
Investigating the Physical Adsorption of DCPD/Furfural and H2 Adsorption–Dissociation Behaviors in RE-MOFs

 ,
, 
Abstract
1. Introduction
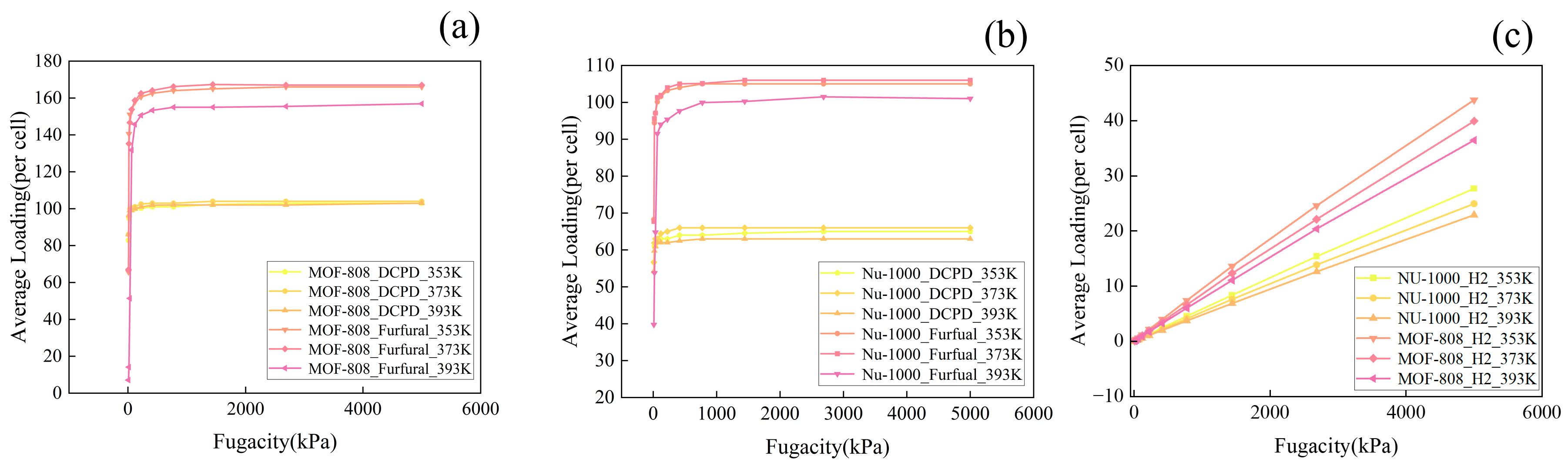
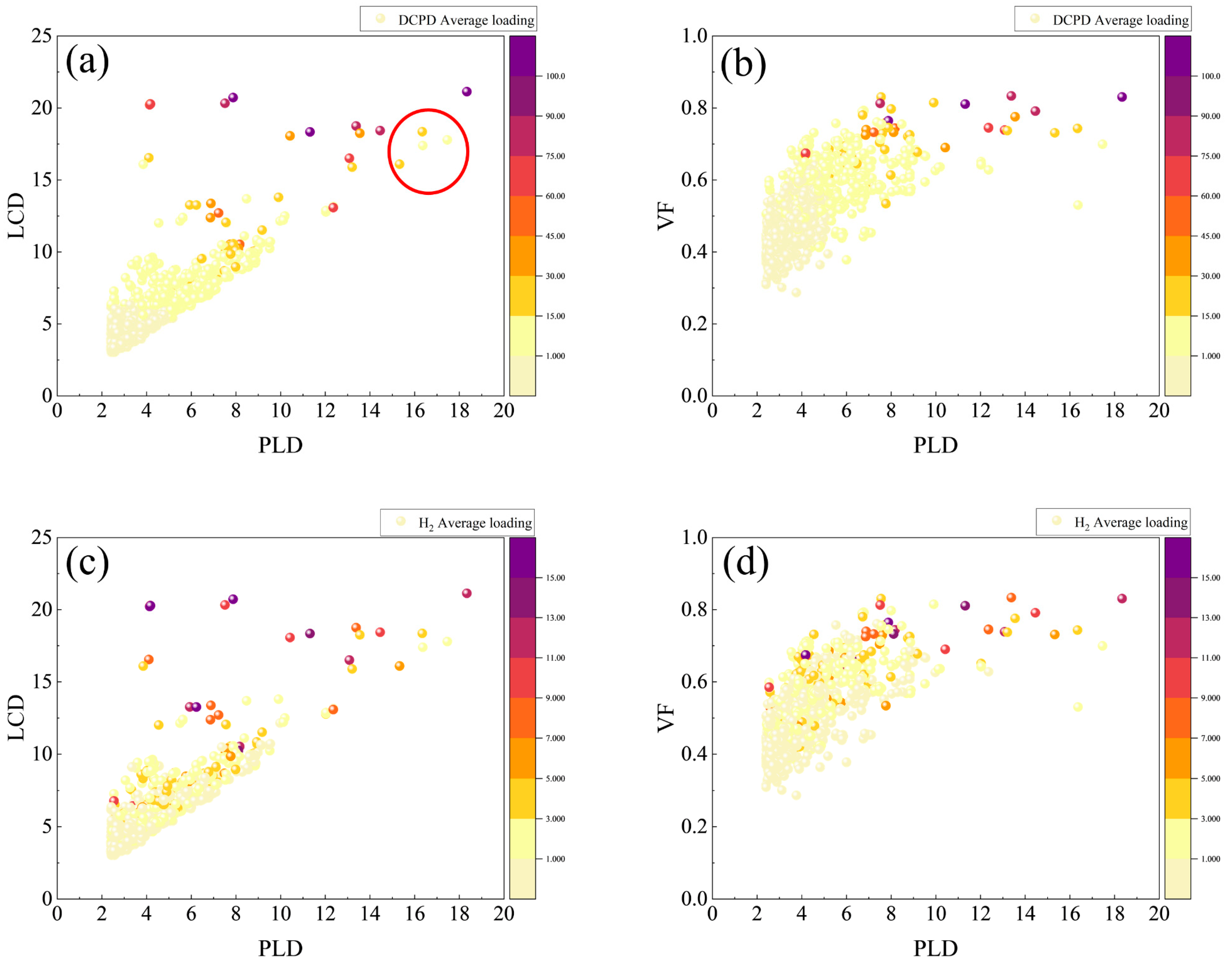
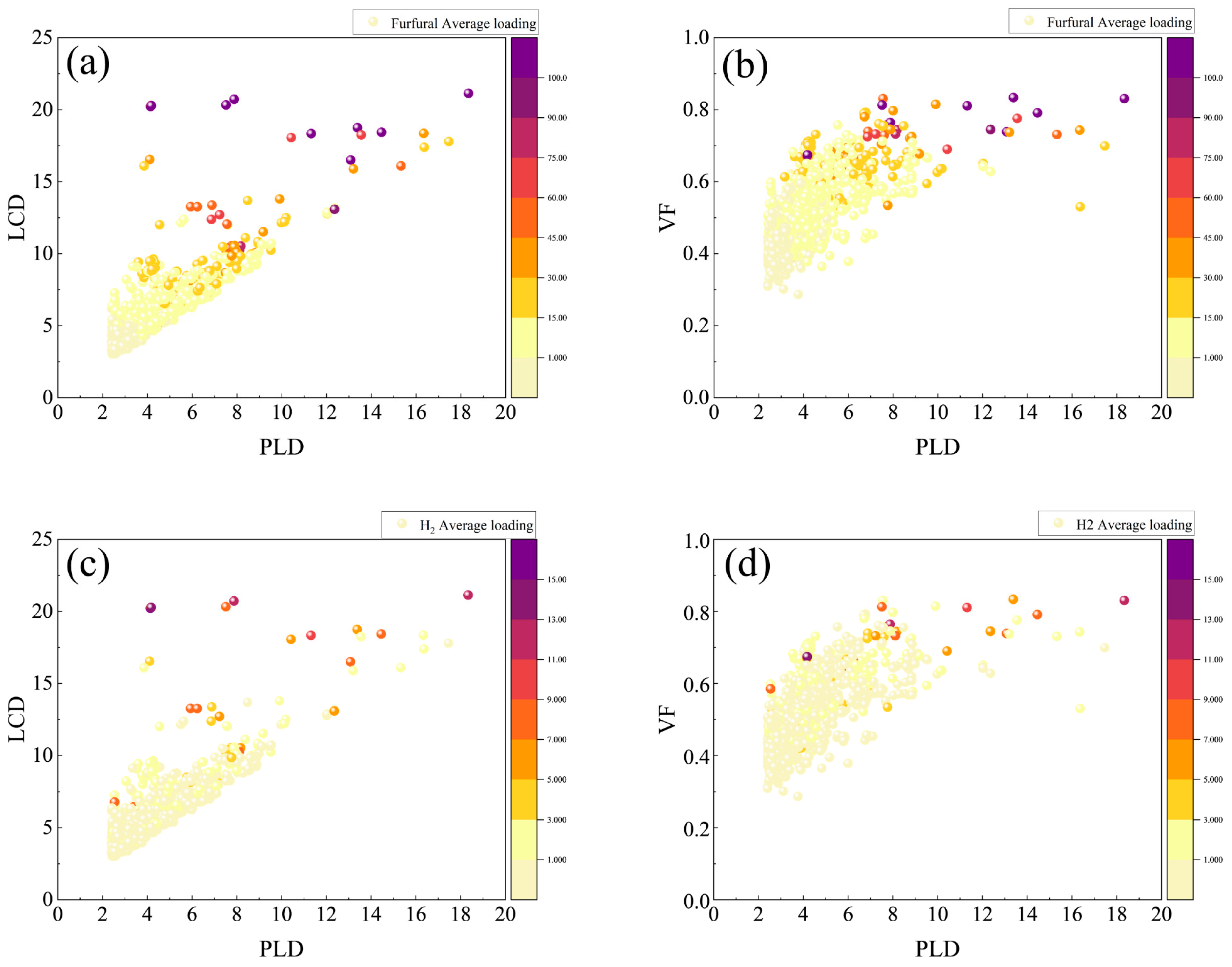
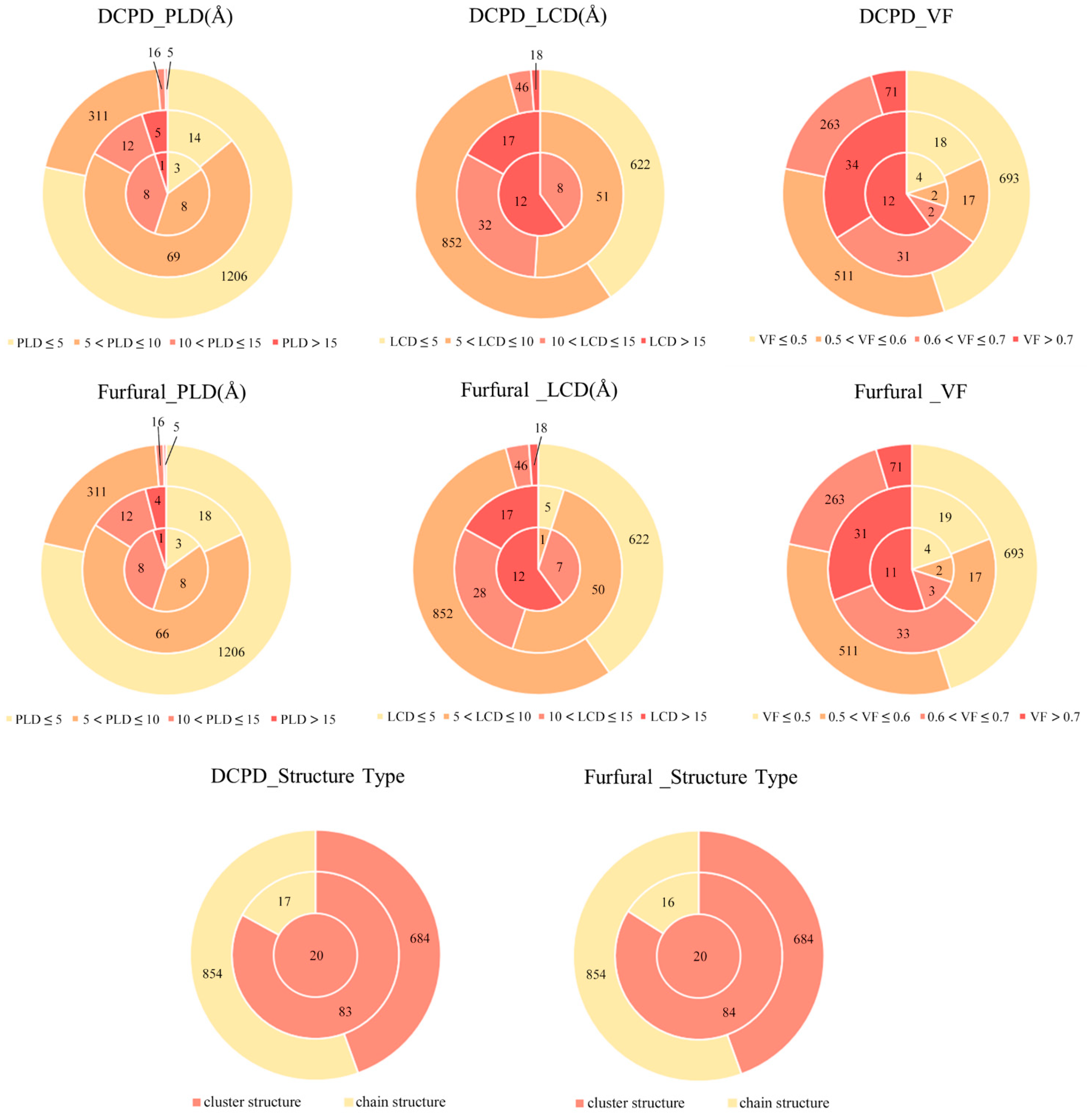
2. Results and Discussions
3. Materials and Methods
3.1. Dynamics
3.2. Porosity Parameter
3.3. DFT Study
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
References
- Bai, Z.; Chen, X.; Liang, C. Hydrotalcite-Based Bimetallic PdNi Catalysts with High Sulfur Tolerance for the Hydrogenation of Dicyclopentadiene Resin. Ind. Eng. Chem. Res. 2023, 62, 6069–6080. [Google Scholar] [CrossRef]
- Vereshchagina, N.V.; Antonova, T.N.; Il’in, A.A.; Chirkova, Z.V. Features of Dicyclopentene Formation during Hydrogenation of Dicyclopentadiene. Pet. Chem. 2016, 56, 38–43. [Google Scholar] [CrossRef]
- Behr, A.; Manz, V.; Lux, A.; Ernst, A. Highly Selective Mono-Hydrogenation of Dicyclopentadiene with Pd-Nanoparticles. Catal. Lett. 2013, 143, 241–245. [Google Scholar] [CrossRef]
- Tamizhdurai, P.; Ramesh, A.; Krishnan, P.S.; Mangesh, V.L.; Umasankar, S.; Narayanan, S.; Ragupathi, C.; Shanthi, K. Hydrogenation of Dicyclopentadiene into Endo-Tetrahydrodicyclopentadie over Supported Different Metal Catalysts. Microporous Mesoporous Mater. 2019, 290, 109678. [Google Scholar] [CrossRef]
- Mironenko, R.M.; Belskaya, O.B.; Likholobov, V.A. Aqueous-Phase Hydrogenation of Furfural in the Presence of Supported Metal Catalysts of Different Types. A Review. Dokl. Phys. Chem. 2023, 509, 33–50. [Google Scholar] [CrossRef]
- Byun, M.Y.; Park, D.-W.; Lee, M.S. Effect of Oxide Supports on the Activity of Pd Based Catalysts for Furfural Hydrogenation. Catalysts 2020, 10, 837. [Google Scholar] [CrossRef]
- Ban, T.; Yu, L.; Li, R.; Wang, C.; Lin, J.; Chen, J.; Xu, X.; Wang, Z.; Gao, H.; Wang, G. Microenvironment Modulation for Electronic Structure of Atomically Dispersed Ir Species in Metal-Organic Frameworks Toward Boosting Catalytic DCPD Hydrogenation Performance. Carbon Neutralization 2025, 4, e70004. [Google Scholar] [CrossRef]
- Xu, D.; Zhang, Q.; Huo, X.; Wang, Y.; Yang, M. Advances in Data-assisted High-throughput Computations for Material Design. Mater. Genome Eng. Adv. 2023, 1, e11. [Google Scholar] [CrossRef]
- Liu, Z.; Wang, C.; Yang, P.; Wang, W.; Gao, H.; An, G.; Liu, S.; Chen, J.; Guo, T.; Xu, X.; et al. Microenvironment and Electronic State Modulation of Pd Nanoparticles within MOFs for Enhancing Low-Temperature Activity towards DCPD Hydrogenation. Chin. J. Catal. 2024, 64, 112–122. [Google Scholar] [CrossRef]
- Khalil, I.E.; Fonseca, J.; Reithofer, M.R.; Eder, T.; Chin, J.M. Tackling Orientation of Metal-Organic Frameworks: The Quest to Enhance MOF Performance. Coord. Chem. Rev. 2023, 481, 215043. [Google Scholar] [CrossRef]
- Gulbalkan, H.C.; Haslak, Z.P.; Altintas, C.; Uzun, A.; Keskin, S. Assessing CH4/N2 Separation Potential of MOFs, COFs, IL/MOF, MOF/Polymer, and COF/Polymer Composites. Chem. Eng. J. 2022, 428, 131239. [Google Scholar] [CrossRef]
- Lu, X.; Song, C.; Qi, X.; Li, D.; Lin, L. Confinement Effects in Well-Defined Metal-Organic Frameworks for Selective CO2 Hydrogenation: A Review. Int. J. Mol. Sci. 2023, 24, 4228. [Google Scholar] [CrossRef]
- Zhao, M.; Yuan, K.; Wang, Y.; Li, G.; Guo, J.; Gu, L.; Hu, W.; Zhao, H.; Tang, Z. Metal-Organic Frameworks as Selectivity Regulators for Hydrogenation Reactions. Nature 2016, 539, 76–80. [Google Scholar] [CrossRef] [PubMed]
- Xie, J. Prospects of Materials Genome Engineering Frontiers. Mater. Genome Eng. Adv. 2023, 1, e17. [Google Scholar] [CrossRef]
- Wang, Y.; Sun, H.; Yang, Z.; Zhu, Y.; Xia, Y. Bismuth-based Metal-organic Frameworks and Derivatives for Photocatalytic Applications in Energy and Environment: Advances and Challenges. Carbon Neutralization 2024, 3, 737–767. [Google Scholar] [CrossRef]
- Li, R.; Wang, L.; Zhou, P.; Lin, J.; Liu, Z.; Chen, J.; Zhao, D.; Huang, X.; Tao, Z.; Wang, G. Electronic State, Abundance and Microenvironment Modulation of Ru Nanoclusters within Hierarchically Porous UiO-66(Ce) for Efficient Hydrogenation of Dicyclopentadiene. Chin. J. Catal. 2024, 56, 150–165. [Google Scholar] [CrossRef]
- Freund, R.; Zaremba, O.; Arnauts, G.; Ameloot, R.; Skorupskii, G.; Dincă, M.; Bavykina, A.; Gascon, J.; Ejsmont, A.; Goscianska, J.; et al. The Current Status of MOF and COF Applications. Angew. Chem. Int. Ed. 2021, 60, 23975–24001. [Google Scholar] [CrossRef]
- Zhao, D.; Hu, F.; Li, R.; Xu, X.; Wang, Z.; Xi, Z.; Tong, J.; Huang, X.; Wang, G. Reverse Modulation of Defect Density of Ce-MOF-808 by Solvent-Assisted Ligand Exchange for Dicyclopentadiene Hydrogenation. Sci. China Chem. 2025, 1–13. [Google Scholar] [CrossRef]
- Orella, C. Work from the Organic Reactions Catalysis Society Meeting 2018. Org. Process Res. Dev. 2018, 22, 1579. [Google Scholar] [CrossRef]
- Fu, Q.; Liu, D.; Niu, W.; Zhang, S.; Chen, R.; Wang, Y.; Zhao, P.; Jiang, H.; Zhao, Y.; Yang, L.; et al. Defect-Engineered MOF-808 with Highly Exposed Zr Sites as Highly Efficient Catalysts for Catalytic Transfer Hydrogenation of Furfural. Fuel 2022, 327, 125085. [Google Scholar] [CrossRef]
- Zhou, S.; Ban, T.; Li, T.; Gao, H.; He, T.; Cheng, S.; Li, H.; Yi, J.; Zhao, F.; Qu, W. Defect Engineering in Ce-Based Metal-Organic Frameworks toward Enhanced Catalytic Performance for Hydrogenation of Dicyclopentadiene. ACS Appl. Mater. Interfaces 2024, 16, 38177–38187. [Google Scholar] [CrossRef] [PubMed]
- Huber, F.; Berwanger, J.; Polesya, S.; Mankovsky, S.; Ebert, H.; Giessibl, F.J. Chemical Bond Formation Showing a Transition from Physisorption to Chemisorption. Science 2019, 366, 235–238. [Google Scholar] [CrossRef]
- Borodin, D.; Rahinov, I.; Shirhatti, P.R.; Huang, M.; Kandratsenka, A.; Auerbach, D.J.; Zhong, T.; Guo, H.; Schwarzer, D.; Kitsopoulos, T.N.; et al. Following the Microscopic Pathway to Adsorption through Chemisorption and Physisorption Wells. Science 2020, 369, 1461–1465. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.-J.; Wang, J.; Han, M.; Jia, J.; Wu, H.-S.; Zhang, X. Adsorption of Multiple H2 Molecules on the Complex TiC6H6: An Unusual Combination of Chemisorption and Physisorption. Energy 2019, 171, 315–325. [Google Scholar] [CrossRef]
- Xu, X.; Wei, Q.; Xi, Z.; Zhao, D.; Chen, J.; Wang, J.; Zhang, X.; Gao, H.; Wang, G. Research Progress of Metal-Organic Frameworks-Based Materials for CO2 Capture and CO2-to-Alcohols Conversion. Coord. Chem. Rev. 2023, 495, 215393. [Google Scholar] [CrossRef]
- Cui, J.; Wu, F.; Zhang, W.; Yang, L.; Hu, J.; Fang, Y.; Ye, P.; Zhang, Q.; Suo, X.; Mo, Y.; et al. Direct Prediction of Gas Adsorption via Spatial Atom Interaction Learning. Nat. Commun. 2023, 14, 7043. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Wan, T.; He, S.; Li, D.; Wang, X.; Xu, H.; Liu, B. Adsorption-photocatalysis for Methylene Blue Dye Removal by Novel Fe-MOFs through Defect Engineering. J. Chin. Chem. Soc. 2025, 72, 51–64. [Google Scholar] [CrossRef]
- An, H.; Tian, W.; Lu, X.; Yuan, H.; Yang, L.; Zhang, H.; Shen, H.; Bai, H. Boosting the CO2 Adsorption Performance by Defect-Rich Hierarchical Porous Mg-MOF-74. Chem. Eng. J. 2023, 469, 144052. [Google Scholar] [CrossRef]
- Gauna, P.S.; Blanco, A.A.G.; Barrera, D.; Villarroel-Rocha, J.; Hinestroza, J.P.; Kimura, M.; Kim, M.L.; Otal, E.H.; Sapag, K. Influence of Defect Engineering on the Hydrogen and Methane Adsorption Capacity in HKUST-1—Like Structure MOF. Adsorption 2023, 29, 351–361. [Google Scholar] [CrossRef]
- Zhang, X.; Shi, X.; Chen, J.; Yang, Y.; Lu, G. The Preparation of Defective UiO-66 Metal Organic Framework Using MOF-5 as Structural Modifier with High Sorption Capacity for Gaseous Toluene. J. Environ. Chem. Eng. 2019, 7, 103405. [Google Scholar] [CrossRef]
- Chen, J.; Gao, H.; Tao, Z.; Wang, L.; Li, R.; Wang, G. Recent Progress in Mixed Rare Earth Metal-Organic Frameworks: From Synthesis to Application. Coord. Chem. Rev. 2023, 485, 215121. [Google Scholar] [CrossRef]
- Lin, J.; Ban, T.; Li, T.; Sun, Y.; Zhou, S.; Li, R.; Su, Y.; Kasemchainan, J.; Gao, H.; Shi, L.; et al. Machine-learning-assisted Intelligent Synthesis of UiO-66(Ce): Balancing the Trade-off between Structural Defects and Thermal Stability for Efficient Hydrogenation of Dicyclopentadiene. Mater. Genome Eng. Adv. 2024, 2, e61. [Google Scholar] [CrossRef]
- Chung, Y.G.; Camp, J.; Haranczyk, M.; Sikora, B.J.; Bury, W.; Krungleviciute, V.; Yildirim, T.; Farha, O.K.; Sholl, D.S.; Snurr, R.Q. Computation-Ready, Experimental Metal–Organic Frameworks: A Tool To Enable High-Throughput Screening of Nanoporous Crystals. Chem. Mater. 2014, 26, 6185–6192. [Google Scholar] [CrossRef]
- Chung, Y.G.; Haldoupis, E.; Bucior, B.J.; Haranczyk, M.; Lee, S.; Zhang, H.; Vogiatzis, K.D.; Milisavljevic, M.; Ling, S.; Camp, J.S.; et al. Advances, Updates, and Analytics for the Computation-Ready, Experimental Metal–Organic Framework Database: CoRE MOF 2019. J. Chem. Eng. Data 2019, 64, 5985–5998. [Google Scholar] [CrossRef]
- Suyetin, M.; Peskov, M.V.; Schwingenschlögl, U. Methane Sorption in a Family of Qzd-MOFs: A Multiscale Computational Study. Chem. Eng. J. 2020, 384, 123296. [Google Scholar] [CrossRef]
- Dong, X.; Fan, Q.; Hao, W.; Chen, Y. Adsorption and Separation of Hexane Isomers in Metal-Organic Frameworks: A Computational Study. Comput. Theor. Chem. 2021, 1197, 113164. [Google Scholar] [CrossRef]
- Oktavian, R.; Schireman, R.; Glasby, L.T.; Huang, G.; Zanca, F.; Fairen-Jimenez, D.; Ruggiero, M.T.; Moghadam, P.Z. Computational Characterization of Zr-Oxide MOFs for Adsorption Applications. ACS Appl. Mater. Interfaces 2022, 14, 56938–56947. [Google Scholar] [CrossRef] [PubMed]
- Willems, T.F.; Rycroft, C.H.; Kazi, M.; Meza, J.C.; Haranczyk, M. Algorithms and Tools for High-Throughput Geometry-Based Analysis of Crystalline Porous Materials. Microporous Mesoporous Mater. 2012, 149, 134–141. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09, Rev. E.01; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Yang, J.; Tan, L.Z.; Rappe, A.M. Hybrid Functional Pseudopotentials. Phys. Rev. B 2018, 97, 085130. [Google Scholar] [CrossRef]
- Hülsen, M.; Weigand, A.; Dolg, M. Quasirelativistic Energy-Consistent 4f-in-Core Pseudopotentials for Tetravalent Lanthanide Elements. Theor. Chem. Acc. 2009, 122, 23–29. [Google Scholar] [CrossRef]
- Grimme, S. Semiempirical GGA-type Density Functional Constructed with a Long-range Dispersion Correction. J. Comput. Chem. 2006, 27, 1787–1799. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, Z.; Gao, H.; Chen, J.; Wang, J.; Yang, P.; Wang, W.; Wang, G. Unraveling Lewis Acid-Base Sites in Defected MOFs for Catalyzing Dicyclopentadiene Hydrogenation via a DFT Study and Descriptor Exploration. J. Catal. 2024, 439, 115747. [Google Scholar] [CrossRef]
- Heshmat, M. Alternative Pathway of CO2 Hydrogenation by Lewis-Pair-Functionalized UiO-66 MOF Revealed by Metadynamics Simulations. J. Phys. Chem. C 2020, 124, 10951–10960. [Google Scholar] [CrossRef] [PubMed]
- Ye, J.; Johnson, J.K. Design of Lewis Pair-Functionalized Metal Organic Frameworks for CO2 Hydrogenation. ACS Catal. 2015, 5, 2921–2928. [Google Scholar] [CrossRef]
- Zhang, J.; An, B.; Li, Z.; Cao, Y.; Dai, Y.; Wang, W.; Zeng, L.; Lin, W.; Wang, C. Neighboring Zn–Zr Sites in a Metal–Organic Framework for CO2 Hydrogenation. J. Am. Chem. Soc. 2021, 143, 8829–8837. [Google Scholar] [CrossRef]
- Ye, J.; Johnson, J.K. Screening Lewis Pair Moieties for Catalytic Hydrogenation of CO2 in Functionalized UiO-66. ACS Catal. 2015, 5, 6219–6229. [Google Scholar] [CrossRef]
- Hicks, K.E.; Rosen, A.S.; Syed, Z.H.; Snurr, R.Q.; Farha, O.K.; Notestein, J.M. Zr6O8 Node-Catalyzed Butene Hydrogenation and Isomerization in the Metal–Organic Framework NU-1000. ACS Catal. 2020, 10, 14959–14970. [Google Scholar] [CrossRef]
- Lu, T.; Chen, Q. Shermo: A General Code for Calculating Molecular Thermochemistry Properties. Comput. Theor. Chem. 2021, 1200, 113249. [Google Scholar] [CrossRef]
- Johnson, R. Computational Chemistry Comparison and Benchmark Database; NIST Standard Reference Database 101; NIST: Gaithersburg, MD, USA, 2002.



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Structure Name | Unconstructed Defect | Constructed Defects | ||
|---|---|---|---|---|
| NDCPD | NFurfural | NDCPD | NFurfural | |
| JALLEQ_clean | 110.095 | 173.179 | 110.658 | 173.023 |
| DUFKAS_clean | 101.137 | 153.937 | 102.321 | 155.248 |
| DOMDAL_clean | 78.269 | 117.318 | 79.786 | 122.779 |
| ALULEZ_clean | 76.632 | 111.336 | 76.651 | 114.25 |
| ZIJSAO_clean | 75.009 | 113.581 | 76.514 | 120.183 |
| XOFGEG_clean | 71.445 | 111.885 | 71.821 | 112.063 |
| MUWRIH_clean | 71.022 | 120.601 | 75.372 | 117.038 |
| PAQMAY_clean | 65.366 | 105.13 | 67.17 | 107.811 |
| ATEYOP_clean | 48.657 | 82.965 | 50.959 | 88.445 |
| c5dt03091a_c5dt03091a2_clean | 45.216 | 70.819 | 45.112 | 75.106 |
| Structure Name | Eads (kcal/mol) | ||
|---|---|---|---|
| ALULEZ_clean | - | 76.6/7.9 | 111.3/6.1 |
| ATEYOP_clean | - | 48.7/11.3 | 82.9/6.9 |
| c5dt03091a_c5dt03091a2_clean | 13.99 | 45.2/7.5 | 70.8/5.1 |
| DOMDAL_clean | 23.34 | 78.3/9.9 | 117.3/7.4 |
| DUFKAS_clean | - | 101.1/13.3 | 153.9/7.4 |
| JALLEQ_clean | 10.41 | 110.1/16.9 | 173.2/12.5 |
| MUWRIH_clean | - | 71.1/12.4 | 120.6/22.1 |
| PAQMAY_clean | 16.29 | 65.4/11.5 | 106.0/8.1 |
| XOFGEG_clean | - | 71.4/18.5 | 111.9/13.9 |
| ZIJSAO_clean | 49.99 | 75.0/10.1 | 113.6/8.3 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Niu, M.; Xi, Z.; He, C.; Ding, W.; Cheng, S.; Zhang, J.; Gao, H. Investigating the Physical Adsorption of DCPD/Furfural and H2 Adsorption–Dissociation Behaviors in RE-MOFs. Molecules 2025, 30, 1954. https://doi.org/10.3390/molecules30091954
Niu M, Xi Z, He C, Ding W, Cheng S, Zhang J, Gao H. Investigating the Physical Adsorption of DCPD/Furfural and H2 Adsorption–Dissociation Behaviors in RE-MOFs. Molecules. 2025; 30(9):1954. https://doi.org/10.3390/molecules30091954
Chicago/Turabian StyleNiu, Muye, Zuoshuai Xi, Chenhui He, Wenting Ding, Shanshan Cheng, Juntao Zhang, and Hongyi Gao. 2025. "Investigating the Physical Adsorption of DCPD/Furfural and H2 Adsorption–Dissociation Behaviors in RE-MOFs" Molecules 30, no. 9: 1954. https://doi.org/10.3390/molecules30091954
APA StyleNiu, M., Xi, Z., He, C., Ding, W., Cheng, S., Zhang, J., & Gao, H. (2025). Investigating the Physical Adsorption of DCPD/Furfural and H2 Adsorption–Dissociation Behaviors in RE-MOFs. Molecules, 30(9), 1954. https://doi.org/10.3390/molecules30091954