2. Results and Discussion
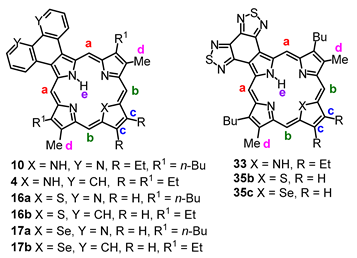
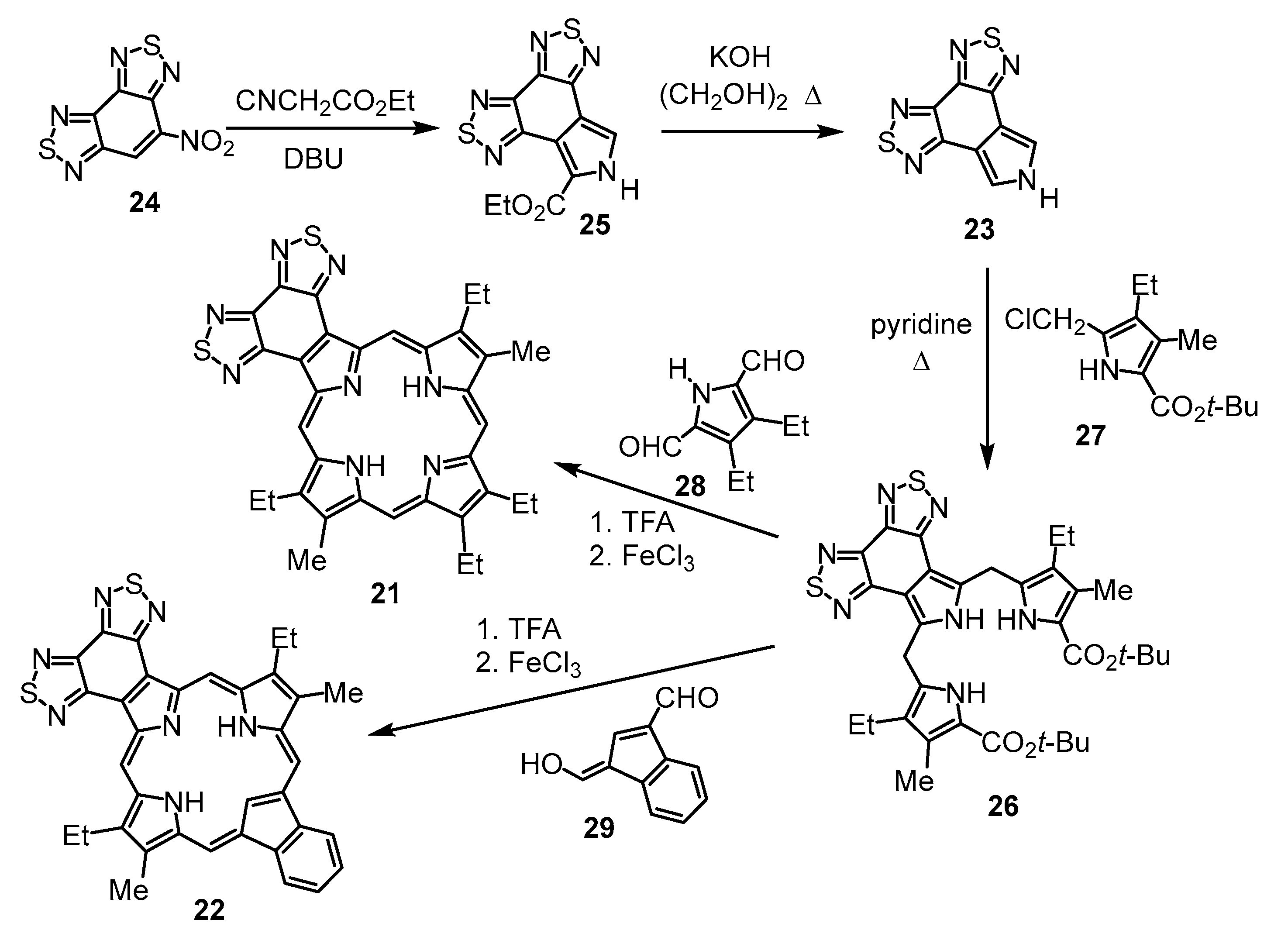
4-Nitrobenzo[1,2-
c:3,4-
c’]bis(1,2,5)thiadiazole (
24) condensed with ethyl isocyanoacetate in the presence of 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) afforded bis-thiadiazolobenzo[4,5-
c]pyrrole
25 in 84% yield [
27] (
Scheme 1). Cleavage of the ethyl ester with KOH in ethylene glycol at 180 °C gave a quantitative yield of the related unsubstituted heterocycle
23. Attempts to form tripyrrane
26 under acidic conditions were unsuccessful. However, when chloromethylpyrrole
27 was treated with pyridine for 1 h and then reacted with
23 under refluxing conditions, tripyrrane
26 could be isolated in 25% yield [
27]. Treatment of
26 with trifluoroacetic acid (TFA) to cleave the terminal
tert-butyl ester groups, followed by condensation with pyrrole dialdehyde
28 or indene dialdehyde
29, afforded porphyrin
21 in 35% yield and carbaporphyrin
22 in 25% yield [
27] (
Scheme 1).
Unfortunately, these products are virtually insoluble in organic solvents, and characterization of the free base forms by NMR spectroscopy was not possible. In an attempt to overcome this problem, a tripyrrane with butyl substituents was targeted (
Scheme 2).
tert-Butyl 4-butyl-3,5-dimethylpyrrole-2-carboxylate (
30) was reacted with
N-chlorosuccinimide (NCS) to give chloromethylpyrrole
31. As was the case for
27 [
14], chloromethylpyrrole
31 proved to be unstable. As it was too soluble in solvents such as hexane to facilitate recrystallization and chromatography led to decomposition,
31 was used without purification. The crude chloromethylpyrrole was stirred with pyridine for 1 h at room temperature to generate a pyridinium derivative,
23 was added, and the resulting mixture was refluxed under nitrogen for 16 h. Following column chromatography on silica, eluting with 1% triethylamine–dichloromethane, crude tripyrrane
32 was isolated in 22% yield.
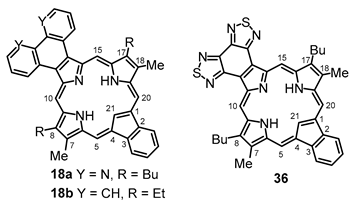
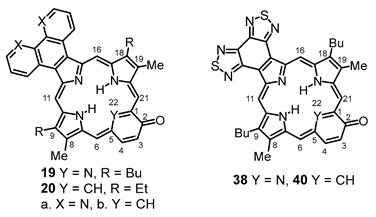
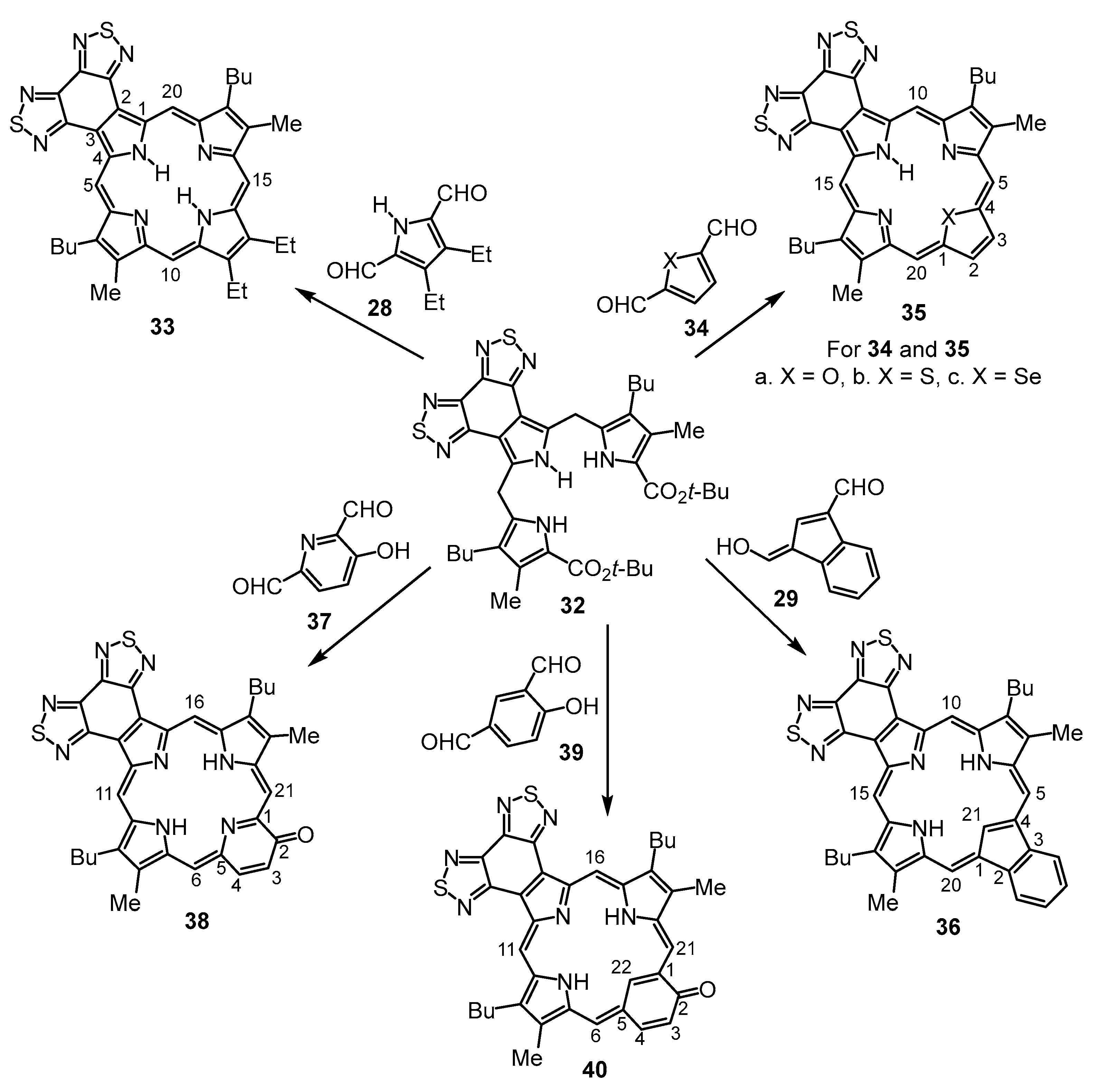
Tripyrrane
32 was deprotected with TFA and reacted with a series of dialdehydes to give, following oxidation with aqueous solutions of ferric chloride, porphyrin
33, and a series of porphyrin analogs (
Scheme 3). Reaction with pyrrole dialdehyde
28 [
29], followed by purification by column chromatography on grade 3 alumina and recrystallization from chloroform–methanol, gave bis(thiadiazolo)benzoporphyrin
33 in 36% yield. Similarly, condensation with furan dialdehyde
34a gave oxaporphyrin
35a, but it was necessary to purify this system in protonated form after washing the solution with hydrochloric acid. The resulting hydrochloride was isolated in 20% yield. Thiophene dialdehyde
34b gave relatively poor results affording thiaporphyrin
35b in only 8% yield. Selenophene dialdehyde gave even worse results, affording only trace amounts of impure selenaporphyrin
35c. It is unclear why these heteroporphyrins are generated in inferior yields, and while the larger chalconide atoms might inhibit cyclizations, this methodology has been utilized to prepare thia- and selenaporphyrins [
30], including phenanthrene- and phenanthroline-fused structures [
9,
22], in good to excellent yields. Reaction of indene dialdehyde
29 [
31] with
32 afforded carbaporphyrin
36 in 24% yield, while reaction with 3-hydroxy-2,6-pyridinedicarbaldehyde (
37) [
32] gave oxypyriporphyrin
38 in 32% yield. Poorer results were obtained with 4-hydroxy-isophthalaldehyde (
39), but oxybenziporphyrin
40 could still be isolated in 12% yield.
The presence of two butyl substituents did improve the solubility of these porphyrinoids, but it remained challenging to obtain proton NMR data for the free base structures. Protonation improved the solubility to a limited extent, but in most cases, only poor-quality
13C{
1H} spectra could be obtained. The free base form of porphyrin
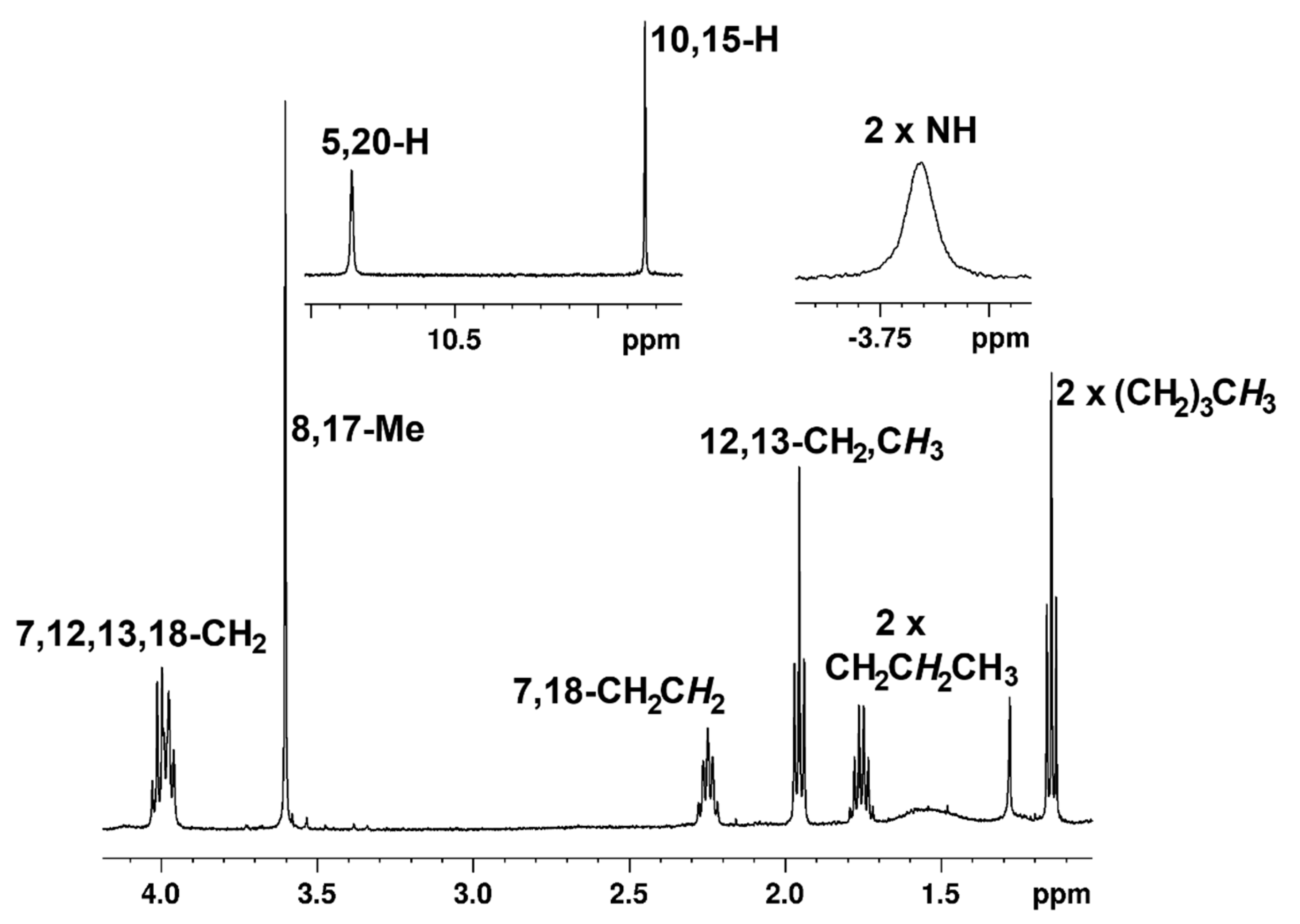
33 gave the best results. At 29 °C, the
meso-proton resonances appeared downfield as two 2H singlets at 10.50 and 9.76 ppm. The methyl substituents gave rise to a 6H singlet at 3.56 ppm, and the internal NHs gave a broad peak at −4.19 ppm (
Table 1). Although some minor shifts were observed at 55 °C (
Table 1,
Figure 5), the data in both cases showed that the global diatropic ring current for
33 is not significantly affected by the presence of the fused heterocycle. A
13C{
1H} spectrum of
33 could not be obtained, but an HSQC spectrum was recorded over a period of approximately 15 h, and this showed the
meso-carbon resonances at 101.8 and 96.2 ppm. In TFA-CDCl
3, the
meso-protons shifted downfield to give two 2H singlets at 12.19 and 10.78, and the methyl substituents could be identified as a 6H singlet at 3.74 ppm. These results demonstrate increased diatropicity, a phenomenon that is commonly observed for protonated porphyrins [
33]. The UV-vis spectrum of
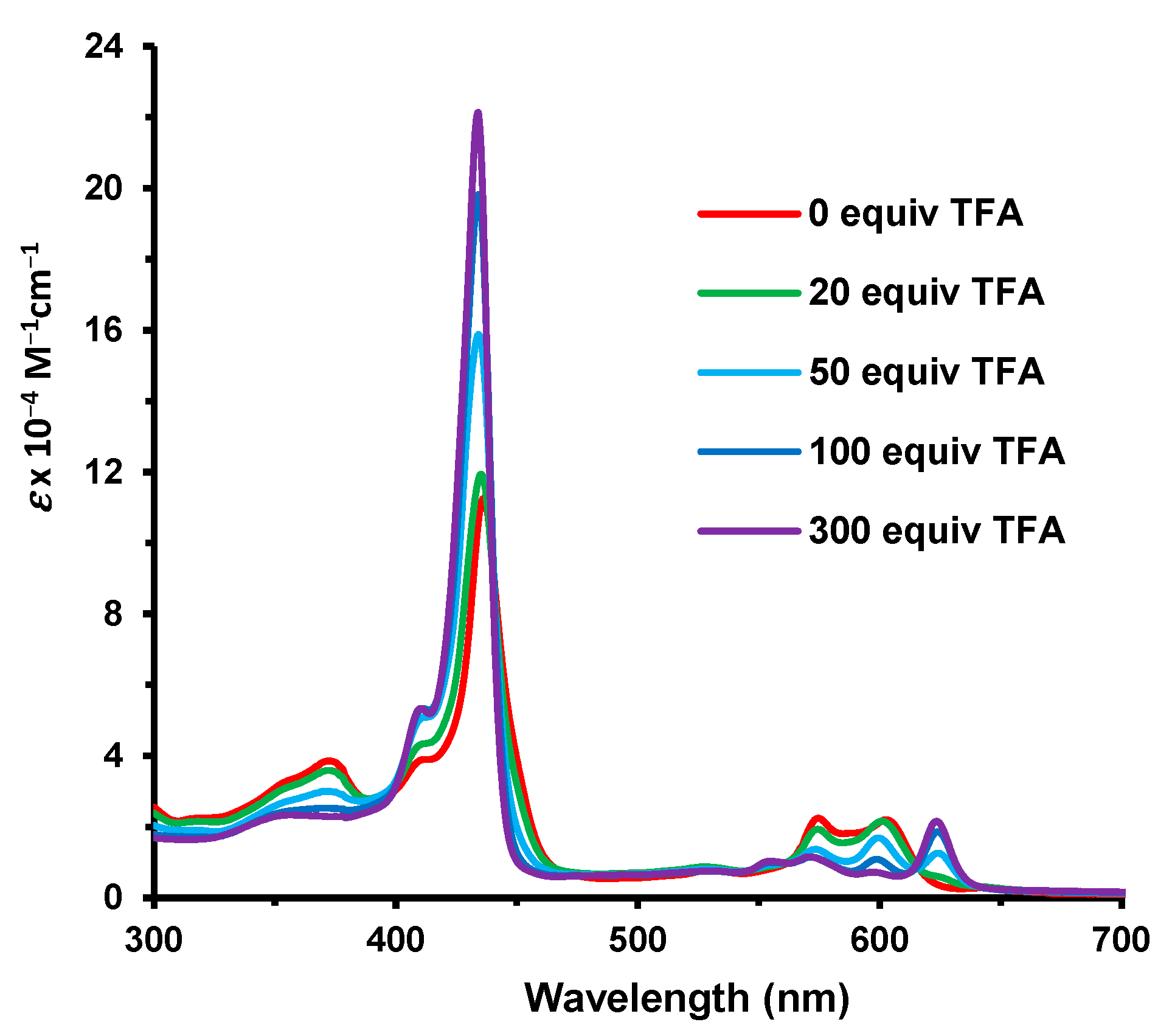
33 is significantly altered as the Soret band is shifted to 430 nm and the visible region is dominated by two Q bands at 575 and 693 nm (
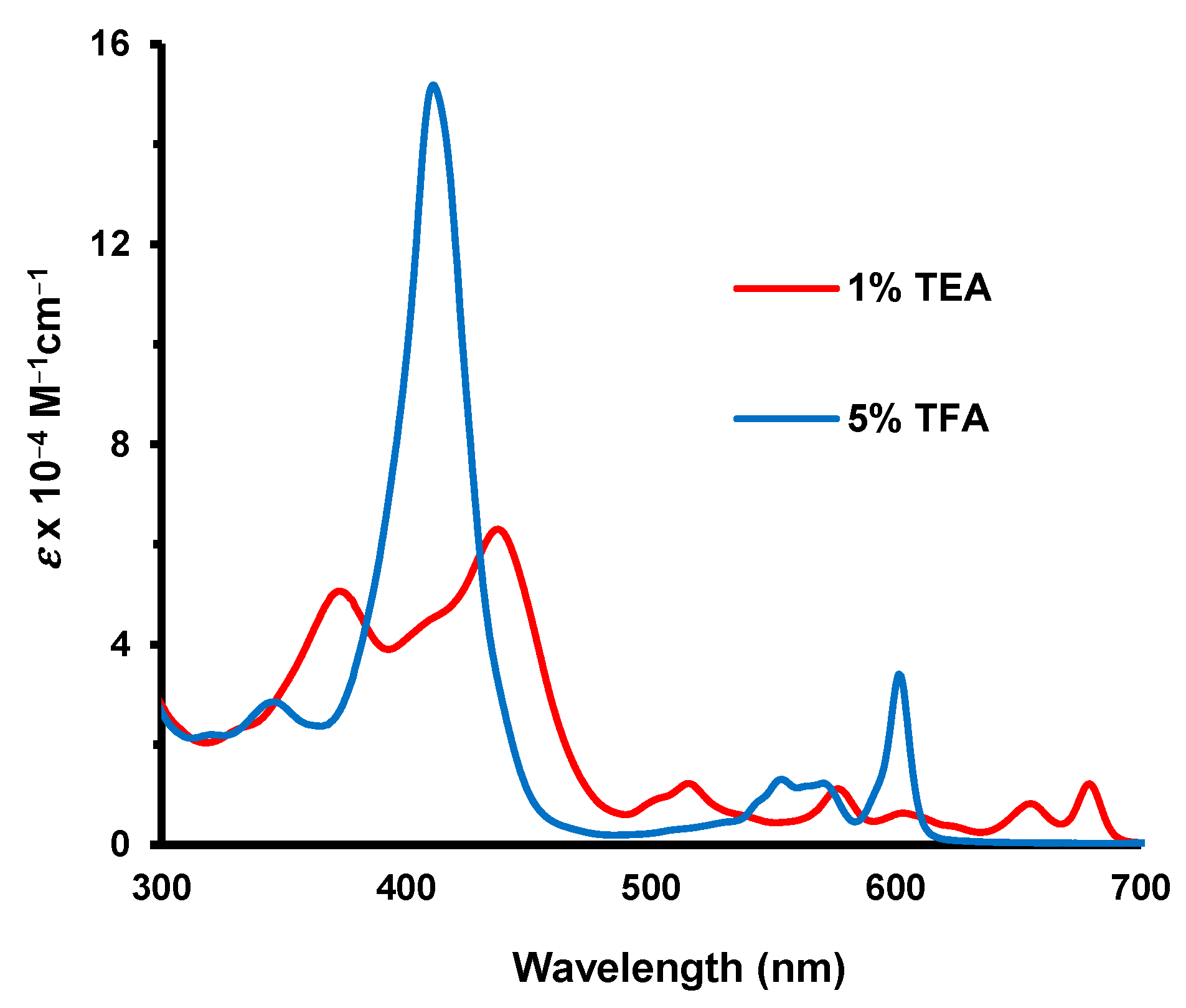
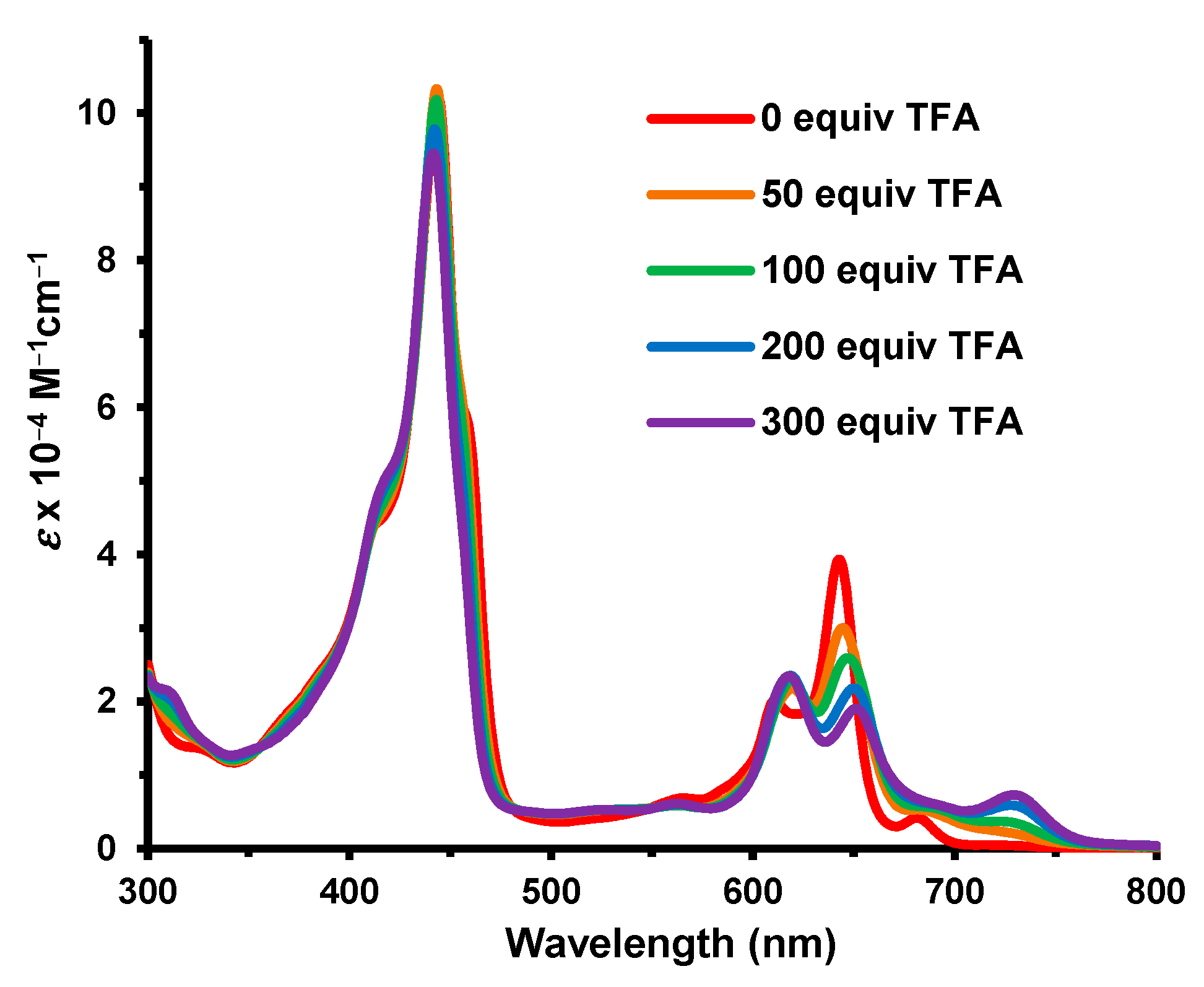
Figure 6). The fused heterocycle drastically reduced the basicity of the porphyrin and approximately 300 equivalents of TFA were required to monoprotonate the system. The Soret band for this species appeared at 434 nm and was greatly enhanced compared to the free base (
Figure 5). At higher concentrations of TFA, only minor spectroscopic changes were noted.
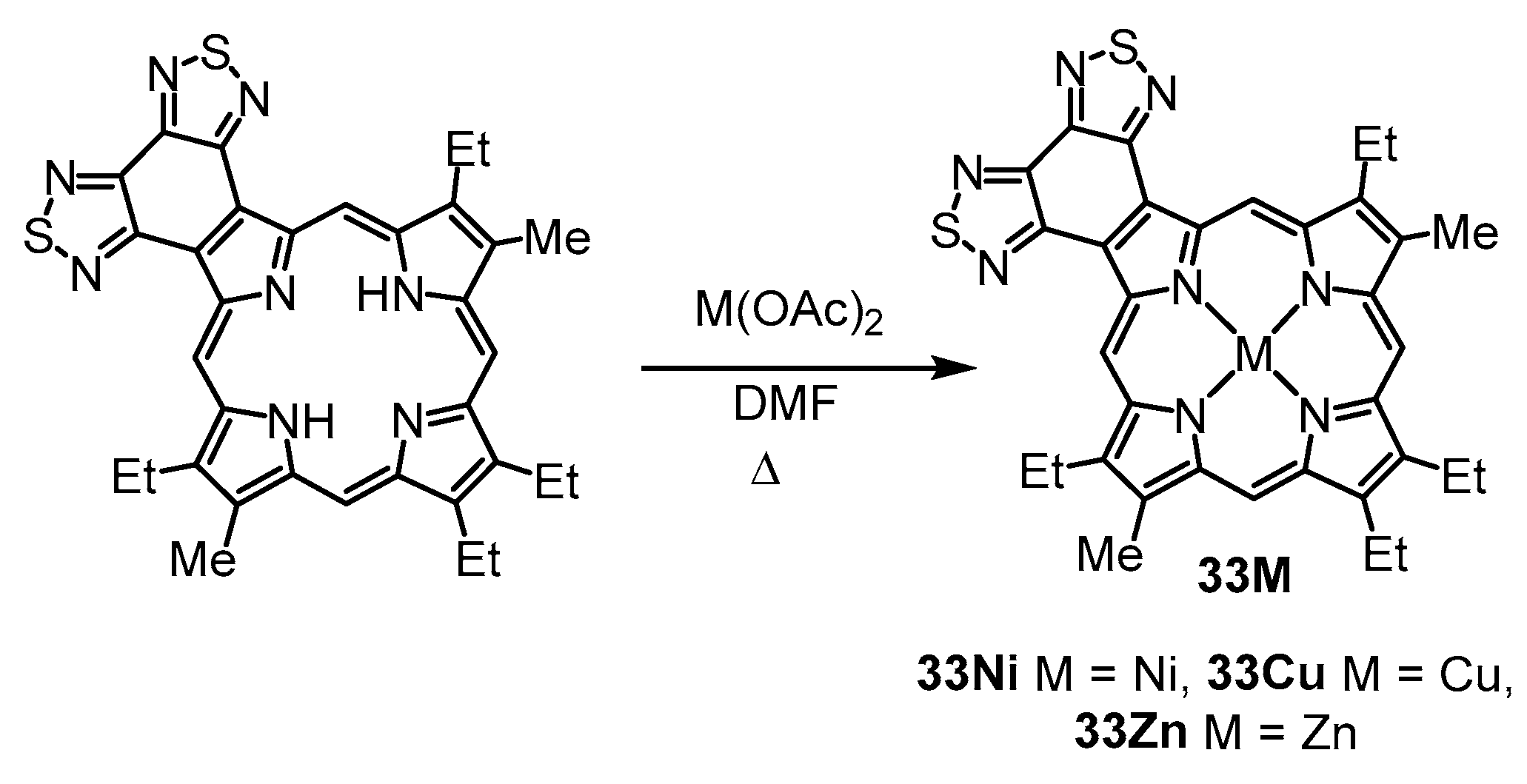
Porphyrin
33 was reacted with nickel(II) acetate, copper(II) acetate, and zinc acetate in refluxing
N,
N-dimethylformamide (DMF), and the related metal complexes
33M were isolated in 67–95% yield (
Scheme 4). The UV-vis spectra for these derivatives gave rise to intense Soret bands and a strong secondary absorption above 600 nm (
Figure 7). Porphyrinoids with strong absorptions in the red or far red are of interest as photosensitizers in photodynamic therapy [
34,
35], and while these bands are not shifted all that far into this region, they show promise. As expected, bathochromic shifts were observed with increasing atomic number for the coordinating metal cations [
8,
36].
33Ni gave a Soret band at 429 nm and a strong Q band at 608 nm, and these appeared at 435 and 615 nm for
33Cu and 444 and 623 nm for
33Zn. The addition of pyrrolidine to solutions of
33Zn led to further shifts giving a Soret band at 458 nm and a long-wavelength Q band at 633 nm. Pyrrolidine coordinates to zinc porphyrins and is well known to improve solubility [
37]. Copper(II) porphyrins are paramagnetic and do not give useful NMR spectra. However,
33Ni was sufficiently soluble in CDCl
3 at 55 °C to give a proton NMR spectrum. The
meso-protons gave rise to two 2H singlets at 10.59 and 9.49 ppm, and the methyl substituents afforded a 6H singlet at 3.34 ppm. Zinc complex
33Zn was far less soluble but a poor-quality NMR spectrum could be obtained at 55 °C that showed the analogous resonances at 9.75, 9.36, and 3.21 ppm. The data indicate that the nickel complex has a slightly reduced diamagnetic ring current compared to
33. However, while this also appears to be the case for
33Zn, the shifts may simply be due to aggregation. The addition of pyrrolidine deaggregates the complex, and this results in the
meso-protons shifting downfield to give two 2H singlets at 11.28 and 9.80 ppm, while the methyl resonance appeared as a 6H singlet at 3.56 ppm, results that indicate retention of a powerful aromatic ring current. The results show similar trends to other metalated porphyrins [
8], and the fused heterocyclic unit does not appear to significantly impact the global aromatic character for these structures.
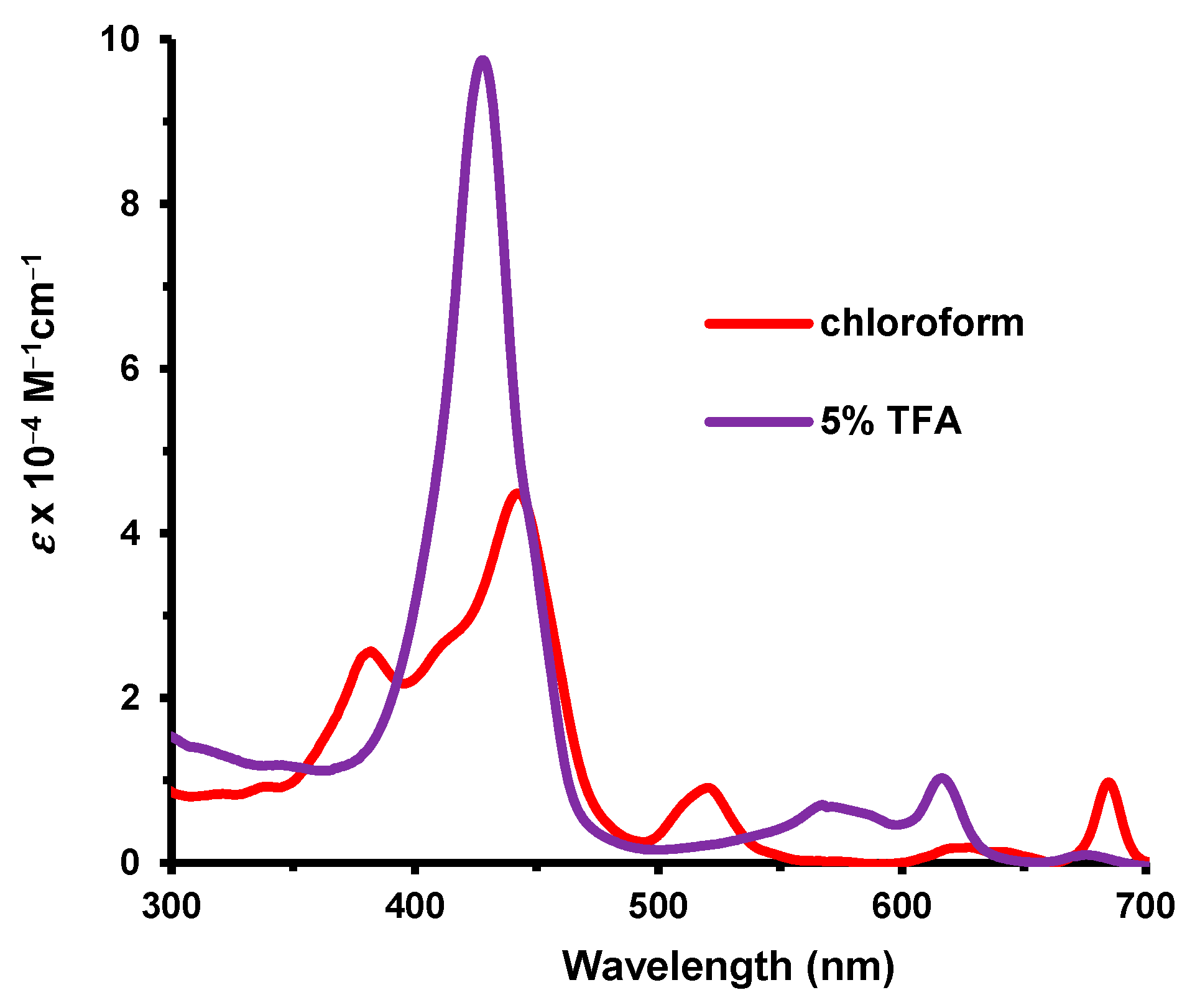
Oxaporphyrin
35a was isolated as a hydrochloride salt. In 1% Et
3N-CHCl
3, the UV-vis spectrum, which corresponded to the free base, showed two broad Soret bands at 373 and 437 nm and a series of minor absorptions in the visible region (
Figure 8). In 5% TFA–chloroform, a strong Soret band emerges at 411 nm that was attributed to a monoprotonated species. A proton NMR spectrum was obtained in TFA-CDCl
3, and this showed the
meso-protons as two 2H singlets at 12.35 and 11.96 ppm, while the methyl groups directly attached to the macrocycle afforded a 6H singlet at 3.91 ppm. Although NMR data could not be obtained for free base
35a, the protonated form clearly has a very strong diamagnetic ring current.
Thiaporphyrin
35b gave a proton NMR spectrum at 50 °C that showed the
meso-protons as two 2H singlets at 10.33 and 10.21 ppm, while the methyl groups gave a 6H singlet at 3.32 ppm (
Table 1). The NH resonance was identified at −4.30 ppm. Although selenaporphyrin
35c was isolated as an impure sample in very low yield, the corresponding resonances were observed at 10.69, 10.53, 3.33, and −3.63 ppm. These spectra show that these heteroporphyrins are strongly diatropic. When contrasting the free base forms of porphyrins, thiaporphyrins, and selenaporphyrins with fused phenanthrene, phenanthroline, and bis(thiadiazolo)benzene rings, only the phenanthroline-fused porphyrinoids show significant upfield shifts to the
meso-protons together with the anomalous upfield shifts to the NH protons [
22], indicating that the previously observed effects are not simply due to the presence of a fused strongly electron-withdrawing unit (
Table 1). The addition of TFA to
35b enhanced the ring current for the protonated form, and the
meso-protons appeared downfield at 12.63 and 11.57 ppm. The UV-vis spectrum for
35b gave a Soret band at 428 nm and a longer-wavelength Q band at 685 nm (
Figure 9). In 5% TFA–chloroform, a strong Soret band emerged at 428 nm.
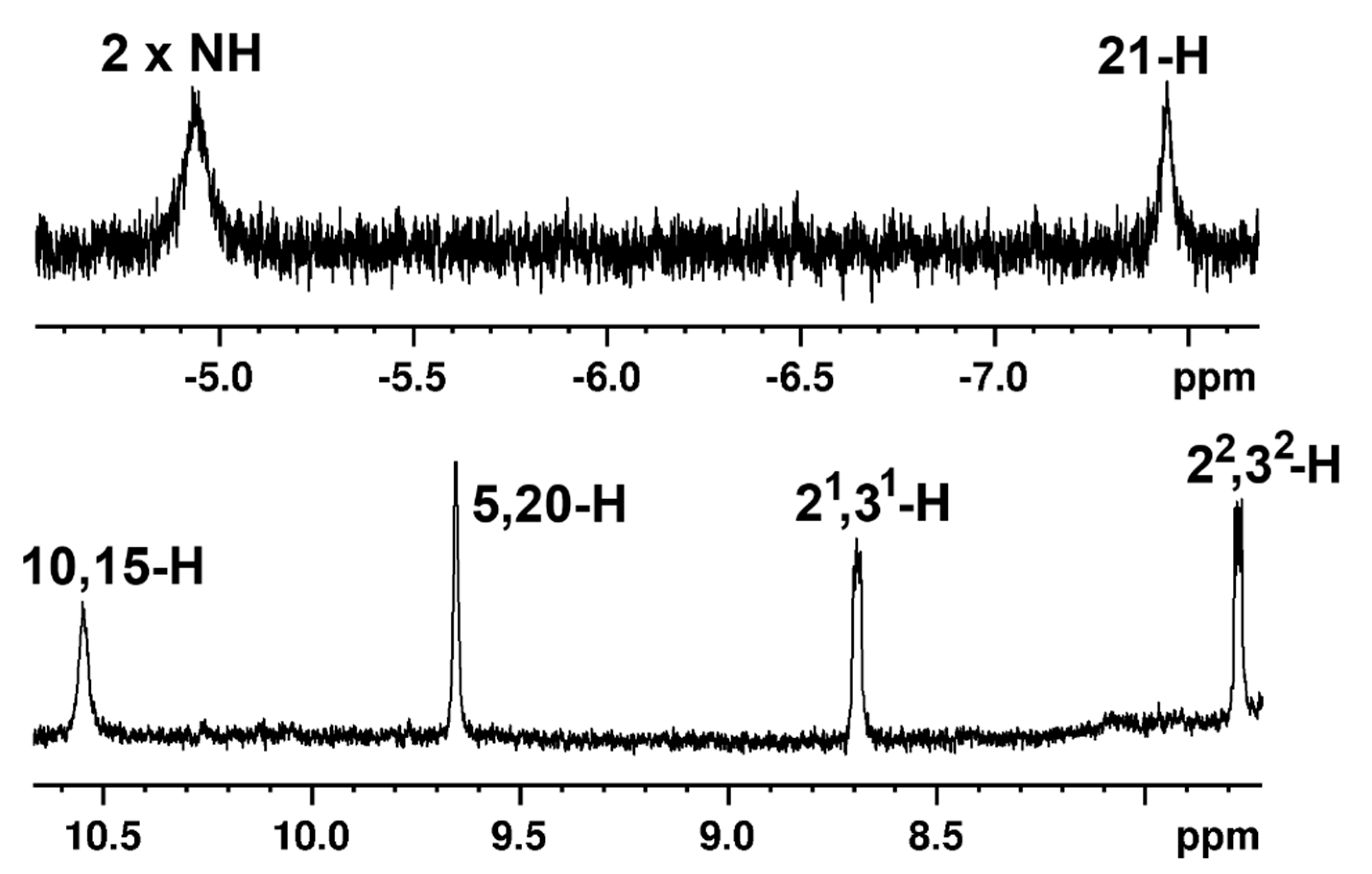
Carbaporphyrin
36 was very poorly soluble, but a low-quality proton NMR spectrum could be obtained in CDCl
3 at 55 °C (
Figure 10). The
meso-protons appeared as two 2H singlets at 10.55 and 9.65 ppm, the methyl groups gave a peak at 3.45 ppm, and the internal NH and 21-H protons showed up at −4.93 and −7.45 ppm. Although the internal and external protons are shifted slightly upfield compared to benzocarbaporphyrins [
25,
38] without the fused heterocycle, the results indicate that the global aromatic properties for this structure have not been significantly altered. In the presence of trace amounts of TFA, the
meso-protons shifted downfield to give two 2H singlets at 11.63 and 10.28 ppm, while the NH and 21-H resonances appeared at −2.57 and −6.51 ppm, respectively. These data indicate that protonation slightly enhances the aromatic properties. The UV-vis spectrum for
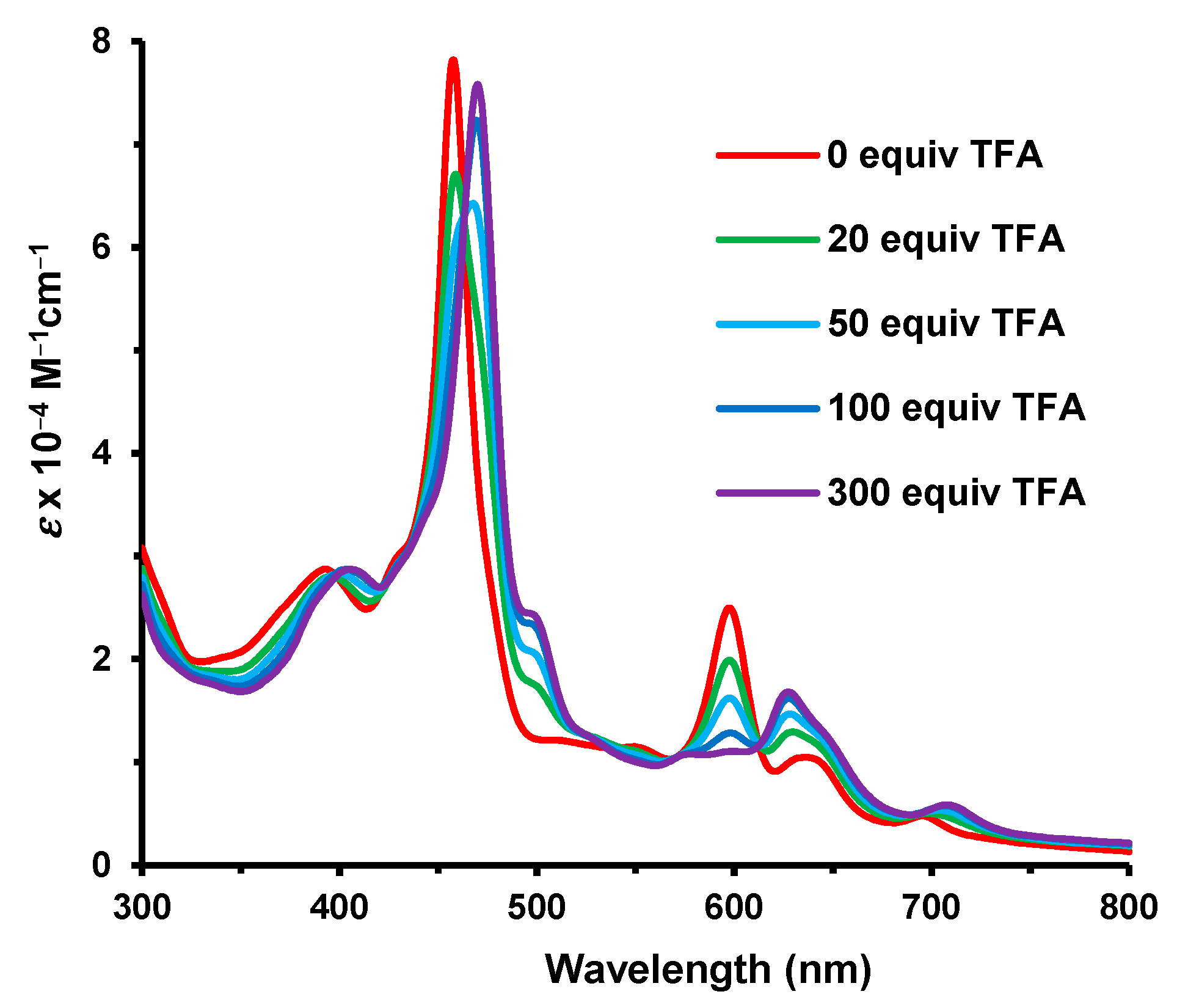
36 gave rise to a Soret band at 458 nm and a series of Q bands that tapered off at 700 nm (
Figure 11). Protonation with TFA required >200 equivalents to complete the formation of the monoprotonated form, and this afforded a Soret band at 471 nm.
Oxypyriporphyrin
38 also has very low solubility, but proton NMR data could be obtained in CDCl
3 at 55 °C. Due to the asymmetry of this system, the
meso-protons gave rise to four 1H singlets, and these appeared at 10.89, 10.83, 10.78, and 9.39 ppm (
Table 3). The inner NH protons produced two broad peaks at −3.91 and −4.01 ppm. These results again show that the bis(thiadiazolo)benzo unit does not significantly impact the aromatic properties of the porphyrinoid system, and the contradictory shifts noted for phenanthroline-fused oxypyriporphyrin
19a are not seen for
38. As expected, the addition of TFA led to the
meso-proton resonances being shifted downfield to give four 1H singlets at 11.82, 11.81, 11.12, and 10.19 ppm. The UV-vis spectrum for
38 in chloroform gave a strong Soret band at 444 nm and a series of Q bands that extend to nearly 700 nm (
Figure 12). Monoprotonation required the addition of ca. 300 equivalents of TFA, but further changes were observed at higher concentrations of TFA. Similar observations were made for
19a.
Oxybenziporphyrin
40 was also rather insoluble, but a proton NMR spectrum could be obtained at 55 °C (
Figure 13 and
Table 3), although this required approximately 1500 transients to obtain an adequate signal-to-noise ratio. The
meso-protons gave signals at 10.52, 10.40, 10.36, and 9.13 ppm; the methyl substituents appeared as two 3H singlets at 3.49 and 3.40 ppm; the internal NH protons gave broad peaks at −3.99 and −4.15 ppm; and the 22-H resonance showed up at −7.08 ppm. The results confirm that the macrocycle is strongly diatropic, but the fused heterocycle does not significantly perturb these results. Once again, the bis(thiadiazolo)benzo unit does not exert the effects observed for tropone-fused or phenanthroline-fused porphyrinoids. Hence, the presence of a strongly electron-withdrawing fused moiety does not in and of itself lead to the effects described for
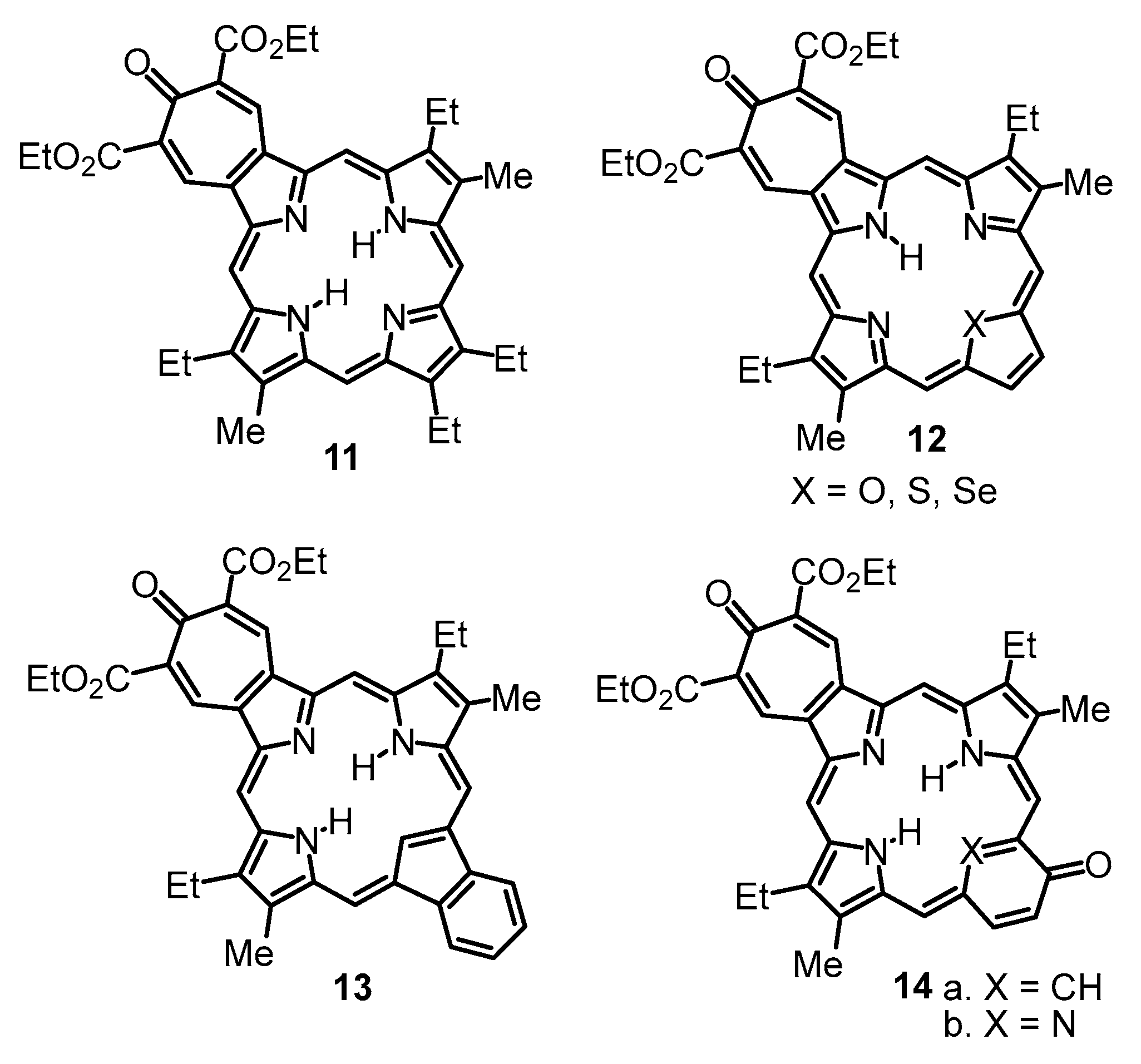
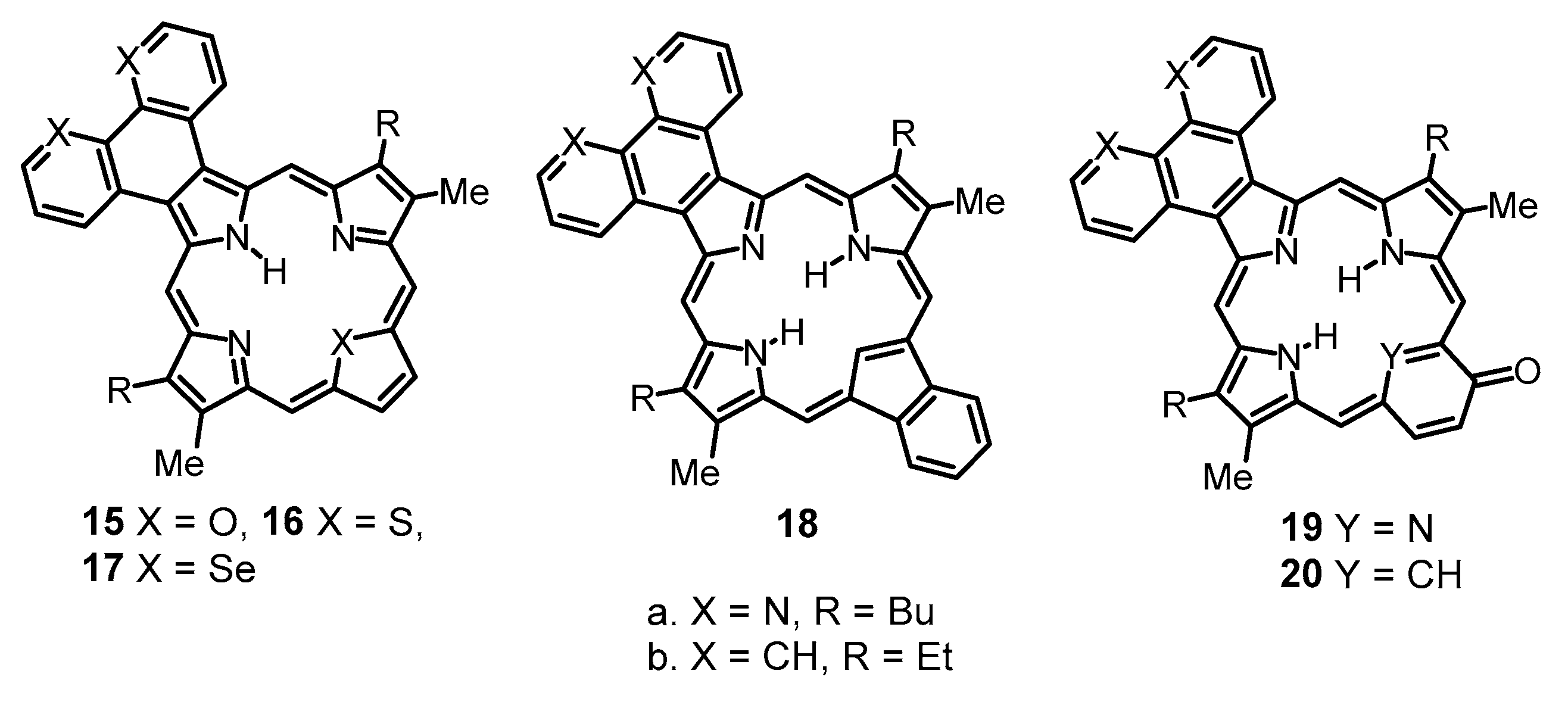
11–
20. The addition of trace amounts of TFA resulted in a slight downfield shift to the
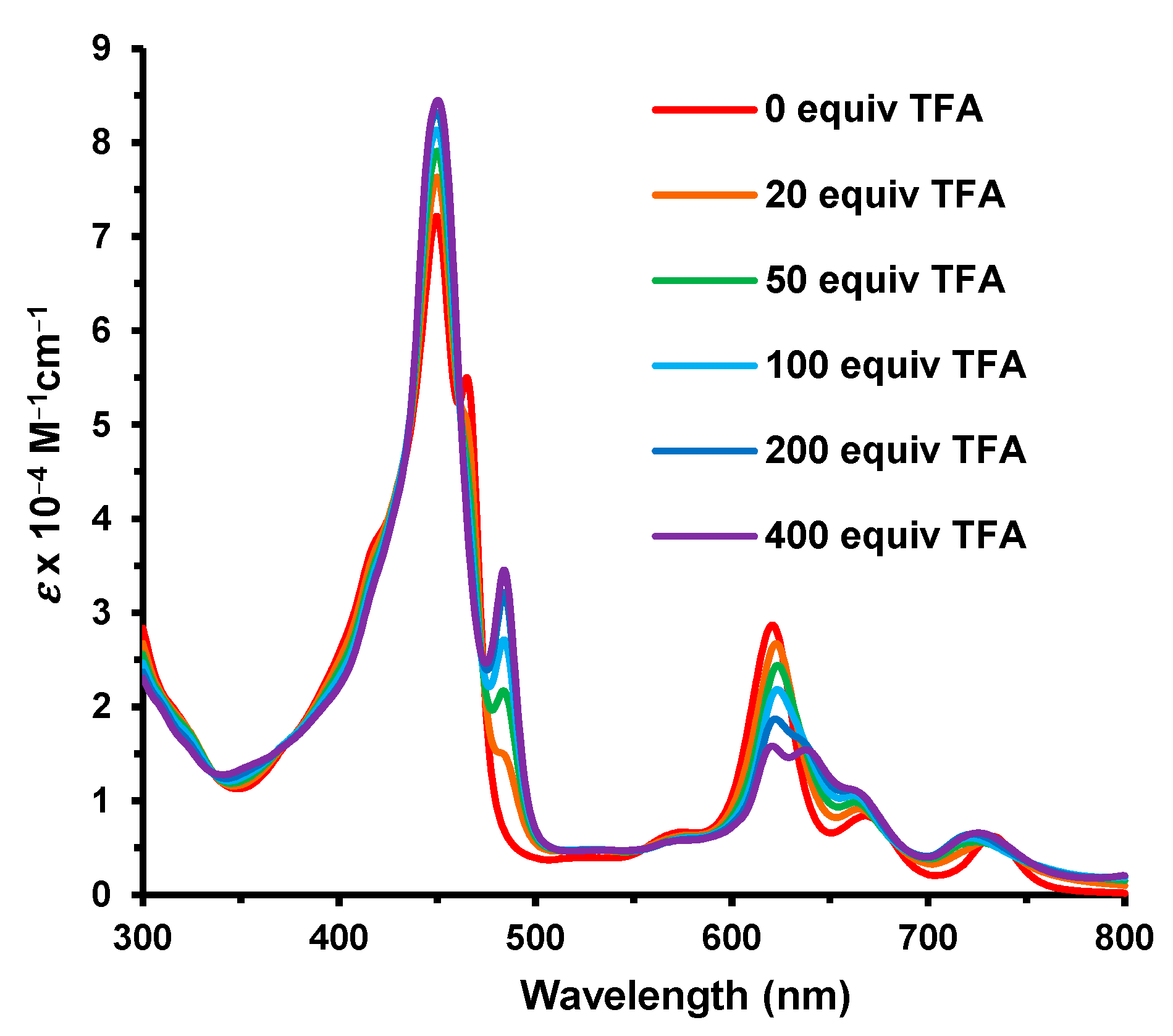
meso-proton resonances, but this is reversed at higher concentrations of TFA. The 22-H shifted downfield towards 0 ppm upon the addition of TFA. This is attributed to protonation onto the carbonyl oxygen as this will result in the arene unit taking on phenolic character, and this interrupts the global delocalization pathways. The UV-vis spectrum of
40 in chloroform gave a Soret band at 450 nm, a secondary absorption at 465 nm, and Q bands at 621, 668, and 733 nm (
Figure 14). Monoprotonation required ca. 400 equivalents of TFA and resulted in an enhancement in the Soret band. At higher concentrations of TFA, this trend was reversed.
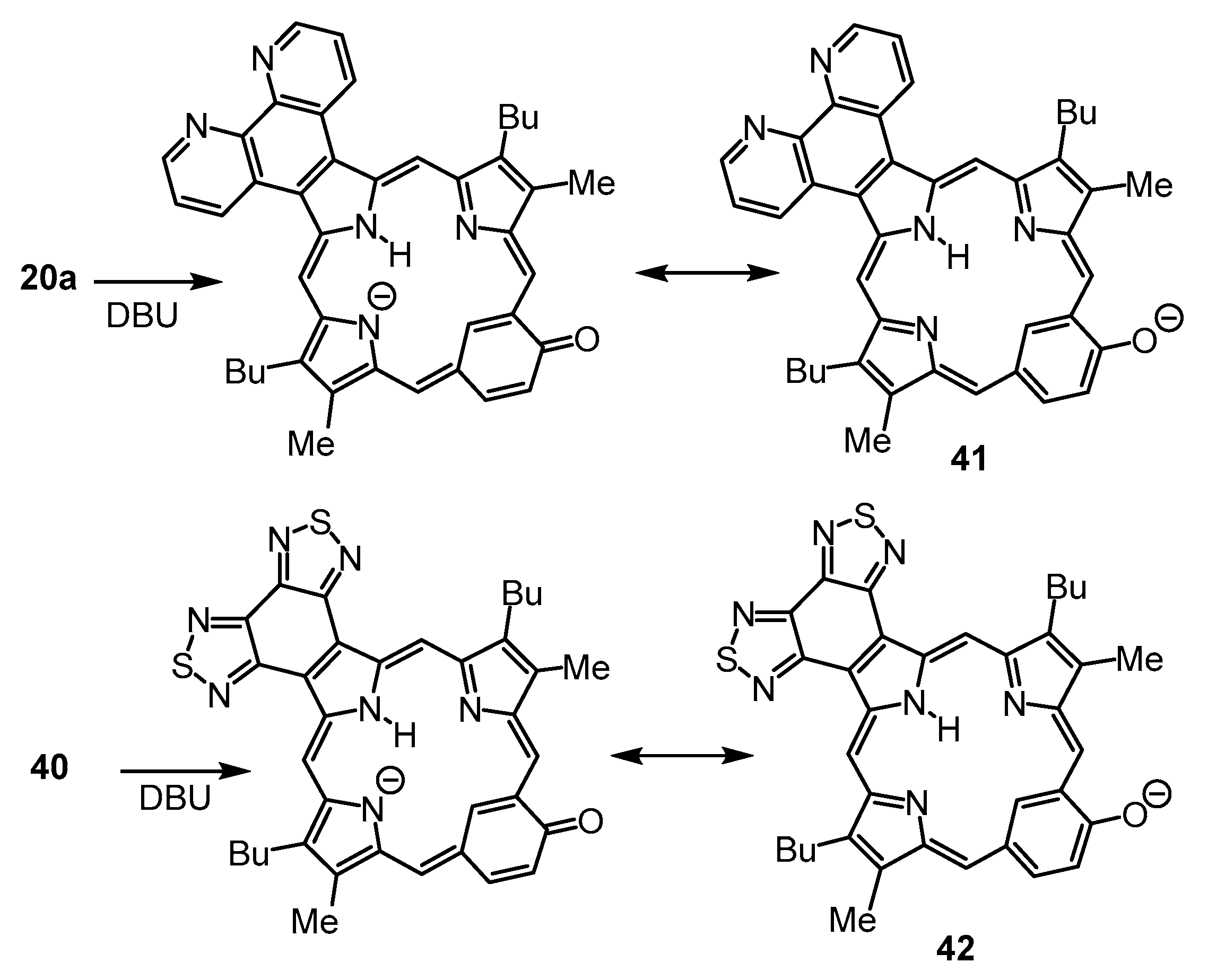
Phenanthrolino-oxybenziporphyrin
20a gave rise to a new species in 1% DBU–chloroform [
22]. This was attributed to deprotonation generating an anionic species
41 (
Scheme 5) that is resonance-stabilized by the carbonyl moiety. We speculated that a similar phenomenon might be observed for oxybenziporphyrin
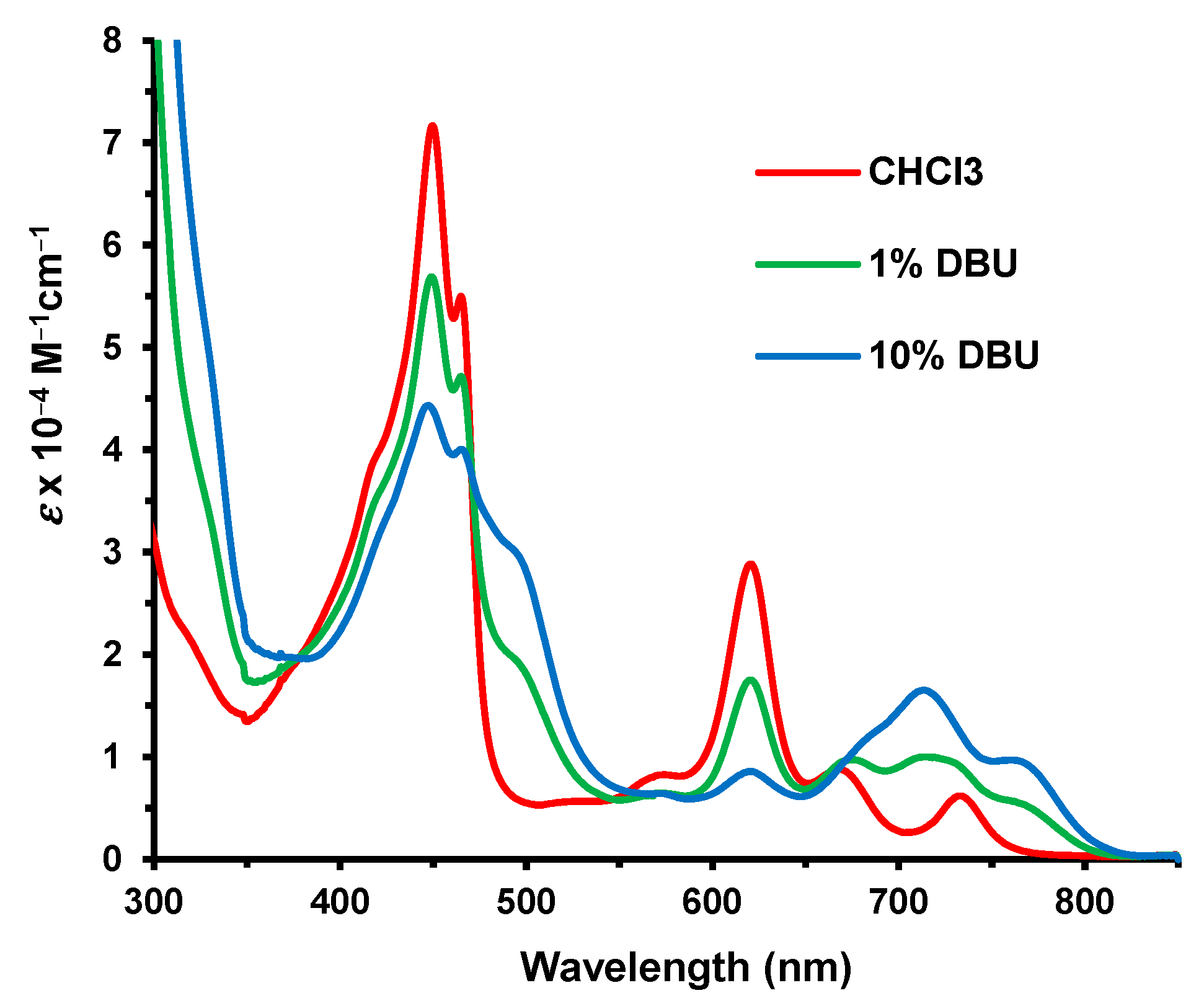
40. Although some spectroscopic changes to the electronic absorption spectra were observed in 1% DBU–chloroform (
Figure 15), even 10% DBU was insufficient to fully convert
40 into a new species (assumed to be
42;
Scheme 5). This result shows that the bis(thiadiazolo)benzo unit is far less effective in stabilizing the negative charge in this type of anionic species compared to 1,10-phenanthroline.
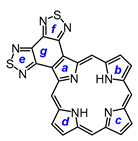
Even though the 1,10-phenanthroline and bis(thiadiazolo)benzene units are both strongly electron-withdrawing, their impact on the aromatic properties of annelated porphyrinoids is quite different. In order to gain insights into these differences, density functional theory (DFT) calculations [
39,
40,
41,
42,
43] were carried out. The structures were optimized using M06-2X with the triple-
ζ basis set 6-311++G(d,p). Four tautomers of unsubstituted bis(thiadiazolo)benzoporphyrin (
S2-BP) were considered, and the two forms with opposite N−H protons were shown to have the lowest energies (
Table 4). Tautomer
S2-BPa, which has an NH on the pyrrole that is fused to the heterocycle, is slightly less stable than
S2-BPb. The aromatic character of these structures was assessed using nucleus-independent chemical shift (NICS) calculations [
44]. Standard NICS calculations include effects due to
σ and
π electrons and are not always accurate, and in this study, NICS(0) and NICS(1)
zz calculations were carried out. In NICS(1)
zz, the calculations were performed 1 Å above the ring, and the results provide a more accurate measure of the diatropic ring currents. Negative values correspond to aromatic species, while positive values are obtained for antiaromatic systems. Positive values may also be observed when the NICS values are measured at points that are external to the aromatic delocalization pathways. All four tautomers gave strongly aromatic NICS(0) and NICS(1)
zz values (
Table 4). The NICS
zz values are much larger than those for standard NICS calculations, but otherwise, the observed trends were similar. The porphyrinoid structures were also assessed using anisotropy of induced current density (AICD) [
45]. The AICD plots for
S2-BPa and S2-BPb show the presence of 18
π-electron circuits within the macrocycle, and the heterocyclic unit appears to be totally disconnected, showing no significant interactions (
Figure 16). This differs from the results for phenanthroline-fused porphyrins as the AICD plot for the tautomer analogous to
S2-BPa shows the presence of a significant 30
π-electron diatropic circuit that extends around the periphery of the fused phenanthroline unit.
NICS and NICS
zz calculations for unsubstituted oxaporphyrin
S2-OxBP, thiaporphyrin
S2-BTP, and selenaporphyrin
S2-BSP all show strongly aromatic values for the two most likely tautomers, although structures with the NH opposite to the chalconide atom are favored (
Table 5). The AICD plots again showed that the fused heterocycle’s
π-system does not interact with the conventional porphyrin-type 18
π-electron pathways. In contrast, heteroporphyrins with fused phenanthroline units show an extended 30
π aromatic circuit through the phenanthroline [
22]. Four tautomers of unsubstituted bis(thiadiazolobenzo)carbaporphyrin (
S2-BCBP) were considered, all of which have 18
π-electron delocalization pathways (
Table 6). Tautomers
S2-BCBPc and
S2-BCBPd with internal methylene units are far higher in energy, and
S2-BCBPa with opposite NHs is favored. NICS and NICS
zz values for
S2-BCBPa show that the macrocycle is strongly aromatic. Ring g gives a low positive value indicating that it lies outside of the aromatic pathways, a feature that is seen in the calculations for all of the bis(thiadiazolobenzo)porphyrinoids. The AICD plot for
S2-BCBPa (
Figure 17) reinforces this interpretation, once again showing that the fused heterocycle does not facilitate extended aromatic conjugation pathways. However, this type of extended aromatic circuit is seen in the AICD plot for the analogous phenanthroline-fused carbaporphyrin [
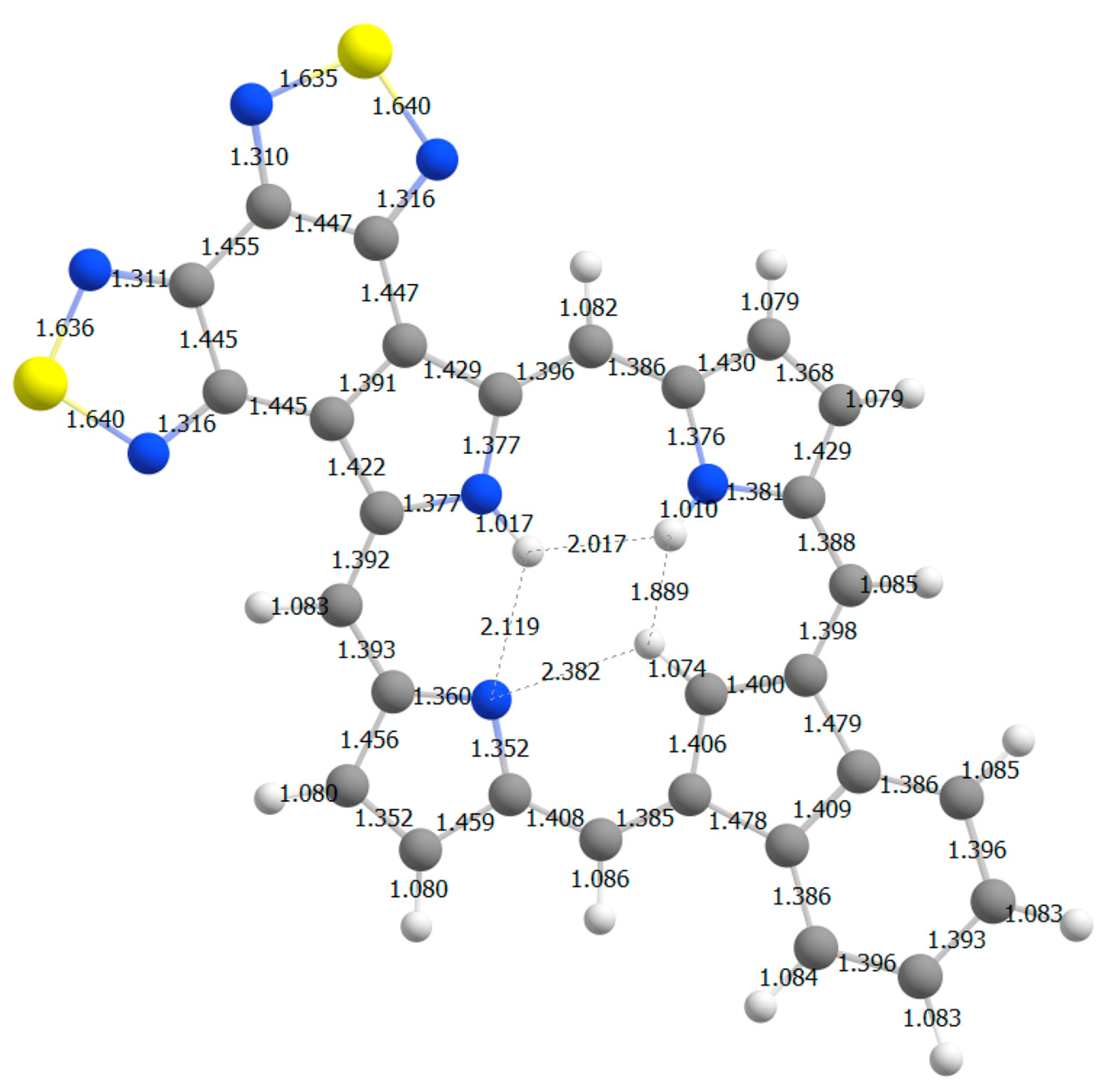
22]. A bond length analysis of
S2-BCBPa is also consistent with this conclusion (
Figure 18). The bonds connecting the thiadiazole units to the porphyrinoid macrocycle are relatively long, as is the case for the bonds connected to the benzo-unit, indicating that both of the fused rings are only weakly interacting with the aromatic
π-system.
Five tautomers of unsubstituted pyriporphyrin
S2-OPBP were considered (
Table 7). Hydroxypyridine tautomer
S2-OPBPd is much less stable, but an unconventional tautomer
S2-OPBPe bearing an NH on the pyridine moiety is only ca. 5 kcal less stable than the favored tautomer
S2-OPBPa, suggesting that this structural arrangement should be easily accessible. Otherwise, as expected, the tautomer with opposite NHs on the pyrrole subunits is the most stable form. NICS and NICS
zz calculations show that all five tautomers are fully aromatic. The AICD plot for
S2-OPBPa shows that the fused heterocycle is disconnected from the macrocycle’s
π-system even though the related phenanthroline-fused system shows the presence of an extended 30
π-electron pathway. Four tautomers of oxybenziporphyrin
S2-OBBP were also investigated (
Table 8). Phenolic tautomer
S2-OBBPd is the least stable for this set, and semiquinone tautomer
S2-OBBPa with opposite NHs is the most stable form. NICS and NICS
zz calculations show that tautomer
S2-OBBPa is at most weakly aromatic, but the remaining tautomers exhibit strongly diatropic properties. For
S2-OBBPa, rings c and g have low positive values, indicating that they lie outside of the global aromatic circuit. AICD plots also show the presence of an 18
π-electron aromatic pathway that does not interact to any extent with the fused heterocycle. The analogous phenanthroline-fused oxybenziporphyrin possesses a 30
π-electron circuit, as is the case for the other phenanthrolinoporphyrinoids discussed in this paper.
The results indicate that the anomalous proton NMR spectra observed for phenanthroline-fused porphyrinoids are triggered by the presence of extended conjugation pathways through the phenanthroline moiety. Extended aromatic circuits do not contribute to the delocalization pathways in porphyrinoids with fused bis(thiadiazolo)benzene units, and this, presumably, is the reason that these two series are so different from one another. While the results for the bis(thiadiazolo)benzene-fused porphyrinoids are self-consistent and the computational data are in good agreement with the experimental results, further studies will be needed to explain the anomalous results for tropone-fused and phenanthroline-fused porphyrinoids.
3. Experiments
Chemicals and solvents were purchased from Fisher (Pittsburgh, PA, USA) or Sigma-Aldrich (Burlington, MA, USA). Melting points were uncorrected. NMR spectra were recorded using a 400 or 500 MHz NMR spectrometer (Bruker, Billerica, MA, USA) and were recorded at 302 K unless otherwise indicated. 1H NMR values are reported as chemical shifts δ, relative integral, multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; m, multiplet; br, broad peak) and coupling constant (J). Chemical shifts are reported in parts per million (ppm) relative to CDCl3 (1H residual CHCl3 singlet δ 7.26 ppm, 13C CDCl3 triplet δ 77.23 ppm), and coupling constants were taken directly from the spectra. NMR assignments were made with the aid of 1H-1H COSY, HSQC, DEPT-135, and nOe difference proton NMR spectroscopy. Two-dimensional NMR experiments were performed using standard software. Mass spectral data were acquired using positive-mode electrospray ionization (ESI+) and a high-resolution time-of-flight mass spectrometer (Agilent, Santa Clara, CA, USA).
Tripyrrane 32. N-Chlorosuccinimide (0.350 g. 2.6 mmol) was added to a solution of
tert-butyl 4-butyl-3,5-dimethyl-pyrrole-2-carboxylate (
30) [
20] (0.650 g, 2.59 mmol) in dichloromethane (50 mL), and the mixture was stirred at room temperature overnight. The solution was washed with water (3 × 50 mL) and evaporated under reduced pressure to give crude chloromethylpyrrole
31 as a yellow-brown oil. The residue was taken up in pyridine (20 mL) and stirred under nitrogen at room temperature for 1 h. Bis-thiadiazolobenzo[4,5-
c]pyrrole
23 [
27] (200 mg, 0.858 mmol) was then added, and the mixture was refluxed with stirring under a nitrogen atmosphere for 16 h. The solution was cooled, diluted with dichloromethane, and washed with water. The organic layer was evaporated, and the residue was purified on a silica column, eluting with 1% triethylamine–dichloromethane, giving the crude tripyrrane as a brown solid (138 mg, 0.188 mmol, 22%).
1H NMR (500 MHz, CDCl
3): δ 9.22 (br s, 2H, 2 × NH), 8.58 (br s, 1H, NH), 4.45 (s, 4H, 2 × bridge-CH
2), 2.38 (t, 4H,
J = 7.5 Hz, 2 × pyrrole-CH
2), 2.23 (s, 6H, 2 × pyrrole-CH
3), 1.39–1.26 (m, 8H), 1.41 (s, 18H, 2 ×
t-Bu), 0.86 (t, 6H,
J = 7.0 Hz, 2 × (CH
2)
3C
H3).
13C{
1H} NMR (CDCl
3): δ 161.4, 154.3, 148.8, 128.4, 127.3, 126.4, 122.9, 119.5, 111.5, 80.5, 33.7, 28.7, 24.0, 23.9, 22.8, 14.1, 10.8. HRMS (ESI):
m/z [M + H]
+ calcd for C
38H
50N
7O
4S
2 732.3360; found 732.3361.
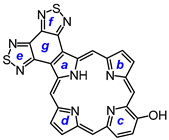
7,18-Dibutyl-12,13-diethyl-8,17-dimethylbis(thiadiazolo)benzo[4,5-c]porphyrin (33). The foregoing crude tripyrrane 32 (50.0 mg, 0.068 mmol) was dissolved in trifluoroacetic acid (1 mL) and stirred at room temperature for 7 min under nitrogen. The mixture was diluted with dichloromethane (20 mL); this was followed by the addition of pyrroledialdehyde 28 (12.2 mg, 0.068 mmol). The mixture was stirred under nitrogen at room temperature for an additional 2 h. The dark solution was diluted with chloroform and vigorously shaken with 0.15% aqueous ferric chloride solution (100 mL) for 5–7 min. The organic solution was washed with water, 5% sodium bicarbonate solution, and water, and the solvent was evaporated under reduced pressure. The residue was purified on a grade 3 alumina column, eluting with dichloromethane. A deep-green fraction was collected and recrystallized from chloroform–methanol to give the title porphyrin (16.6 mg, 0.0247 mmol, 36%) as deep purple crystals, mp > 260 °C. UV-vis (CHCl3): λmax/nm (log ε) 372 (4.58), 410 (sh, 4.59), 436 (5.05), 530 (3.89), 575 (4.35), 603 (4.34), 642 (3,45). UV-vis (300 eq TFA-CHCl3): λmax/nm (log ε) 410 (4.73), 434 (5.34), 554 (4.00), 571 (4.06), 598 (3.86), 623 (4.33). UV-vis (1% TFA-CHCl3): λmax/nm (log ε) 353 (4.00), 409 (4.33), 433 (5.37), 570 (4.05), 622 (4.33). UV-vis (5% TFA-CHCl3): λmax/nm (log ε) 408 (4.75), 431 (5.39), 569 (4.05), 622 (3.31). 1H NMR (500 MHz, CDCl3, 32 °C): δ 10.50 (s, 2H, 5,20-H), 9.76 (s, 2H, 10,15-H), 4.00 (q, 4H, J = 7.7 Hz, 12,13-CH2), 3.85 (t, 4H, J = 7.8 Hz, 7,18-CH2), 3.56 (s, 6H, 8,17-Me), 2.15 (pentet, 4H, J = 7.5 Hz, 7,19-CH2CH2), 1.95 (t, 6H, J = 7.7 Hz, 12,13-CH2CH3), 1.68 (sextet, 4H, J = 7.4 Hz, 2 × CH2CH2CH3), 1.09 (t, 6H, J = 7.4 Hz, 2 × (CH2)3CH3), −4.19 (br s, 2H, 2 × NH). 1H NMR (500 MHz, CDCl3, 55 °C): δ 10.86 (s, 2H, 5,20-H), 9.85 (s, 2H, 10,15-H), 4.03–3.96 (m, 8H, 7,12,13,18-CH2), 3.60 (s, 6H, 8,17-Me), 2.25 (pentet, 4H, J = 7.5 Hz, 7,19-CH2CH2), 1.95 (t, 6H, J = 7.7 Hz, 12,13-CH2CH3), 1.75 (sextet, 4H, J = 7.4 Hz, 2 × CH2CH2CH3), 114 (t, 6H, J = 7.4 Hz, 2 × (CH2)3CH3), −3.77 (br s, 2H, 2 × NH). Partial 13C NMR derived from HSQC spectrum (CDCl3, 55 °C): δ 101.8 (5,20-CH), 96.2 (10.15-CH), 34.8 (7,18-CH2CH2), 26.0 (7,18-CH2), 23.0 (2 × CH2CH2CH3), 19.8 (12,13-CH2), 18.2 (12,13-CH2CH3), 14.0 (2 × (CH2)3CH3), 11.3 (8,17-Me). 1H NMR (500 MHz, TFA-CDCl3): δ 12.19 (s, 2H, 5,20-H), 10.76 (s, 2H, 10,15-H), 4.31 (t, 4H, J = 7.8 Hz, 7,18-CH2), 4.20 (q, 4H, J = 7.7 Hz, 12,13-CH2), 3.74 (s, 6H, 8,17-CH3), 2.31–2.35 (m, 4H, 8,17-CH2CH2), 1.84–1.74 (m, 10H, 12,13-CH2CH3 and 7,18-CH2CH2CH2), 1.15 (t, J = 7.4 Hz, 7,18-(CH2)3CH3), −3.32 (br s), −3.37 (br s) (3H). 13C{1H} NMR (TFA-CDCl3): δ 152.6, 150.3, 146.3, 145.8, 144.8, 143.8, 143.5, 139.8, 136.4, 126.4, 103.2 (5,20-CH), 99.8 (10,15-CH), 34.7 (7,18-CH2CH2), 27.1 (7,18-CH2), 23.4 (2 × CH2CH2CH3), 20.2 (12,13-CH2), 17.3 (12,13-CH2CH3), 13.8 (2 × (CH2)3CH3), 12.1 (8,17-Me). HRMS (ESI+): m/z [M + H]+ calcd for C38H41N8S2: 673.2890; found 673.2875.
Metal complexes of 33 were prepared by refluxing the porphyrin (9.0 mg, 0.0134 mmol) with 20 mg of a zinc, copper(II), or nickel(II) acetate in DMF (10 mL) for 3–16 h. After diluting the mixture with chloroform, washing with water, and evaporating the solvent under reduced pressure, the residues were recrystallized from chloroform–methanol to give purple crystals. 33Ni: 6.6 mg, 0.0090 mmol, 67%, mp > 260 °C. UV-vis (CHCl3): λmax/nm (log ε) 372 (4.21), 429 (4.65), 608 (4.32). 1H NMR (500 MHz, CDCl3, 55 °C): δ 10.59 (s, 2H, 5,20-H), 9.49 (s, 2H, 10,15-H), 3.91–3.86 (m, 4H, 12,13-CH2), 3.72–3.68 (m, 4H, 7,18-CH2), 3.34 (s, 6H, 8,17-Me), 2.14–2.07 (m, 4H, 7,19-CH2CH2), 1.85 (t, 6H, J = 7.5 Hz, 12,13-CH2CH3), 1.72–1.68 (m, 4H, 2 × CH2CH2CH3), 1.11 (t, 6H, J = 7.4 Hz, 2 × (CH2)3CH3). HRMS (EI): m/z M+ calcd for C38H38N8NiS2 728.2014; found 728.2020. 33Cu: 9.4 mg, 0.0128 mg, 95%, mp > 260 °C. UV-vis (CHCl3): λmax (log ε) 374 (4.30), 435 (4.76), 574 (3.77), 615 (4.25). HRMS (MALDI): m/z M+ calcd for C38H38CuN8S2 733.1957; found 733.1940. 33Zn: 7.3 mg, 0.0099 mmol, 74%, mp > 260 °C. UV-vis (CHCl3): λmax/nm (log ε) 378 (4.40), 444 (4.72), 534 (sh, 3.77), 569 (3.77), 623 (4.33). UV-vis (1% pyrrolidine-CHCl3): λmax/nm (log ε) 383 (4.38), 432 (sh, 4.40), 458 (4.81), 540 (3.75), 580 (3.77), 633 (4.38). 1H NMR (500 MHz, CDCl3, 55 °C, meso-protons only): δ 9.75 (br s, 2H, 5,20-H), 9.36 (s, 2H, 10,15-H). 1H NMR (500 MHz, pyrrolidine-CDCl3): δ 11.28 (s, 2H, 5,20-H), 9.80 (s, 2H, 10,15-H), 4.09 (t, 4H, J = 7.5 Hz, 7,18-CH2), 4.02 (q, 4H, J = 7.5 Hz, 12,13-CH2), 3.56 (s, 6H, 8,17-Me), 2.36–2.30 (m, 4H, 7,19-CH2CH2), 1.89 (t, 6H, obscured by pyrrolidine, 12,13-CH2CH3), 1.53–1.47 (m, 4H, 2 × CH2CH2CH3), 1.16 (t, 6H, J = 7.5 Hz, 2 × (CH2)3CH3). HRMS (ESI): m/z [M + H]+ calcd for C38H39N8S2Zn 735.2025; found 735.2037.
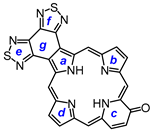
8,17-Dibutyl-7,18-dimethyl-benzo[b]bis(thiadiazolo)benzo[4,5-l]-21-carbaporphyrin (36). Tripyrrane 32 (50.0 mg, 0.068 mmol) was dissolved in trifluoroacetic acid (1 mL) and stirred at room temperature for 7 min under nitrogen. The mixture was diluted with dichloromethane (20 mL); this was followed by the addition of indenedialdehyde 29 (11.7 mg, 0.068 mmol). The mixture was stirred under nitrogen at room temperature for an additional 2 h. The dark solution was diluted with chloroform and vigorously shaken with 0.15% aqueous ferric chloride solution (100 mL) for 5–7 min. The organic solution was washed with water, 5% sodium bicarbonate solution, and water, and the solvent evaporated under reduced pressure. The residue was purified on a grade 3 alumina column, eluting with dichloromethane. Recrystallization from chloroform–methanol gave the carbaporphyrin (11.0 mg, 0.0165 mmol, 24%) as a dark solid, mp > 260 °C. UV-vis (CHCl3): λmax/nm (log ε) 393 (4.46), 458 (4.89), 597 (4.40), 635 (4.02), 696 (3.68). UV-vis (1% TFA-CHCl3): λmax/nm (log ε) 411 (4.43), 471 (4.83), 500 (sh, 4.36), 629 (4.21), 708 (3.79). 1H NMR (500 MHz, CDCl3, 55 °C, partial data): δ 10.55 (s, 2H, 5,20-H), 9.65 (s, 2H, 10,15-H), 8.70–8.68 (m, 2H, 21,31-H), 7.79–7.77 (m, 2H, 22,32-H), 3.81 (br t, 4H), 3.45 (s, 6H), −4.93 (br s, 2H, 2 × NH), −7.45 (s, 1H, 21-H). 1H NMR (500 MHz, 5 mL TFA-CDCl3): δ 11.63 (s, 2H, 5,20-H), 10.28 (s, 2H, 10,15-H), 8.65–8.63 (m, 2H, 21,31-H), 7.72–7.70 (m, 2H, 22,32-H), 4.18 (t, 4H, J = 7.4 Hz, 8,17-CH2), 3,59 (s, 6H, 7,18-Me), 2.26–2.19 (m, 4H, 8,17-CH2CH2), 1.79–1.72 (m, 4H, 2 × CH2CH2CH3), 1.14 (t, 6H, J = 7.2 Hz, 2 × (CH2)3CH3), −2.57 (s, 2H, 2 × NH), −6.51 (s, 1H, 21-H). 13C{1H} NMR (5 mL TFA-CDCl3, 125 MHz): δ 143.8, 142.3, 142.1, 139.6, 136.1, 128.9, 121.9, 105.2, 99.1, 34.7, 26.6, 23.3, 14.2, 12.0. HRMS (ESI): m/z [M + H]+ calcd for C39H36N7S2 666.2468; found 666.2472.
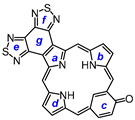
8,17-Dibutyl-7,18-dimethyl-bis(thiadiazolo)benzo[4,5-l]-21-oxaporphyrin hydrochloride (35a.HCl). Tripyrrane 32 (50.0 mg, 0.068 mmol) was dissolved in trifluoroacetic acid (1 mL) and stirred at room temperature for 7 min under nitrogen. The mixture was diluted with dichloromethane (20 mL); this was followed by the addition of furandialdehyde 34a (8.4 mg, 0.068 mmol). The mixture was stirred under nitrogen at room temperature for an additional 2 h. The dark solution was diluted with chloroform and vigorously shaken with 0.15% aqueous ferric chloride solution (100 mL) for 5–7 min. The organic solution was washed with water, 5% sodium bicarbonate solution, water, and 10% hydrochloric acid, and the solvent evaporated under reduced pressure. The residue was purified on a silica column, eluting with chloroform and then 5% methanol–chloroform. Recrystallization from chloroform-hexanes gave the title compound (9.0 mg 0.0137 mmol, 20%) as a dark solid, mp > 260 °C. UV-vis (1% Et3N-CHCl3): λmax/nm (log ε) 373 (4.70), 437 (4.80), 515 (4.09), 576 (4.05), 603 (3.80), 655 (3.91), 679 (4.08). UV-vis (CHCl3): λmax/nm (log ε) 348 (4.43), 413 (5.13), 554 (4.05), 570 (sh, 4.01), 602 (4.45), 645 (2.87), UV-vis (5% TFA-CHCl3): λmax/nm (log ε) 345 (4.45), 411 (5.18), 470 (sh, 4.09), 553 (4.12), 570 (4.09), 601 (4.53). 1H NMR (500 MHz, TFA-CDCl3): δ 12.35 (s, 2H, 5,20-H), 11.06 (s, 2H, 10,15-H), 10.57 (s, 2H, 2,3-H), 4.45 (t, 4H, J = 7.8 Hz, 8,17-CH2), 3,91 (s, 6H, 7,18-Me), 2.52–2.46 (m, 4H, 8,17-CH2CH2), 1.96–1.89 (m, 4H, 2 × CH2CH2CH3), 1.26 (t, 6H, J = 7.4 Hz, 2 × (CH2)3CH3), −4.7 (v br, 2H). 13C{1H} NMR (TFA-CDCl3): δ 154.6, 153.0, 150.0, 146.2, 141.4, 141.3, 139.7, 132.6), 107.9, 97.8, 35.4, 26.9, 23.5, 14.2, 12.1. HRMS (ESI): m/z [M + H]+ calcd for C34H32N7OS2 618.2104; found 618.2096.
8,17-Dibutyl-7,18-dimethyl-bis(thiadiazolo)benzo[4,5-l]-21-thiaporphyrin (35b). Tripyrrane 32 (50.0 mg, 0.068 mmol) was dissolved in trifluoroacetic acid (1 mL) and stirred at room temperature for 7 min under nitrogen. The mixture was diluted with dichloromethane (20 mL); this was followed by the addition of 2,5-thiophenedicarbaldehyde 34b (9.5 mg; 0.068 mmol). The mixture was stirred under nitrogen at room temperature for an additional 2 h. The dark solution was diluted with chloroform and vigorously shaken with 0.15% aqueous ferric chloride solution (100 mL) for 5–7 min. The organic solution was washed with water, 5% sodium bicarbonate solution, and water, and the solvent evaporated under reduced pressure. The residue was purified on a grade 3 alumina column, eluting with dichloromethane. Recrystallization from chloroform–methanol gave the thiaporphyrin (3.6 mg 0.0057 mmol, 8.3%) as purple crystals, mp > 260 °C. UV-vis (CHCl3): λmax/nm (log ε) 382 (4.41), 442 (4.65), 520 (3.96), 628 (3.28), 685 (3.99). UV-vis (1% TFA-CHCl3): λmax/nm (log ε) 428 (4.96), 571 (3.76), 616 (4.00). 1H NMR (500 MHz, CDCl3, 50 °C): δ 10.33 (s, 2H, 5,20-H), 10.21 (s, 2H, 10,15-H), 9.94 (s, 2H, 2,3-H), 3.52 (t, 4H, J = 7.8 Hz, 8,17-CH2), 3,32 (s, 6H, 7,18-Me), 2.09–2.03 (m, 4H, 8,17-CH2CH2), 1.71–1.64 (m, 4H, 2 × CH2CH2CH3), 1.08 (t, 6H, J = 7.3 Hz, 2 × (CH2)3CH3), −4.30 (br s, 1H, NH). 1H NMR (500 MHz, TFA-CDCl3): δ 12.63 (s, 2H, 5,20-H), 11.57 (s, 2H, 10,15-H), 10.61 (s, 2H, 2,3-H), 4.45 (t, 4H, J = 7.8 Hz, 8,17-CH2), 3,91 (s, 6H, 7,18-Me), 2.56–2.52 (m, 4H, 8,17-CH2CH2), 1.98–1.90 (m, 4H, 2 × CH2CH2CH3), 1.26 (t, 6H, J = 7.2 Hz, 2 × (CH2)3CH3). 13C{1H} NMR (TFA-CDCl3): δ 153.0, 150.1, 146.4, 145.6, 140.9, 137.8, 111.6, 108.5, 35.2, 26.7, 23.4, 14.2, 12.1. HRMS (ESI): m/z [M + H]+ calcd for C34H31N7S3 634.1876; found 634.1876.
9,18-Dibutyl-8,19-dimethyl-bis(thiadiazolo)benzo[4,5-m]oxypyriporphyrin (38). Tripyrrane 32 (50.0 mg, 0.068 mmol) was dissolved in trifluoroacetic acid (1 mL) and stirred at room temperature for 7 min under nitrogen. The mixture was diluted with dichloromethane (20 mL); this was followed by the addition of 3-hydroxy-2,6-pyridinedicarbaldehyde 37 (10.3 mg, 0.068 mmol). The mixture was stirred under nitrogen at room temperature for an additional 2 h. The dark solution was diluted with chloroform and vigorously shaken with 0.15% aqueous ferric chloride solution (100 mL) for 5–7 min. The organic solution was washed with water, 5% sodium bicarbonate solution, and water, and the solvent evaporated under reduced pressure. The residue was purified on a grade 3 alumina column, eluting with chloroform. Recrystallization from chloroform–methanol gave the oxypyriporphyrin (14.1 mg. 0.0219 mmol, 32%) as purple crystals, mp > 260 °C. UV-vis (CHCl3): λmax/nm (log ε) 412 (sh, 4.64), 444 (5.01), 459 (sh, 4.76), 611 (4.30), 643 (4.59), 681 (3.62). UV-vis (200 equivalents TFA-CHCl3): λmax/nm (log ε) 413 (sh, 4.66), 442 (4.99), 619 (4.37), 650 (4.34), 727 (3.77). UV-vis (1% TFA-CHCl3): λmax/nm (log ε) 437 (4.92), 448 (4.97), 610 (4.33), 672 (4.14), 736 (4.03). UV-vis (5% TFA-CHCl3): λmax/nm (log ε) 450 (5.30), 580 (3.98), 612 (3.97), 675 (4.38). 1H NMR (500 MHz, CDCl3, 55 °C, partial data): δ 10.90 (s, 1H), 10.84 (s, 1H), 10.78 (s, 1H), 9.39 (s, 1H), 9.21–9.19 (m, 1H), 7.89 (d, 1H, J = 8.5 Hz), −3.91–−4.01 (br, 2H). 1H NMR (500 MHz, TFA-CDCl3): δ 11.82 (s, 1H), 11.81 (s, 1H), 11.12 (s, 1H), 10.19 (s, 1H), 9.86 (d, 1H, J = 9.7 Hz), 8.72 (d, 1H, J = 9.7 Hz), 4.14 (t, 4H, J = 7.8 Hz, 9,18-CH2), 3,60 (s, 3H), 3.56 (s, 3H) (8,19-Me), 2.22–2.16 (m, 4H, 9,18-CH2CH2), 1.76–1.68 (m, 4H, 2 × CH2CH2CH3), 1.13–1.10 (2 overlapping triplets, 6H, 2 × (CH2)3CH3). 13C{1H} NMR (TFA-CDCl3, 125 MHz): δ 151.9, 150.4, 146.4, 146.2, 145.3, 144.5, 143.6, 142.2, 141.1, 136.3, 134.7, 134.1, 107.6, 105.0, 104.7, 103.6, 34.5, 26.7, 23.2, 13.9, 12.2, 12.0. HRMS (ESI): m/z [M + H]+ calcd for C39H33N8OS2 645.2213; found 645.2236.
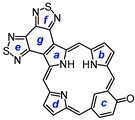
9,18-Dibutyl-8,19-dimethyl-bis(thiadiazolo)benzo[4,5-m]oxybenziporphyrin (40). Tripyrrane 32 (50.0 mg, 0.068 mmol) was dissolved in trifluoroacetic acid (1 mL) and stirred at room temperature for 7 min under nitrogen. The mixture was diluted with dichloromethane (20 mL); this was followed by the addition of 4-hydroxyisophthalaldehyde 39 (10.2 mg, 0.068 mmol). The mixture was stirred under nitrogen at room temperature for an additional 2 h. The dark solution was diluted with chloroform and vigorously shaken with 0.15% aqueous ferric chloride solution (100 mL) for 5–7 min. The organic solution was washed with water, 5% sodium bicarbonate solution, and water, and the solvent evaporated under reduced pressure. The residue was purified on a grade 3 alumina column, eluting with chloroform. Recrystallization from chloroform–methanol gave the oxybenziporphyrin (5.4 mg. 0.0084 mmol, 12%) as purple crystals, mp > 260 °C. UV-vis (CHCl3): λmax/nm (log ε) 417 (sh, 4.57), 450 (4.86), 465 (4.74), 570 (sh, 4.11), 621 (4.46), 668 (3.93), 723 (3.79). UV-vis (400 equivalents TFA-CHCl3): λmax/nm (log ε) 450 (4.93), 484 (4.54), 620 (4.20), 637 (4.19), 663 (sh, 4.04), 726 (3.82). UV-vis (5% TFA-CHCl3): λmax/nm (log ε) 378 (4.48), 448 (4.85), 576 (3.87), 627 (4.22), 712 (3.74), 783 (3.78). UV-vis (10% DBU-CHCl3): λmax/nm (log ε) 447 (4.65), 465 (4.60), 497 (4.47), 620 (3.93), 713 (4.22), 764 (3.99). 1H NMR (500 MHz, CDCl3, 55 °C): δ 10.52 (s, 1H), 10.40 (s, 1H), 10.36 (s, 1H), 9.13 (s, 1H), 8.67 (d, 1H, J = 9.2 Hz), 7.44 (d, 1H, J = 9.2 Hz), 3.86–3.80 (m, 4H), 3.49 (s, 3H), 3.40 (s, 3H), 2.17–2.11 (m, 4H), 1.74–1.67 (m, 4H), 1.14 (t, 6H, J = 7.4 Hz), −3.99 (br s, 1H), −4.16 (br s, 1H), −7.08 (s, 1H). 1H NMR (500 MHz, TFA-CDCl3): δ 10.42 (s, 1H), 10.39 (s, 1H), 10.14 (s, 1H), 9.38 (s, 1H), 8.79 (dd, 1H, J = 2.1, 8.8 Hz), 7.71 (d, 1H, J = 9.0 Hz), 3.74–3.69 (t, 4H, 9,18-CH2), 3,25 (s, 3H), 3.23 (s, 3H) (8,19-Me), 2.04–1.97 (m, 4H, 9,18-CH2CH2), 1.67–1.61 (m, 4H, 2 × CH2CH2CH3), 1.11–1.07 (2 overlapping triplets, 6H, 2 × (CH2)3CH3), −6.59 (s, 1H, 22-H). 13C{1H} NMR (TFA-CDCl3, 125 MHz): δ 151.2, 150.3, 150.2, 142.2, 141.2, 129.1, 124.7, 100.1, 98.6, 33.4, 33.3, 25.5, 23.0, 14.0, 11.6. HRMS (ESI): m/z [M + H]+ calcd for C36H34N7OS2 644.2261; found 644.2256.
Computational Studies. All calculations were performed using Gaussian 16, Revision C.01 [
46]. Geometry optimizations were performed using the M06-2X functional and the 6-311++G(d,p) basis set [
47,
48,
49,
50]. Vibrational frequencies were computed to confirm the absence of imaginary frequencies and derive zero-point energy and vibrational entropy corrections from unscaled frequencies. Single-point energy calculations were performed on the optimized minima using M06-2X/cc-PVTZ [
51]. NICS values were calculated using the GIAO method [
52] using CAM-B3LYP/6-31+G(d,p), and AICD plots were obtained from CGST calculations using B3LYP/6-31+G(d) [
53]. NICS(0) was calculated at the mean position of all four heavy atoms in the middle of the macrocycle. NICS(a), NICS(b), NICS(c), NICS(d), NICS(e), NICS(f), and NICS(g) values were obtained by applying the same method to the mean position of the heavy atoms that comprise the individual rings of each macrocycle. In addition, NICS(1)
zz, NICS(1a)
zz, NICS(1b)
zz, NICS(1c)
zz, NICS(1d)
zz, NICS(1e)
zz, NICS(1f)
zz, NICS(1f)
zz, and NICS(1h)
zz were obtained by applying the same method to ghost atoms placed 1 Å above each of the corresponding NICS(0) points and extracting the
zz contribution of the magnetic tensor. The resulting energies, Cartesian coordinates, and AICD plots can be found in the
Supporting Information.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}