
Ceria–Zirconia-Supported Pt as an Efficient Catalyst for the Sustainable Synthesis of Hydroxylamines and Primary Amines via the Hydrogenation of Oximes Under Ambient Conditions



Abstract

1. Introduction
2. Results and Discussion
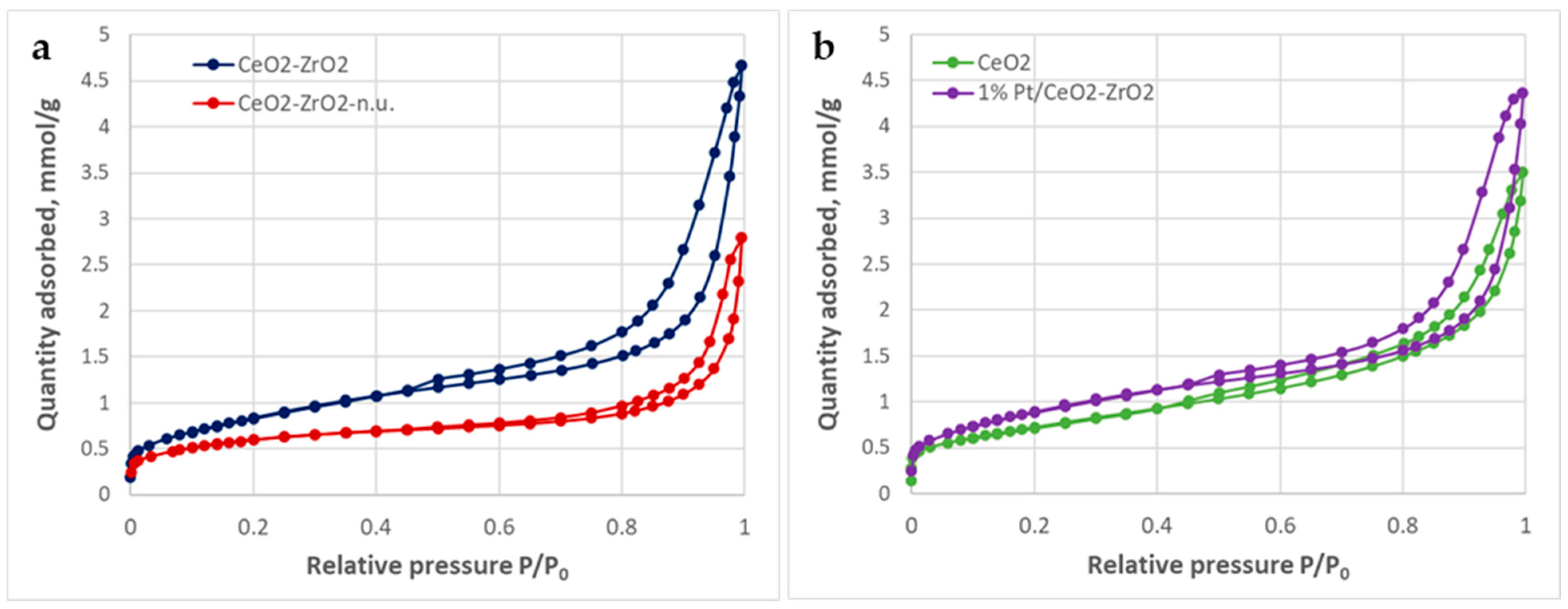
2.1. Catalyst Characterization
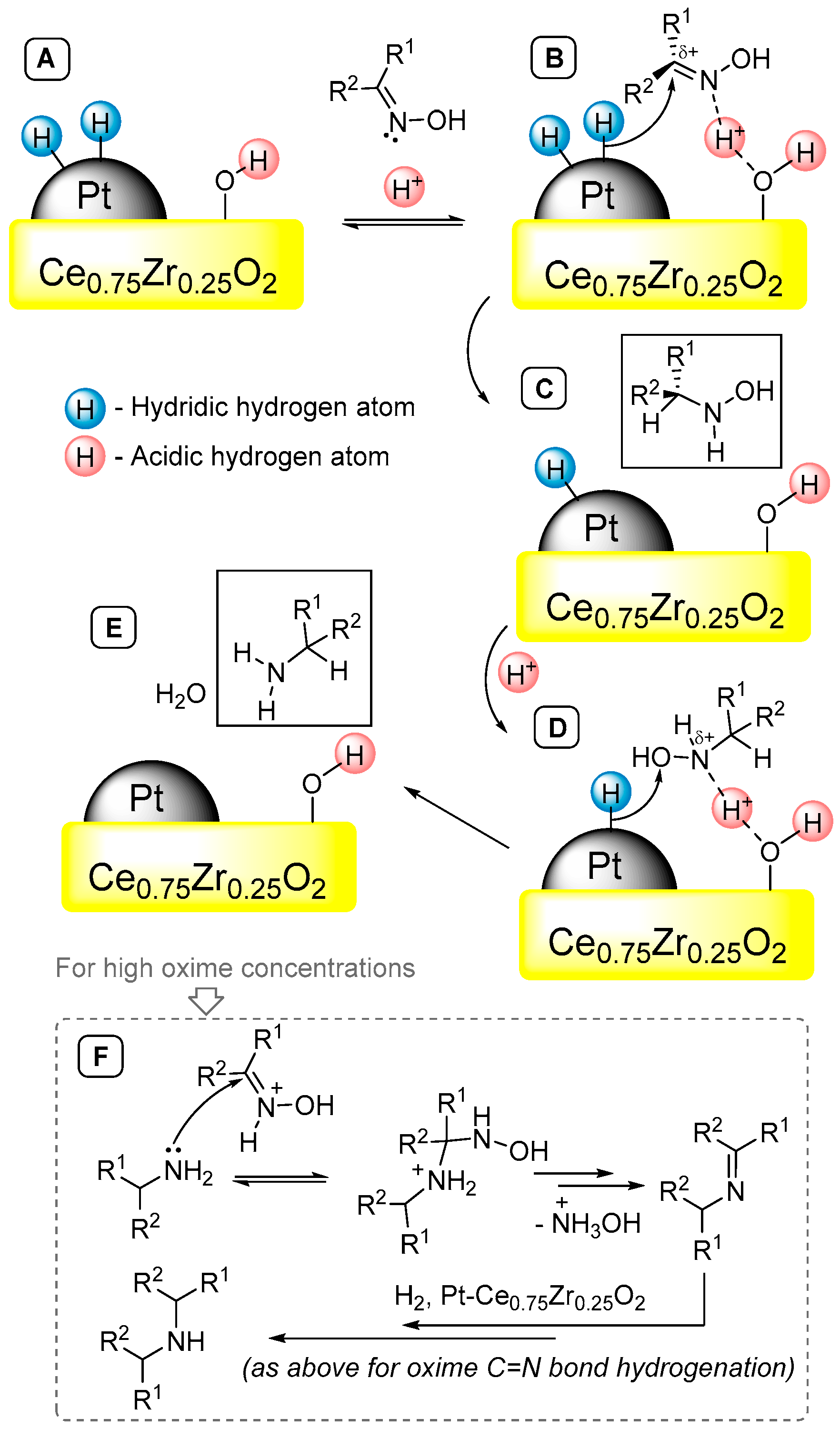
2.2. Catalytic Results: Hydrogenation of Oximes
3. Materials and Methods
3.1. Catalyst Preparation
3.1.1. Support Synthesis
3.1.2. Pt Deposition on the Support
3.2. XRD
3.3. N2 Adsorption−Desorption
3.4. TPR-H2
3.5. SEM, EDX-SEM, and TEM
3.6. DRIFTS-CO
3.7. Catalytic Reaction
3.8. NMR and HRMS
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ozsvár, D.; Nagy, V.; Zupkó, I.; Szakonyi, Z. Synthesis and Biological Application of Isosteviol-Based 1,3-Aminoalcohols. Int. J. Mol. Sci. 2021, 22, 11232. [Google Scholar] [CrossRef]
- Zhang, X.-L.; Ma, Y.; Pan, Q.; Bai, Z.-G.; Qi, H.; Zhang, Q.-Z. Synthesis of (5,6-Dihydro-4H-Pyrrolo[1,2-b]Pyrazol-3-Yl)-Methanamine. HETEROCYCLES 2017, 94, 1923. [Google Scholar] [CrossRef]
- Pospelov, E.V.; Sukhorukov, A.Y. Building Up a Piperazine Ring from a Primary Amino Group via Catalytic Reductive Cyclization of Dioximes. Int. J. Mol. Sci. 2023, 24, 11794. [Google Scholar] [CrossRef]
- Srinivasan, A.; Banerjee, S.; Pachore, S.; Syam Kumar, U. Three-Component Coupling–Oxidative Amidation–Heterocycloannulation: Synthesis of the Indole Alkaloids Hamacanthin A and Trans-2,5-Bis(3′-Indolyl)Piperazine. Synlett 2017, 28, 1057–1064. [Google Scholar] [CrossRef]
- da Silva, F.P.; Fiorio, J.L.; Rossi, L.M. Tuning the Catalytic Activity and Selectivity of Pd Nanoparticles Using Ligand-Modified Supports and Surfaces. ACS Omega 2017, 2, 6014–6022. [Google Scholar] [CrossRef]
- Uruş, S.; Keleş, M.; Köşker Akkaya, S. Synthesis and Characterization of Pd(II) and Ru(II) Complexes of Tetradentate N,N,N,N-(Diphosphinomethyl)Amine Ligands: Catalytic Properties in Transfer Hydrogenation and Heck Coupling Reactions. HETEROCYCLES 2020, 100, 1019. [Google Scholar] [CrossRef]
- Rajmane, A.; Jadhav, S.; Kumbhar, A. N, O-Polydentate Ligands for Palladium-Catalyzed Cross-Coupling Reactions (Part III). J. Organomet. Chem. 2022, 957, 122147. [Google Scholar] [CrossRef]
- Kim, K.; Singstock, N.R.; Childress, K.K.; Sinha, J.; Salazar, A.M.; Whitfield, S.N.; Holder, A.M.; Stansbury, J.W.; Musgrave, C.B. Rational Design of Efficient Amine Reductant Initiators for Amine–Peroxide Redox Polymerization. J. Am. Chem. Soc. 2019, 141, 6279–6291. [Google Scholar] [CrossRef]
- Huang, Y.; Liu, L.; Zheng, G.; Zhang, X.; Wang, Z. Efficient Production of Red Monascus Pigments with Single Non-Natural Amine Residue by in Situ Chemical Modification. World J. Microbiol. Biotechnol. 2019, 35, 13. [Google Scholar] [CrossRef]
- Nagao, Y. Synthesis and Properties of Perylene Pigments. Prog. Org. Coat. 1997, 31, 43–49. [Google Scholar] [CrossRef]
- Politzer, P.; Murray, J.S. Some Intrinsic Features of Hydroxylamines, Oximes and Hydroxamic Acids: Integration of Theory and Experiment. In PATAI’S Chemistry of Functional Groups; Rappoport, Z., Liebman, J.F., Eds.; Wiley: Hoboken, NJ, USA, 2008; pp. 1–27. ISBN 978-0-470-51261-6. [Google Scholar]
- Bailey, P.D.; Morgan, K.M. Synthesis of Amines. In Organonitrogen Chemistry; Oxford University Press: Oxford, UK, 2022; ISBN 978-0-19-855775-3. [Google Scholar]
- Marvin, C.C. 6.02 Synthesis of Amines and Ammonium Salts. In Comprehensive Organic Synthesis II; Elsevier: Amsterdam, The Netherlands, 2014; pp. 34–99. ISBN 978-0-08-097743-0. [Google Scholar]
- Zeynizadeh, B.; Zabihzadeh, M. Rapid and Green Reduction of Aromatic/Aliphatic Nitro Compounds to Amines with NaBH4 and Additive Ni2B in H2O. J. Iran. Chem. Soc. 2015, 12, 1221–1226. [Google Scholar] [CrossRef]
- Prasad, A.S.B.; Kanth, J.V.B.; Periasamy, M. Convenient Methods for the Reduction of Amides, Nitriles, Carboxylic Esters, Acids and Hydroboration of Alkenes Using NaBH4/I2system. Tetrahedron 1992, 48, 4623–4628. [Google Scholar] [CrossRef]
- Zhenjiang, L. Sodium Borohydride—A Versatile Reducing Agent. Synlett 2004, 2005, 182–183. [Google Scholar] [CrossRef]
- Pohland, A.; Sullivan, H.R. Lithium Aluminum Hydride Reduction of Grignard—Nitrile Adducts to Primary Amines. J. Am. Chem. Soc. 1953, 75, 5898–5899. [Google Scholar] [CrossRef]
- Newman, M.S.; Fukunaga, T. The Reduction of Amides to Amines via Nitriles by Lithium Aluminum Hydride 1. J. Am. Chem. Soc. 1960, 82, 693–696. [Google Scholar] [CrossRef]
- Di Gioia, M.L.; Leggio, A.; Guarino, I.F.; Leotta, V.; Romio, E.; Liguori, A. A Simple Synthesis of Anilines by LiAlH4/TiCl4 Reduction of Aromatic Nitro Compounds. Tetrahedron Lett. 2015, 56, 5341–5344. [Google Scholar] [CrossRef]
- Bäumler, C.; Bauer, C.; Kempe, R. The Synthesis of Primary Amines through Reductive Amination Employing an Iron Catalyst. ChemSusChem 2020, 13, 3110–3114. [Google Scholar] [CrossRef]
- Dong, C.; Wu, Y.; Wang, H.; Peng, J.; Li, Y.; Samart, C.; Ding, M. Facile and Efficient Synthesis of Primary Amines via Reductive Amination over a Ni/Al2 O3 Catalyst. ACS Sustain. Chem. Eng. 2021, 9, 7318–7327. [Google Scholar] [CrossRef]
- Hahn, G.; Kunnas, P.; De Jonge, N.; Kempe, R. General Synthesis of Primary Amines via Reductive Amination Employing a Reusable Nickel Catalyst. Nat. Catal. 2018, 2, 71–77. [Google Scholar] [CrossRef]
- Sahyoun, T.; Arrault, A.; Schneider, R. Amidoximes and Oximes: Synthesis, Structure, and Their Key Role as NO Donors. Molecules 2019, 24, 2470. [Google Scholar] [CrossRef] [PubMed]
- Krylov, I.B.; Paveliev, S.A.; Budnikov, A.S.; Terent’ev, A.O. Oxime Radicals: Generation, Properties and Application in Organic Synthesis. Beilstein J. Org. Chem. 2020, 16, 1234–1276. [Google Scholar] [CrossRef] [PubMed]
- Krylov, I.B.; Segida, O.O.; Budnikov, A.S.; Terent’ev, A.O. Oxime-Derived Iminyl Radicals in Selective Processes of Hydrogen Atom Transfer and Addition to Carbon-Carbon π-Bonds. Adv. Synth. Catal. 2021, 363, 2502–2528. [Google Scholar] [CrossRef]
- Rykaczewski, K.A.; Wearing, E.R.; Blackmun, D.E.; Schindler, C.S. Reactivity of Oximes for Diverse Methodologies and Synthetic Applications. Nat. Synth. 2022, 1, 24–36. [Google Scholar] [CrossRef]
- Rosen, W.E.; Green, M.J. The Reduction of 2-Indanone Oxime to 2-Aminoindane. Methods and Mechanisms. J. Org. Chem. 1963, 28, 2797–2804. [Google Scholar] [CrossRef]
- Nakamura, Y. Asymmetrische Synteese. III. Versuch Zur Darstellung Eines Optisch-Aktiven Amins Durch Die Reduktion Des Ketoxims Beim Vorhandensein von Optisch-Aktiver Säure. Bull. Chem. Soc. Jpn. 1941, 16, 367–370. [Google Scholar] [CrossRef]
- Gebauer-Henke, E.; Leitner, W.; Prokofieva, A.; Vogt, H.; Müller, T.E. Controlling Selectivity in the Reaction Network of Aldoxime Hydrogenation to Primary Amines. Catal. Sci. Technol. 2012, 2, 2539. [Google Scholar] [CrossRef]
- Baucom, K.; Guram, A.; Borths, C. Effective Conversion of Heteroaromatic Ketones into Primary Amines via Hydrogenation of Intermediate Ketoximes. Synlett 2014, 26, 201–204. [Google Scholar] [CrossRef]
- Haadsma-Svensson, S.R.; Cleek, K.A.; Dinh, D.M.; Duncan, J.N.; Haber, C.L.; Huff, R.M.; Lajiness, M.E.; Nichols, N.F.; Smith, M.W.; Svensson, K.A.; et al. Dopamine D3 Receptor Antagonists. 1. Synthesis and Structure−Activity Relationships of 5,6-Dimethoxy-N-Alkyl- and N-Alkylaryl-Substituted 2-Aminoindans. J. Med. Chem. 2001, 44, 4716–4732. [Google Scholar] [CrossRef]
- Mas-Roselló, J.; Smejkal, T.; Cramer, N. Iridium-Catalyzed Acid-Assisted Asymmetric Hydrogenation of Oximes to Hydroxylamines. Science 2020, 368, 1098–1102. [Google Scholar] [CrossRef]
- Wang, F.; Chen, Y.; Yu, P.; Chen, G.-Q.; Zhang, X. Asymmetric Hydrogenation of Oximes Synergistically Assisted by Lewis and Brønsted Acids. J. Am. Chem. Soc. 2022, 144, 17763–17768. [Google Scholar] [CrossRef]
- Pospelov, E.V.; Boyko, Y.D.; Ioffe, S.L.; Sukhorukov, A.Y. Synthesis of Bis(β-Oximinoalkyl)Malonates and Their Catalytic Reductive Cyclization to Piperidines. Adv. Synth. Catal. 2022, 364, 2557–2564. [Google Scholar] [CrossRef]
- Redina, E.A.; Ivanova, I.I.; Arkhipova, N.Y.; Kustov, L.M. Heterogeneous Catalysis as an Efficient Tool for Selective Hydrogenation of Oximes to Amines and Hydroxylamines. Catalysts 2022, 12, 1614. [Google Scholar] [CrossRef]
- Sanz-Cervera, J.F.; Blasco, R.; Piera, J.; Cynamon, M.; Ibáñez, I.; Murguía, M.; Fustero, S. Solution versus Fluorous versus Solid-Phase Synthesis of 2,5-Disubstituted 1,3-Azoles. Preliminary Antibacterial Activity Studies. J. Org. Chem. 2009, 74, 8988–8996. [Google Scholar] [CrossRef] [PubMed]
- Laufer, S.A.; Liedtke, A.J. A Concise and Optimized Four-Step Approach toward 2-(Aryl-)Alkylsulfanyl-, 4(5)-Aryl-, 5(4)-Heteroaryl-Substituted Imidazoles Using Alkyl- or Arylalkyl Thiocyanates. Tetrahedron Lett. 2006, 47, 7199–7203. [Google Scholar] [CrossRef]
- Mas-Roselló, J.; Cramer, N. Catalytic Reduction of Oximes to Hydroxylamines: Current Methods, Challenges and Opportunities. Chem. Eur. J. 2022, 28, e202103683. [Google Scholar] [CrossRef]
- Matthews, J.M.; Chen, X.; Cryan, E.; Hlasta, D.J.; Rybczynski, P.J.; Strauss, K.; Tang, Y.; Xu, J.Z.; Yang, M.; Zhou, L.; et al. Design and Synthesis of Indane-Ureido-Thioisobutyric Acids: A Novel Class of PPARα Agonists. Bioorg. Med. Chem. Lett. 2007, 17, 6773–6778. [Google Scholar] [CrossRef]
- Borszeky, K.; Mallat, T.; Aeschiman, R.; Schweizer, W.B.; Baiker, A. Enantioselective Hydrogenation of Pyruvic Acid Oxime to Alanine on Pd/Alumina. J. Catal. 1996, 161, 451–458. [Google Scholar] [CrossRef]
- Muramatsu, W.; Tsuji, H.; Yamamoto, H. Catalytic Peptide Synthesis: Amidation of N -Hydroxyimino Esters. ACS Catal. 2018, 8, 2181–2187. [Google Scholar] [CrossRef]
- Ignatov, A.V.; Varakutin, A.E.; Solov’eva, I.N.; Karmanova, I.B.; Kozlov, I.A.; Semenova, M.N.; Semenov, V.V. Efficient Hydrogenation of Benzaldoximes and Schiff Bases on Ceramic High-Porosity Palladium Catalysts. Russ. Chem. Bull. 2018, 67, 1394–1400. [Google Scholar] [CrossRef]
- Mas-Roselló, J.; Cope, C.J.; Tan, E.; Pinson, B.; Robinson, A.; Smejkal, T.; Cramer, N. Iridium-Catalyzed Acid-Assisted Hydrogenation of Oximes to Hydroxylamines. Angew. Chem. Int. Ed. 2021, 60, 15524–15532. [Google Scholar] [CrossRef]
- Li, B.; Chen, J.; Liu, D.; Gridnev, I.D.; Zhang, W. Nickel-Catalysed Asymmetric Hydrogenation of Oximes. Nat. Chem. 2022, 14, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Ou, W.; Espinosa, S.; Meléndez, H.J.; Farré, S.M.; Alvarez, J.L.; Torres, V.; Martínez, I.; Santiago, K.M.; Ortiz-Marciales, M. Asymmetric Synthesis of Nonracemic Primary Amines via Spiroborate-Catalyzed Reduction of Pure (E)- and (Z)-O-Benzyloximes: Applications toward the Synthesis of Calcimimetic Agents. J. Org. Chem. 2013, 78, 5314–5327. [Google Scholar] [CrossRef] [PubMed]
- Mohr, J.; Porwal, D.; Chatterjee, I.; Oestreich, M. Extending the Scope of the B(C6F5)3-Catalyzed C=N Bond Reduction: Hydrogenation of Oxime Ethers and Hydrazones. Chem. Eur. J. 2015, 21, 17583–17586. [Google Scholar] [CrossRef]
- Hübner, S.; de Vries, J.G.; Farina, V. Why Does Industry Not Use Immobilized Transition Metal Complexes as Catalysts? Adv. Synth. Catal. 2016, 358, 3–25. [Google Scholar] [CrossRef]
- Demidova, Y.S.; Mozhaitsev, E.S.; Suslov, E.V.; Nefedov, A.A.; Saraev, A.A.; Volcho, K.P.; Salakhutdinov, N.F.; Simakov, A.; Simakova, I.L.; Murzin, D.Y. Menthylamine Synthesis via Gold-Catalyzed Hydrogenation of Menthone Oxime. Appl. Catal. Gen. 2020, 605, 117799. [Google Scholar] [CrossRef]
- Demidova, Y.S.; Mozhaitsev, E.S.; Munkuev, A.A.; Suslov, E.V.; Saraev, A.A.; Volcho, K.P.; Salakhutdinov, N.F.; Simakova, I.L.; Murzin, D.Y. Monoterpenoid Oximes Hydrogenation Over Platinum Catalysts. Top. Catal. 2020, 63, 187–195. [Google Scholar] [CrossRef]
- Liu, Y.; Quan, Z.; He, S.; Zhao, Z.; Wang, J.; Wang, B. Heterogeneous Palladium-Based Catalyst Promoted Reduction of Oximes to Amines: Using H2 at 1 Atm in H2O under Mild Conditions. React. Chem. Eng. 2019, 4, 1145–1152. [Google Scholar] [CrossRef]
- Benington, F.; Morin, R.D.; Clark, L.C. Behavioral and Neuropharmacological Actions of N-Aralkylhydroxylamines and Their O-Methyl Ethers. J. Med. Chem. 1965, 8, 100–104. [Google Scholar] [CrossRef]
- Redina, E.A.; Vikanova, K.V.; Kapustin, G.I.; Mishin, I.V.; Tkachenko, O.P.; Kustov, L.M. Selective Room-Temperature Hydrogenation of Carbonyl Compounds under Atmospheric Pressure over Platinum Nanoparticles Supported on Ceria-Zirconia Mixed Oxide. Eur. J. Org. Chem. 2019, 2019, 4159–4170. [Google Scholar] [CrossRef]
- Redina, E.A.; Vikanova, K.V. Highly Efficient Pt-Catalyst Supported on Mesoporous Ceria-Zirconia Oxide for Hydrogenation of Nitroaromatic Compounds to Anilines. Russ. J. Phys. Chem. A 2018, 92, 2374–2378. [Google Scholar] [CrossRef]
- Redina, E.A.; Krylov, I.B.; Novikov, R.A.; Kapustin, G.I.; Tkachenko, O.P.; Vikanova, K.V.; Ivanova, I.I.; Dmitrenok, A.S.; Kustov, L.M. High-Performance Pt/CeO2-ZrO2 Catalysts for Selective Hydrogenation of α,β-Unsaturated Aldehydes to Unsaturated Alcohols under Mild Reaction Conditions: “Giant” Hydrogen Spillover behind the Activity Enhancement. J. Catal. 2024, 429, 115231. [Google Scholar] [CrossRef]
- Vikanova, K.V.; Redina, E.A.; Kapustin, G.I.; Mishin, I.V.; Davshan, N.A.; Kustov, L.M. Selective Hydrogenation of α,β-Unsaturated Aldehydes over Pt Supported on Cerium–Zirconium Mixed Oxide of Different Composition. Mendeleev Commun. 2022, 32, 488–491. [Google Scholar] [CrossRef]
- Redina, E.A.; Vikanova, K.V.; Kapustin, G.I. Monometallic Copper Catalysts for the Hydrogenation of 5-Hydroxymethylfurfural. Russ. J. Phys. Chem. A 2020, 94, 2558–2562. [Google Scholar] [CrossRef]
- Sharma, A.; Pandey, A.; Patle, V.K.; Sharma, N.; Jain, H.; Khare, A.; Dwivedi, N.; Gupta, G.; Mondal, D.P.; Srivastava, A.K.; et al. Sustainable Lightweight Multifunctional Carbon Foams Derived from Coal Tar Pitch Using Urea as a Pore-Forming Agent. J. Anal. Appl. Pyrolysis 2023, 174, 106145. [Google Scholar] [CrossRef]
- Li, Z.H.; Cheng, C.; Zhan, X.Y.; Wu, Y.P.; Zhou, X.D. A Foaming Process to Prepare Porous Polymer Membrane for Lithium Ion Batteries. Electrochim. Acta 2009, 54, 4403–4407. [Google Scholar] [CrossRef]
- Xiao, W.; Miao, C.; Yin, X.; Zheng, Y.; Tian, M.; Li, H.; Mei, P. Effect of Urea as Pore-Forming Agent on Properties of Poly(Vinylidene Fluoride-Co-Hexafluoropropylene)-Based Gel Polymer Electrolyte. J. Power Sources 2014, 252, 14–20. [Google Scholar] [CrossRef]
- Thommes, M.; Kaneko, K.; Neimark, A.V.; Olivier, J.P.; Rodriguez-Reinoso, F.; Rouquerol, J.; Sing, K.S.W. Physisorption of Gases, with Special Reference to the Evaluation of Surface Area and Pore Size Distribution (IUPAC Technical Report). Pure Appl. Chem. 2015, 87, 1051–1069. [Google Scholar] [CrossRef]
- Garnier, A.; Sall, S.; Garin, F.; Chetcuti, M.J.; Petit, C. Site Effects in the Adsorption of Carbon Monoxide on Real 1.8nm Pt Nanoparticles: An Infrared Investigation in Time and Temperature. J. Mol. Catal. Chem. 2013, 373, 127–134. [Google Scholar] [CrossRef]
- Vikanova, K.V.; Chernova, M.S.; Redina, E.A.; Kapustin, G.I.; Tkachenko, O.P.; Kustov, L.M. Heterogeneous Additive-free Highly Selective Synthesis of 2,5-bis(Hydroxymethyl)Furan over Catalysts with Ultra-low Pt Content. J. Chem. Technol. Biotechnol. 2021, 96, 2421–2425. [Google Scholar] [CrossRef]
- Wang, H.; Liu, J.-X.; Allard, L.F.; Lee, S.; Liu, J.; Li, H.; Wang, J.; Wang, J.; Oh, S.H.; Li, W.; et al. Surpassing the Single-Atom Catalytic Activity Limit through Paired Pt-O-Pt Ensemble Built from Isolated Pt1 Atoms. Nat. Commun. 2019, 10, 3808. [Google Scholar] [CrossRef]
- Gibson, E.K.; Crabb, E.M.; Gianolio, D.; Russell, A.E.; Thompsett, D.; Wells, P.P. Understanding the Role of Promoters in Catalysis: Operando XAFS/DRIFTS Study of CeOx/Pt/Al2O3 during CO Oxidation. Catal. Struct. React. 2017, 3, 5–12. [Google Scholar] [CrossRef]
- Lentz, C.; Jand, S.P.; Melke, J.; Roth, C.; Kaghazchi, P. DRIFTS Study of CO Adsorption on Pt Nanoparticles Supported by DFT Calculations. J. Mol. Catal. Chem. 2017, 426, 1–9. [Google Scholar] [CrossRef]
- Bolis, V.; Morterra, C.; Volante, M.; Orio, L.; Fubini, B. Development and Suppression of Surface Acidity on Monoclinic Zirconia: A Spectroscopic and Calorimetric Investigation. Langmuir 1990, 6, 695–701. [Google Scholar] [CrossRef]
- Vikanova, K.; Redina, E.; Kapustin, G.; Chernova, M.; Tkachenko, O.; Nissenbaum, V.; Kustov, L. Advanced Room-Temperature Synthesis of 2,5-Bis(Hydroxymethyl)Furan—A Monomer for Biopolymers—From 5-Hydroxymethylfurfural. ACS Sustain. Chem. Eng. 2021, 9, 1161–1171. [Google Scholar] [CrossRef]
- Vikanova, K.V.; Redina, E.A.; Kustov, L.M. Hydrogen Spillover on Cerium-Based Catalysts. Russ. Chem. Bull. 2022, 71, 1579–1592. [Google Scholar] [CrossRef]
- Stuckert, A.N.; Wang, L.; Yang, R.T. Characteristics of Hydrogen Storage by Spillover on Pt-Doped Carbon and Catalyst-Bridged Metal Organic Framework. Langmuir 2010, 26, 11963–11971. [Google Scholar] [CrossRef]
- Damljanović, I.; Vukićević, M.; Vukićević, R.D. A Simple Synthesis of Oximes. Monatshefte Chem. Chem. Mon. 2006, 137, 301–305. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Sinha, A.S.; Epa, K.N.; Spartz, C.L.; Desper, J. A Versatile and Green Mechanochemical Route for Aldehyde–Oxime Conversions. Chem. Commun. 2012, 48, 11289. [Google Scholar] [CrossRef]
- Aakeröy, C.B.; Sinha, A.S. Synthesis of Ketoximes via a Solvent-Assisted and Robust Mechanochemical Pathway. RSC Adv. 2013, 3, 8168. [Google Scholar] [CrossRef]
- Kachala, V.V.; Khemchyan, L.L.; Kashin, A.S.; Orlov, N.V.; Grachev, A.A.; Zalesskiy, S.S.; Ananikov, V.P. Target-Oriented Analysis of Gaseous, Liquid and Solid Chemical Systems by Mass Spectrometry, Nuclear Magnetic Resonance Spectroscopy and Electron Microscopy. Russ. Chem. Rev. 2013, 82, 648–685. [Google Scholar] [CrossRef]
- An, Q.; Wang, Z.; Chen, Y.; Wang, X.; Zhang, K.; Pan, H.; Liu, W.; Zuo, Z. Cerium-Catalyzed C–H Functionalizations of Alkanes Utilizing Alcohols as Hydrogen Atom Transfer Agents. J. Am. Chem. Soc. 2020, 142, 6216–6226. [Google Scholar] [CrossRef] [PubMed]
- De Jong, A.P.; Fesik, S.W.; Makriyannis, A. Conformational Requirements for Norepinephrine Uptake Inhibition by Phenethylamines in Brain Synaptosomes. Effects of .Alpha.-Alkyl Substitution. J. Med. Chem. 1982, 25, 1438–1441. [Google Scholar] [CrossRef]
- Orekhov, D. Research and Development on the Manufacturing Process of Dexmedetomidine from Chiral Precursors: Resolution–Racemization–Recycle Synthesis Strategy. Org. Process Res. Dev. 2024, 28, 1032–1054. [Google Scholar] [CrossRef]
- Chowdhury, D.; Sutradhar, R.; Paul, A.; Mukherjee, A. Insight into the MOtBu (M=Na, K)-Mediated Dehydrogenation of Dimethylamine-Borane and Transfer Hydrogenation of Nitriles to Primary Amines. Chem. Eur. J. 2024, 30, e202400942. [Google Scholar] [CrossRef]
- Jackson, D.M.; Ashley, R.L.; Brownfield, C.B.; Morrison, D.R.; Morrison, R.W. Rapid Conventional and Microwave-Assisted Decarboxylation of L-Histidine and Other Amino Acids via Organocatalysis with R-Carvone Under Superheated Conditions. Synth. Commun. 2015, 45, 2691–2700. [Google Scholar] [CrossRef]
- Wilson, A.J.; Masuda, M.; Sijbesma, R.P.; Meijer, E.W. Chiral Amplification in the Transcription of Supramolecular Helicity into a Polymer Backbone. Angew. Chem. Int. Ed. 2005, 44, 2275–2279. [Google Scholar] [CrossRef]
- Behnke, N.E.; Kielawa, R.; Kwon, D.-H.; Ess, D.H.; Kürti, L. Direct Primary Amination of Alkylmetals with NH-Oxaziridine. Org. Lett. 2018, 20, 8064–8068. [Google Scholar] [CrossRef] [PubMed]
- Guizzetti, S.; Michaut, A.; Federspiel, G.; Eymard, J.; Caron, I.; Quatrevaux, S.; Daras, E.; Jolly, S.; Guillemont, J.; Lançois, D. A Fit-for-Purpose Synthesis of (R)-2-Methylazepane. Org. Process Res. Dev. 2020, 24, 729–733. [Google Scholar] [CrossRef]
- Chen, Y.; Cantillo, D.; Kappe, C.O. Visible Light-Promoted Beckmann Rearrangements: Separating Sequential Photochemical and Thermal Phenomena in a Continuous Flow Reactor. Eur. J. Org. Chem. 2019, 2019, 2163–2171. [Google Scholar] [CrossRef]
- Yu, J.; Lu, M. Copper(II)-Promoted Direct Conversion of Methylarenes into Aromatic Oximes. Org. Biomol. Chem. 2015, 13, 7397–7401. [Google Scholar] [CrossRef]
- Betke, T.; Rommelmann, P.; Oike, K.; Asano, Y.; Gröger, H. Cyanide-Free and Broadly Applicable Enantioselective Synthetic Platform for Chiral Nitriles through a Biocatalytic Approach. Angew. Chem. Int. Ed. 2017, 56, 12361–12366. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, A.; Tanaka, K.; Hashimoto, Y.; Morita, N.; Tamura, O. Intermolecular 1,3-Dipolar Cycloaddition Reaction of N-Carbamoyl Nitrones Generated by N-Selective Carbamoylation of Oximes with Isocyanates. Chem. Eur. J. 2024, 30, e202303790. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | a, Å | a Table, Å | Phase | D, nm |
|---|---|---|---|---|
| CeO2-ZrO2 | 5.354 | 5.349 | Ce0.75Zr0.25O2 Cerium Zirconium oxide | 10 |
| CeO2 | 5.410 | 5.411 | CeO2 Cerianite | 14 |
| CeO2-ZrO2—n.u. | 5.410 | 5.411 | CeO2 Cerianite ZrO2 amorphous | 10 n.d. |
| Sample | ABET m2/g | Vtotal [a] cm3/g | Vmeso, cm3/g | Vmicro, DFT cm3/g | D av. pore, nm |
|---|---|---|---|---|---|
| CeO2-ZrO2 | 70 | 0.162 | 0.158 | 0.006 | 8.9 (2–3, 3–5, 5–50) |
| 1% Pt/CeO2-ZrO2 | 74 | 0.151 | 0.142 | 0.009 | 8.1 (1–2, 2–5, 5–50) |
| CeO2 | 58 | 0.121 | 0.109 | 0.009 | 8.0 (1–2, 2–15) |
| CeO2-ZrO2—n.u. | 48 | 0.097 | 0.091 | 0.006 | 7.5 (2–5, 5–20) |
| Sample | H2/Pt, mol/mol −100–25 °C | H2/Pt, mol/mol 24–300 °C |
|---|---|---|
| 1% Pt/CeO2-ZrO2 | 8.3 | 4.6 |
| 1% Pt/CeO2 | 6.6 | 1.8 |
| 1% Pt/CeO2-ZrO2—n.u. | 5.9 | 2.6 |
| 1% Pt/ZrO2 | 0.2 | 0 |
| 1% Pt/SiO2 | 0.2 | 0 |
 | ||||||
|---|---|---|---|---|---|---|
| № | Catalyst | Product | τ, h | X, % | Yield, % [a] | TOF, h−1 [b] |
| 1 | 1% Pt/ZrO2 | 2a | 1 | 20 | 20 | 39 |
| 2 | 1% Pt/SiO2 | 2a | 1 | 21 | 21 | 40 |
| 3 | 1% Pt/CeO2 | 2a | 0.25 | 60 | 60 | 428 |
| 4 | 1% Pt/CeO2-ZrO2 | 2a | 0.25 | 75 | 75 | 554 |
| 5 | 1% Pt/CeO2-ZrO2 | 2a | 0.5 | >99 | >99 | - |
| 6 | 1% Pt/CeO2-ZrO2 | 2a | 1 | >99 | >99 | - |
| 7 | 1% Pt/CeO2 | 2b | 1 | 85 | 85 | - |
| 8 | 1% Pt/CeO2-ZrO2-n.u. | 2a + 2b | 1 | >99 | 29 + 49 | - |
| 9 [c] | 1% Pd/AC-comm | - | 2 | 0 | 0 | - |
| 10 | 1% Pt/C-comm | 2a | 4.5 | 68 | 48 | - |
| 11 [d] | 1% Pt/CeO2-ZrO2 | 2a | 1 | 70 | 70 | - |
 | |||
|---|---|---|---|
| Recycle | Product | X, % | Yield, % [a] |
| 1 | 2b | >99 | >99 |
| 2 | 2b | >99 | >99 |
| 3 | 2a | >99 | >99 |
 | ||||||
|---|---|---|---|---|---|---|
| № | Substrate | n Sub, mmol | Solvent, ml | τ, h | Product | Yield, % [a] |
| 1 | 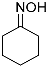 1 1 | 0.46 | 2 | 1 | 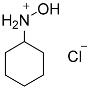 2a 2a | 91 |
| 2 |  1 1 | 0.23 | 4 | 2 | 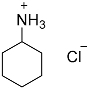 2b 2b | 99 |
| 3 | 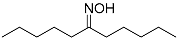 3 3 | 0.46 | 2 | 4.5 | 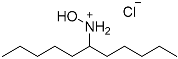 4 4 | 77 |
| 4 |  5 5 | 0.46 | 2 | 4.5 |  6 6 | 74 |
| 5 | 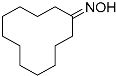 7 7 | 0.23 | 4 | 4.5 | 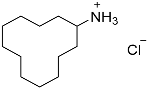 8 8 | 97 |
| 6 | 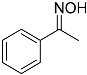 9 9 | 0.46 | 2 | 2 |  10 10 | 74 |
| 7 | 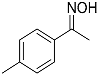 11 11 | 0.46 | 4 | 2 | 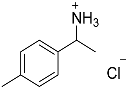 12 12 | 38 [b] |
| 8 | 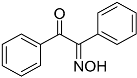 13 13 | 0.23 | 4 | 6 |  14 14 | 89 |
 | ||||||
|---|---|---|---|---|---|---|
| № | Substrate | n Sub, mmol | Solvent, ml | τ, h | Product | Yield, % [a] |
| 1 | 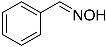 15 15 | 0.46 | 2 | 1 | 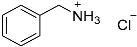 16 16 | 45 [b] |
| 2 |  17 17 | 0.46 | 2 | 1 |  18 18 | 98 |
| 3 | 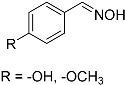 19 19 | 0.46 | 2 | 1 | 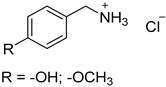 20 20 | <5 [b] |
| 4 |  21 21 | 0.23 | 2 | 2 | 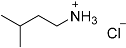 22 22 | 95 |
| 5 | 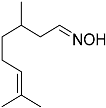 23 23 | 0.23 | 4 | 4 |  24 24 | 84 |
| 6 |  25 25 | 0.23 | 4 | 6 | 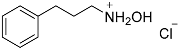 26a 26a 26b 26b | 70 [b] + 30 [b] |
| 7 | 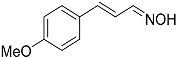 27 27 | 0.23 | 4 | 6 | 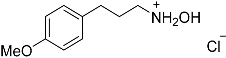 28a 28a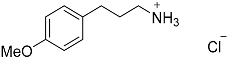 28b 28b | 55 [b] + 45 [b] |
| 8 | 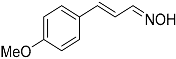 27 27 | 0.23 | 4 | 4 |  28b 28b | 48 [c] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Redina, E.; Ivanova, I.; Tkachenko, O.; Kapustin, G.; Mishin, I.; Kustov, L. Ceria–Zirconia-Supported Pt as an Efficient Catalyst for the Sustainable Synthesis of Hydroxylamines and Primary Amines via the Hydrogenation of Oximes Under Ambient Conditions. Molecules 2025, 30, 1926. https://doi.org/10.3390/molecules30091926
Redina E, Ivanova I, Tkachenko O, Kapustin G, Mishin I, Kustov L. Ceria–Zirconia-Supported Pt as an Efficient Catalyst for the Sustainable Synthesis of Hydroxylamines and Primary Amines via the Hydrogenation of Oximes Under Ambient Conditions. Molecules. 2025; 30(9):1926. https://doi.org/10.3390/molecules30091926
Chicago/Turabian StyleRedina, Elena, Inna Ivanova, Olga Tkachenko, Gennady Kapustin, Igor Mishin, and Leonid Kustov. 2025. "Ceria–Zirconia-Supported Pt as an Efficient Catalyst for the Sustainable Synthesis of Hydroxylamines and Primary Amines via the Hydrogenation of Oximes Under Ambient Conditions" Molecules 30, no. 9: 1926. https://doi.org/10.3390/molecules30091926
APA StyleRedina, E., Ivanova, I., Tkachenko, O., Kapustin, G., Mishin, I., & Kustov, L. (2025). Ceria–Zirconia-Supported Pt as an Efficient Catalyst for the Sustainable Synthesis of Hydroxylamines and Primary Amines via the Hydrogenation of Oximes Under Ambient Conditions. Molecules, 30(9), 1926. https://doi.org/10.3390/molecules30091926