Lightweight waste-based gypsum composites for building temperature and moisture control using coal fly ash and plant fibers. Constr. Build. Mater. 2023, 393, 132092. [Google Scholar] [CrossRef]
- Pedrajas, D.L.; Franco, M.C.; Saenz, I.G.; Mellado, F.J.R.; Romero, J.F.R.; Simon, A.M.B. Polystyrene nanoparticles slurry as an additive for developing insulating and waterproof gypsum composites. Appl. Therm. Eng. 2022, 217, 119271. [Google Scholar] [CrossRef]
- Mroz, P.; Mucha, M. Hydroxyethyl methyl cellulose as a modifier of gypsum properties. J. Therm. Anal. Calorim. 2018, 134, 1083–1089. [Google Scholar] [CrossRef]
- Thompson, B.R.; Horozov, T.S.; Stoyanov, S.D.; Paunov, V.N. Hierarchically porous composites fabricated by hydrogel templating and viscous trapping techniques. Mater. Des. 2018, 137, 384–393. [Google Scholar] [CrossRef]
- Chen, Y.; Mi, Z.; Yang, J.; Zheng, X.; Wang, H.; Record, M.C.; Boulet, P.; Wang, J.; Albina, J.M.; Huang, Y. Synthesis and Characterisation of Hemihydrate Gypsum-Polyacrylamide Composite: A Novel Inorganic/Organic Cementitious Material. Materials 2024, 17, 1510. [Google Scholar] [CrossRef]
- Zhao, H.; Hu, G.H.; Ye, G.B.; Ren, X.M.; Zhang, Q.C.; Jiang, T. Effects of superplasticisers on hydration process, structure and properties of α-hemihydrate calcium sulfate. Adv. Cem. Res. 2018, 30, 37–44. [Google Scholar] [CrossRef]
- Guan, B.; Ye, Q.; Zhang, J.; Lou, W.; Wu, Z. Interaction between α-calcium sulfate hemihydrate and superplasticizer from the point of adsorption characteristics, hydration and hardening process. Cem. Concr. Res. 2010, 40, 253–259. [Google Scholar] [CrossRef]
- Beaugnon, F.; Preturlan, J.G.D.; Fusseis, F.; Gouillart, E.; Quiligotti, S.; Wallez, G. From atom level to macroscopic scale: Structural mechanism of gypsum dehydration. Solid State Sci. 2022, 126, 106845. [Google Scholar] [CrossRef]
- Kyere-Yeboah, K.; Qiao, X.c. Application of cold plasma technology for the simultaneous degradation and viscosity removal of polyacrylamide and its copolymers from contaminated wastewater. J. Environ. Manag. 2024, 370, 122861. [Google Scholar] [CrossRef]
- Gong, J.P.; Katsuyama, Y.; Kurokawa, T.; Osada, Y. Double-network hydrogels with extremely high mechanical strength. Adv. Mater. 2003, 15, 1155–1158. [Google Scholar] [CrossRef]
- GB/T (17669.3-1999); Gypsum Plasters—Determination of Mechanical Properties. National Standard of the People’s Republic of China: Beijing, China, 1999.
- GB/T (17669.5-1999); Gypsum Plasters—Determination of Physical Properties of Powder. National Standard of the People’s Republic of China: Beijing, China, 1999.
- Kresse, G.; Hafner, J. Ab Initio Molecular Dynamics for Liquid Metals. Phys. Rev. B 1993, 47, 558–561. [Google Scholar] [CrossRef]
- Kresse, G.; Furthmüller, J. Efficiency of Ab-Initio Total Energy Calculations for Metals and Semiconductors Using a Plane-Wave Basis Set. Comput. Mater. Sci. 1996, 6, 15–50. [Google Scholar] [CrossRef]
- Dudarev, S.L.; Botton, G.A.; Savrasov, S.Y.; Humphreys, C.J.; Sutton, A.P. Electron-Energy-Loss Spectra and the Structural Stability of Nickel Oxide: An LSDA+U Study. Phys. Rev. B 1998, 57, 1505–1509. [Google Scholar] [CrossRef]
- Perdew, J.P.; Burke, K.; Ernzerhof, M. Generalized Gradient Approximation Made Simple. Phys. Rev. Lett. 1996, 77, 3865–3868. [Google Scholar] [CrossRef]
- Momma, K.; Izumi, F. VESTA3 for three-dimensional visualization of crystal, volumetric and morphology data. J. Appl. Crystallogr. 2011, 44, 1272–1276. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| H2O/-HHG | PAM/-HHG | CPO/PAM/-HHG-L10 | |
|---|---|---|---|
| Softening Coefficient | 0.44 | 0.21 | 0.20 |
| Wet Compressive Strength (MPa) | 15.46 | 12.55 | 14.03 |
| Precursor Solutions | AM (g) | APS (g) | MBA (g) | TEMED (mL) | Deionised Water (mL) | CPO (g) |
|---|---|---|---|---|---|---|
| PAM-L | 8.8850 | 0.0285 | 0.0193 | 0.0645 | 50 | 0 |
| CPO/PAM-L2.5 | 8.8850 | 0.0285 | 0.0193 | 0.0645 | 50 | 0.0250 |
| CPO/PAM-L5 | 8.8850 | 0.0285 | 0.0193 | 0.0645 | 50 | 0.0500 |
| CPO/PAM-L7.5 | 8.8850 | 0.0285 | 0.0193 | 0.0645 | 50 | 0.0750 |
| CPO/PAM-L10 | 8.8850 | 0.0285 | 0.0193 | 0.0645 | 50 | 0.1000 |
| CPO/PAM-L12.5 | 8.8850 | 0.0285 | 0.0193 | 0.0645 | 50 | 0.1250 |
| CPO/PAM-L15 | 8.8850 | 0.0285 | 0.0193 | 0.0645 | 50 | 0.1500 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, Y.; Chen, L.; Li, H.; Zhang, B.; Record, M.-C.; Boulet, P.; Wang, J.; Albina, J.-M.; Yang, Y.; Ma, W. Triethylamine-Capped Calcium Phosphate Oligomers/Polyacrylamide Synergistically Reinforced α-Hemihydrate Gypsum Composites: A Mechanistic Study on Mechanical Strengthening via Organic/Inorganic Interpenetrating Networks. Molecules 2025, 30, 2002. https://doi.org/10.3390/molecules30092002
Chen Y, Chen L, Li H, Zhang B, Record M-C, Boulet P, Wang J, Albina J-M, Yang Y, Ma W. Triethylamine-Capped Calcium Phosphate Oligomers/Polyacrylamide Synergistically Reinforced α-Hemihydrate Gypsum Composites: A Mechanistic Study on Mechanical Strengthening via Organic/Inorganic Interpenetrating Networks. Molecules. 2025; 30(9):2002. https://doi.org/10.3390/molecules30092002
Chicago/Turabian StyleChen, Yuan, Li Chen, Hao Li, Bin Zhang, Marie-Christine Record, Pascal Boulet, Juan Wang, Jan-Michael Albina, Yi Yang, and Weiliang Ma. 2025. "Triethylamine-Capped Calcium Phosphate Oligomers/Polyacrylamide Synergistically Reinforced α-Hemihydrate Gypsum Composites: A Mechanistic Study on Mechanical Strengthening via Organic/Inorganic Interpenetrating Networks" Molecules 30, no. 9: 2002. https://doi.org/10.3390/molecules30092002
APA StyleChen, Y., Chen, L., Li, H., Zhang, B., Record, M.-C., Boulet, P., Wang, J., Albina, J.-M., Yang, Y., & Ma, W. (2025). Triethylamine-Capped Calcium Phosphate Oligomers/Polyacrylamide Synergistically Reinforced α-Hemihydrate Gypsum Composites: A Mechanistic Study on Mechanical Strengthening via Organic/Inorganic Interpenetrating Networks. Molecules, 30(9), 2002. https://doi.org/10.3390/molecules30092002