


Recent Progress and Potential of G4 Ligands in Cancer Immunotherapy

 and
and
Abstract
:1. Introduction
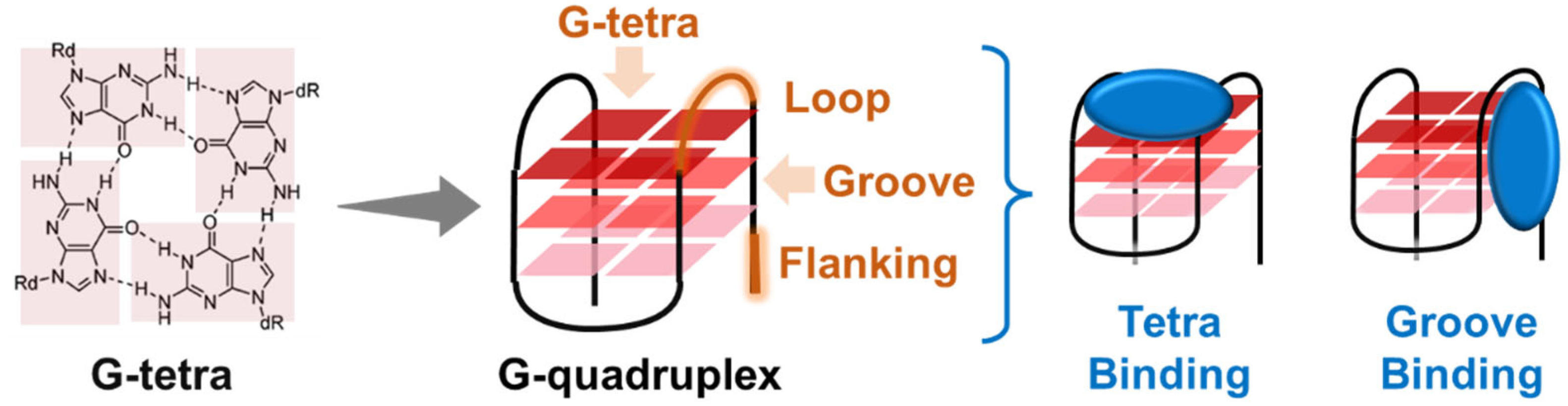
2. Overview of G4 Structures and Biological Roles
3. Overview of G4 Ligands as Anticancer Agents
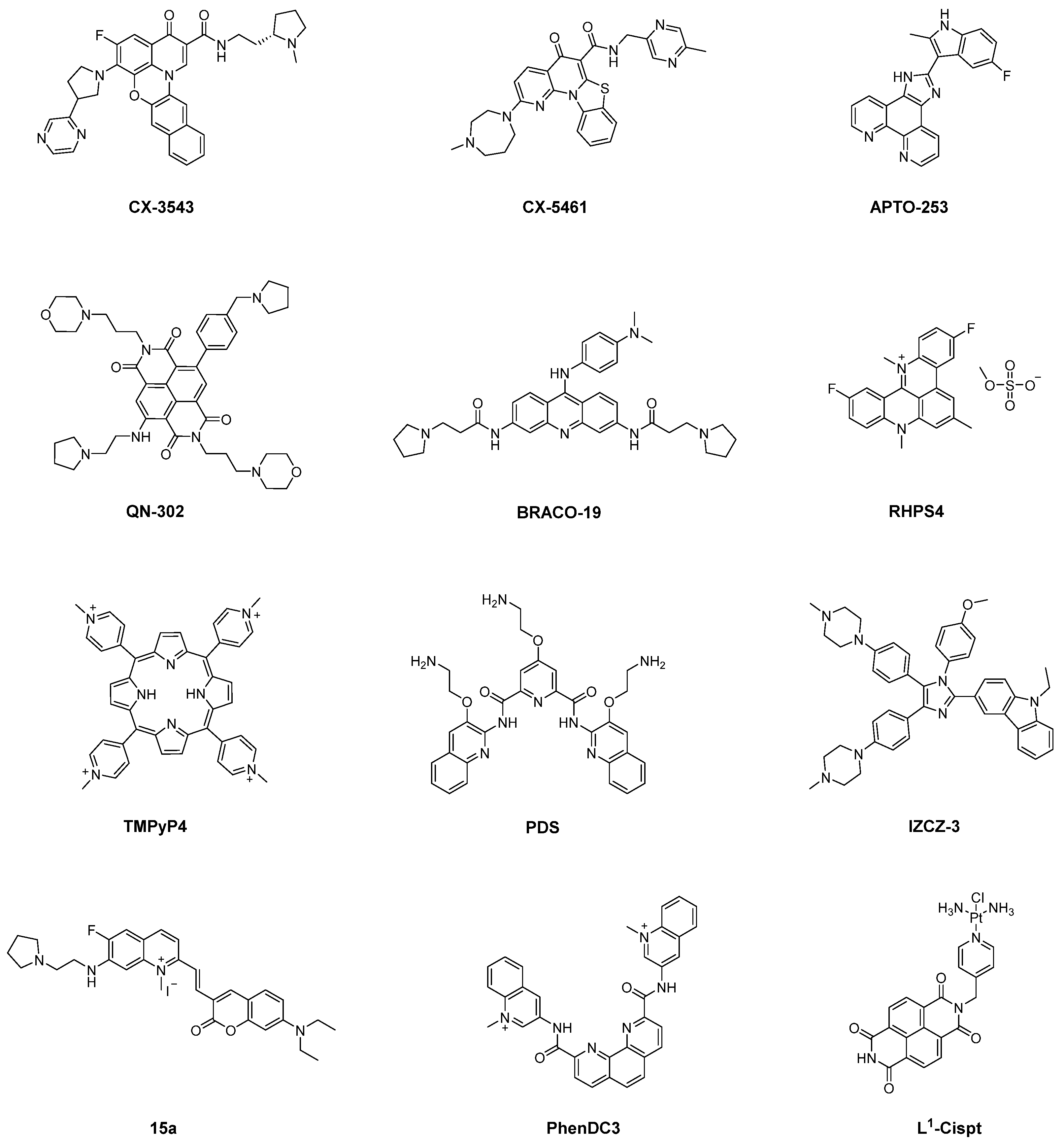
3.1. Representative G4 Ligands
3.2. Pharmacologic Effect of G4 Ligands
3.2.1. Inducing Telomere Shortening
3.2.2. Inducing DNA Damage
3.2.3. Modulating Oncogene Expression
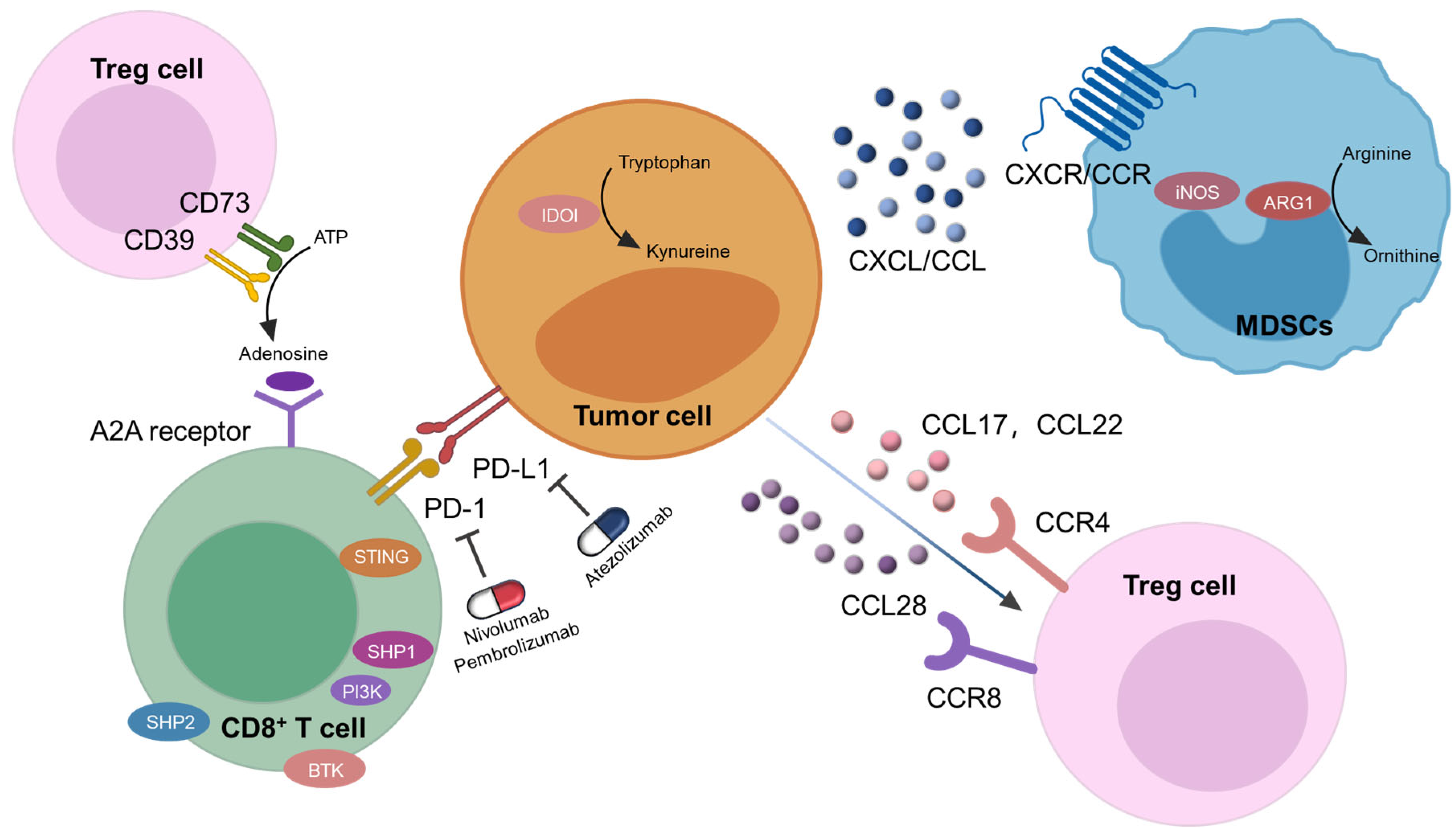
4. Potential of G4 Ligands in Cancer Immunotherapy

4.1. DNA Damage Inducing G4 Ligands
4.2. Telomere Shortening Inducing G4 Ligands

4.3. Oncogene Expression Modulating G4 Ligands
4.3.1. c-MYC Inhibiting G4 Ligands

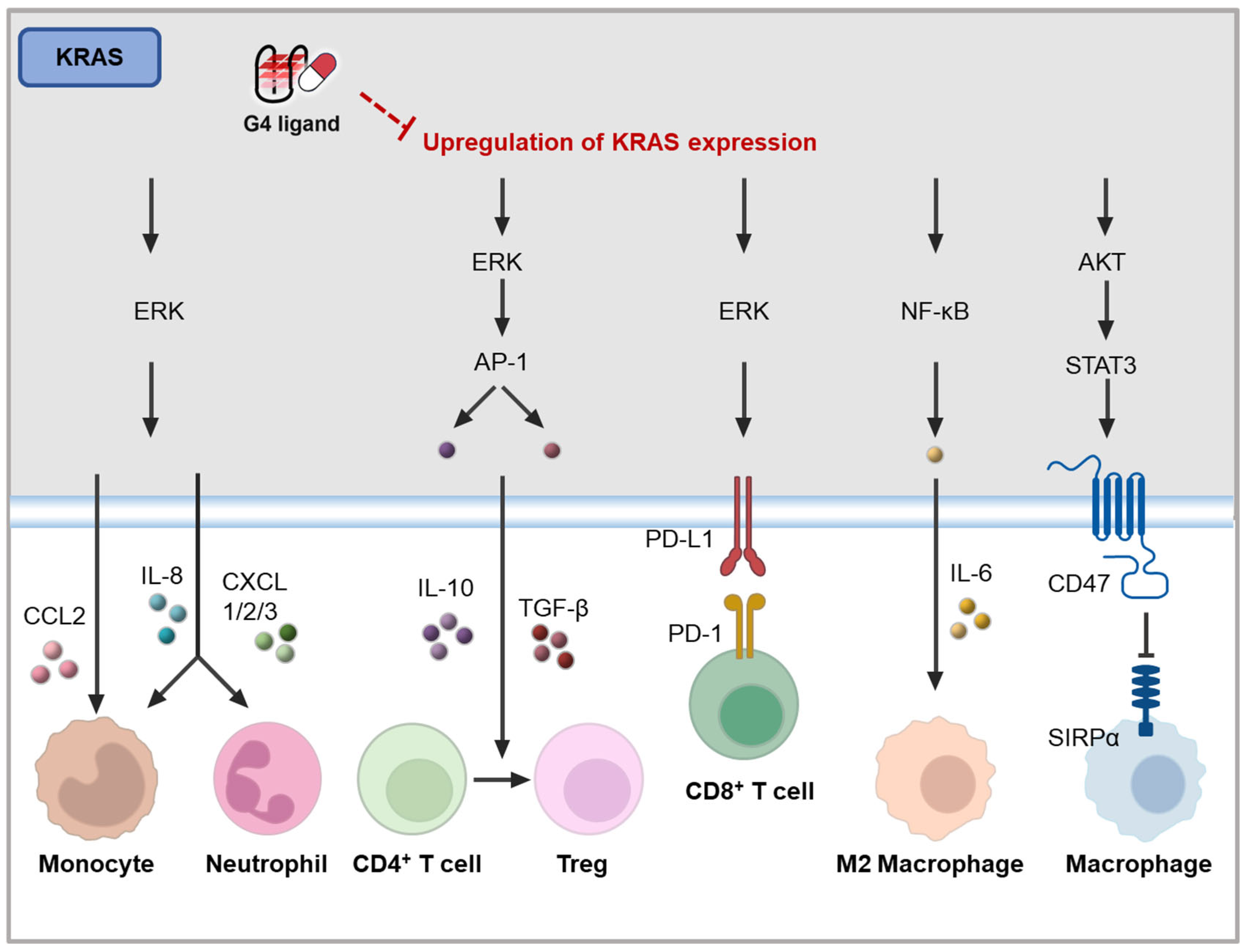
4.3.2. KRAS Inhibiting G4 Ligands
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
4.3.3. Immune Genes Modulating G4 Ligands
5. Discussion and Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Galassi, C.; Chan, T.A.; Vitale, I.; Galluzzi, L. The hallmarks of cancer immune evasion. Cancer Cell 2024, 42, 1825–1863. [Google Scholar] [CrossRef] [PubMed]
- Mantovani, A.; Allavena, P.; Sica, A.; Balkwill, F. Cancer-related inflammation. Nature 2008, 454, 436–444. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef] [PubMed]
- Ai, L.; Xu, A.; Xu, J. Roles of PD-1/PD-L1 Pathway: Signaling, Cancer, and Beyond. Adv. Exp. Med. Biol. 2020, 1248, 33–59. [Google Scholar] [CrossRef]
- Han, Y.; Liu, D.; Li, L. PD-1/PD-L1 pathway: Current researches in cancer. Am. J. Cancer Res. 2020, 10, 727–742. [Google Scholar]
- Kleffel, S.; Posch, C.; Barthel, S.R.; Mueller, H.; Schlapbach, C.; Guenova, E.; Elco, C.P.; Lee, N.; Juneja, V.R.; Zhan, Q.; et al. Melanoma Cell-Intrinsic PD-1 Receptor Functions Promote Tumor Growth. Cell 2015, 162, 1242–1256. [Google Scholar] [CrossRef]
- Yegutkin, G.G.; Boison, D. ATP and Adenosine Metabolism in Cancer: Exploitation for Therapeutic Gain. Pharmacol. Rev. 2022, 74, 797–822. [Google Scholar] [CrossRef]
- Sugiyama, D.; Nishikawa, H.; Maeda, Y.; Nishioka, M.; Tanemura, A.; Katayama, I.; Ezoe, S.; Kanakura, Y.; Sato, E.; Fukumori, Y.; et al. Anti-CCR4 mAb selectively depletes effector-type FoxP3+CD4+ regulatory T cells, evoking antitumor immune responses in humans. Proc. Natl. Acad. Sci. USA 2013, 110, 17945–17950. [Google Scholar] [CrossRef]
- Facciabene, A.; Peng, X.; Hagemann, I.S.; Balint, K.; Barchetti, A.; Wang, L.P.; Gimotty, P.A.; Gilks, C.B.; Lal, P.; Zhang, L.; et al. Tumour hypoxia promotes tolerance and angiogenesis via CCL28 and T(reg) cells. Nature 2011, 475, 226–230. [Google Scholar] [CrossRef]
- Mantovani, A.; Allavena, P.; Sozzani, S.; Vecchi, A.; Locati, M.; Sica, A. Chemokines in the recruitment and shaping of the leukocyte infiltrate of tumors. Semin. Cancer Biol. 2004, 14, 155–160. [Google Scholar] [CrossRef]
- Kitamura, T.; Qian, B.Z.; Soong, D.; Cassetta, L.; Noy, R.; Sugano, G.; Kato, Y.; Li, J.; Pollard, J.W. CCL2-induced chemokine cascade promotes breast cancer metastasis by enhancing retention of metastasis-associated macrophages. J. Exp. Med. 2015, 212, 1043–1059. [Google Scholar] [CrossRef] [PubMed]
- Ma, R.Y.; Zhang, H.; Li, X.F.; Zhang, C.B.; Selli, C.; Tagliavini, G.; Lam, A.D.; Prost, S.; Sims, A.H.; Hu, H.Y.; et al. Monocyte-derived macrophages promote breast cancer bone metastasis outgrowth. J. Exp. Med. 2020, 217, e20191820. [Google Scholar] [CrossRef] [PubMed]
- Waldman, A.D.; Fritz, J.M.; Lenardo, M.J. A guide to cancer immunotherapy: From T cell basic science to clinical practice. Nat. Rev. Immunol. 2020, 20, 651–668. [Google Scholar] [CrossRef]
- Zheng, Z.; Liu, J.; Ma, J.; Kang, R.; Liu, Z.; Yu, J. Advances in new targets for immunotherapy of small cell lung cancer. Thorac. Cancer 2024, 15, 3–14. [Google Scholar] [CrossRef]
- Friedlaender, A.; Addeo, A.; Banna, G. New emerging targets in cancer immunotherapy: The role of TIM3. ESMO Open 2019, 4, e000497. [Google Scholar] [CrossRef]
- Cho, B.C.; Abreu, D.R.; Hussein, M.; Cobo, M.; Patel, A.J.; Secen, N.; Lee, K.H.; Massuti, B.; Hiret, S.; Yang, J.C.H.; et al. Tiragolumab plus atezolizumab versus placebo plus atezolizumab as a first-line treatment for PD-L1-selected non-small-cell lung cancer (CITYSCAPE): Primary and follow-up analyses of a randomised, double-blind, phase 2 study. Lancet Oncol. 2022, 23, 781–792. [Google Scholar] [CrossRef]
- Burge, S.; Parkinson, G.N.; Hazel, P.; Todd, A.K.; Neidle, S. Quadruplex DNA: Sequence, topology and structure. Nucleic Acids Res. 2006, 34, 5402–5415. [Google Scholar] [CrossRef]
- Varshney, D.; Spiegel, J.; Zyner, K.; Tannahill, D.; Balasubramanian, S. The regulation and functions of DNA and RNA G-quadruplexes. Nat. Rev. Mol. Cell Biol. 2020, 21, 459–474. [Google Scholar] [CrossRef]
- Bhattacharyya, D.; Mirihana Arachchilage, G.; Basu, S. Metal Cations in G-Quadruplex Folding and Stability. Front. Chem. 2016, 4, 38. [Google Scholar] [CrossRef]
- Ma, Y.; Iida, K.; Nagasawa, K. Topologies of G-quadruplex: Biological functions and regulation by ligands. Biochem. Biophys. Res. Commun. 2020, 531, 3–17. [Google Scholar] [CrossRef]
- Maity, A.; Winnerdy, F.R.; Chang, W.D.; Chen, G.; Phan, A.T. Intra-locked G-quadruplex structures formed by irregular DNA G-rich motifs. Nucleic Acids Res. 2020, 48, 3315–3327. [Google Scholar] [CrossRef] [PubMed]
- Rhodes, D.; Lipps, H.J. G-quadruplexes and their regulatory roles in biology. Nucleic Acids Res. 2015, 43, 8627–8637. [Google Scholar] [CrossRef] [PubMed]
- Todd, A.K.; Johnston, M.; Neidle, S. Highly prevalent putative quadruplex sequence motifs in human DNA. Nucleic Acids Res. 2005, 33, 2901–2907. [Google Scholar] [CrossRef]
- Guédin, A.; Gros, J.; Alberti, P.; Mergny, J.L. How long is too long? Effects of loop size on G-quadruplex stability. Nucleic Acids Res. 2010, 38, 7858–7868. [Google Scholar] [CrossRef]
- Xiao, S.; Zhang, J.Y.; Wu, J.; Wu, R.Y.; Xia, Y.; Zheng, K.W.; Hao, Y.H.; Zhou, X.; Tan, Z. Formation of DNA:RNA hybrid G-quadruplexes of two G-quartet layers in transcription: Expansion of the prevalence and diversity of G-quadruplexes in genomes. Angew. Chem. Int. Ed. Engl. 2014, 53, 13110–13114. [Google Scholar] [CrossRef]
- Li, X.M.; Zheng, K.W.; Zhang, J.Y.; Liu, H.H.; He, Y.D.; Yuan, B.F.; Hao, Y.H.; Tan, Z. Guanine-vacancy-bearing G-quadruplexes responsive to guanine derivatives. Proc. Natl. Acad. Sci. USA 2015, 112, 14581–14586. [Google Scholar] [CrossRef]
- Mukundan, V.T.; Phan, A.T. Bulges in G-quadruplexes: Broadening the definition of G-quadruplex-forming sequences. J. Am. Chem. Soc. 2013, 135, 5017–5028. [Google Scholar] [CrossRef]
- Chambers, V.S.; Marsico, G.; Boutell, J.M.; Di Antonio, M.; Smith, G.P.; Balasubramanian, S. High-throughput sequencing of DNA G-quadruplex structures in the human genome. Nat. Biotechnol. 2015, 33, 877–881. [Google Scholar] [CrossRef]
- Kwok, C.K.; Marsico, G.; Sahakyan, A.B.; Chambers, V.S.; Balasubramanian, S. rG4-seq reveals widespread formation of G-quadruplex structures in the human transcriptome. Nat. Methods 2016, 13, 841–844. [Google Scholar] [CrossRef]
- Robinson, J.; Raguseo, F.; Nuccio, S.P.; Liano, D.; Di Antonio, M. DNA G-quadruplex structures: More than simple roadblocks to transcription? Nucleic Acids Res. 2021, 49, 8419–8431. [Google Scholar] [CrossRef]
- Korsakova, A.; Phan, A.T. Prediction of G4 formation in live cells with epigenetic data: A deep learning approach. NAR Genom. Bioinform. 2023, 5, lqad071. [Google Scholar] [CrossRef] [PubMed]
- Belotserkovskii, B.P.; Liu, R.; Tornaletti, S.; Krasilnikova, M.M.; Mirkin, S.M.; Hanawalt, P.C. Mechanisms and implications of transcription blockage by guanine-rich DNA sequences. Proc. Natl. Acad. Sci. USA 2010, 107, 12816–12821. [Google Scholar] [CrossRef] [PubMed]
- Estep, K.N.; Butler, T.J.; Ding, J.; Brosh, R.M. G4-Interacting DNA Helicases and Polymerases: Potential Therapeutic Targets. Curr. Med. Chem. 2019, 26, 2881–2897. [Google Scholar] [CrossRef] [PubMed]
- Henderson, E.; Hardin, C.C.; Walk, S.K.; Tinoco, I., Jr.; Blackburn, E.H. Telomeric DNA oligonucleotides form novel intramolecular structures containing guanine-guanine base pairs. Cell 1987, 51, 899–908. [Google Scholar] [CrossRef]
- Zhou, J.M.; Zhu, X.F.; Lu, Y.J.; Deng, R.; Huang, Z.S.; Mei, Y.P.; Wang, Y.; Huang, W.L.; Liu, Z.C.; Gu, L.Q.; et al. Senescence and telomere shortening induced by novel potent G-quadruplex interactive agents, quindoline derivatives, in human cancer cell lines. Oncogene 2006, 25, 503–511. [Google Scholar] [CrossRef]
- Che, T.; Chen, S.B.; Tu, J.L.; Wang, B.; Wang, Y.Q.; Zhang, Y.; Wang, J.; Wang, Z.Q.; Zhang, Z.P.; Ou, T.M.; et al. Discovery of Novel Schizocommunin Derivatives as Telomeric G-Quadruplex Ligands That Trigger Telomere Dysfunction and the Deoxyribonucleic Acid (DNA) Damage Response. J. Med. Chem. 2018, 61, 3436–3453. [Google Scholar] [CrossRef]
- Redon, S.; Reichenbach, P.; Lingner, J. The non-coding RNA TERRA is a natural ligand and direct inhibitor of human telomerase. Nucleic Acids Res. 2010, 38, 5797–5806. [Google Scholar] [CrossRef]
- Wang, W.; Hu, S.; Gu, Y.; Yan, Y.; Stovall, D.B.; Li, D.; Sui, G. Human MYC G-quadruplex: From discovery to a cancer therapeutic target. Biochim. Biophys. Acta Rev. Cancer 2020, 1874, 188410. [Google Scholar] [CrossRef]
- Islam, M.A.; Thomas, S.D.; Murty, V.V.; Sedoris, K.J.; Miller, D.M. c-Myc quadruplex-forming sequence Pu-27 induces extensive damage in both telomeric and nontelomeric regions of DNA. J. Biol. Chem. 2014, 289, 8521–8531. [Google Scholar] [CrossRef]
- Siddiqui-Jain, A.; Grand, C.L.; Bearss, D.J.; Hurley, L.H. Direct evidence for a G-quadruplex in a promoter region and its targeting with a small molecule to repress c-MYC transcription. Proc. Natl. Acad. Sci. USA 2002, 99, 11593–11598. [Google Scholar] [CrossRef]
- Esain-Garcia, I.; Kirchner, A.; Melidis, L.; Tavares, R.C.A.; Dhir, S.; Simeone, A.; Yu, Z.; Madden, S.K.; Hermann, R.; Tannahill, D.; et al. G-quadruplex DNA structure is a positive regulator of MYC transcription. Proc. Natl. Acad. Sci. USA 2024, 121, e2320240121. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, J.; Mergny, J.L.; Cruz, C. G-quadruplex ligands in cancer therapy: Progress, challenges, and clinical perspectives. Life Sci. 2024, 340, 122481. [Google Scholar] [CrossRef] [PubMed]
- Li, Q.; Xiang, J.F.; Yang, Q.F.; Sun, H.X.; Guan, A.J.; Tang, Y.L. G4LDB: A database for discovering and studying G-quadruplex ligands. Nucleic Acids Res. 2013, 41, D1115–D1123. [Google Scholar] [CrossRef]
- Yang, Q.F.; Wang, X.R.; Wang, Y.H.; Wu, X.H.; Shi, R.Y.; Wang, Y.X.; Zhu, H.N.; Yang, S.; Tang, Y.L.; Li, F. G4LDB 3.0: A database for discovering and studying G-quadruplex and i-motif ligands. Nucleic Acids Res. 2025, 53, D91–D98. [Google Scholar] [CrossRef]
- Duan, W.; Rangan, A.; Vankayalapati, H.; Kim, M.Y.; Zeng, Q.; Sun, D.; Han, H.; Fedoroff, O.Y.; Nishioka, D.; Rha, S.Y.; et al. Design and synthesis of fluoroquinophenoxazines that interact with human telomeric G-quadruplexes and their biological effects. Mol. Cancer Ther. 2001, 1, 103–120. [Google Scholar]
- Drygin, D.; Siddiqui-Jain, A.; O’Brien, S.; Schwaebe, M.; Lin, A.; Bliesath, J.; Ho, C.B.; Proffitt, C.; Trent, K.; Whitten, J.P.; et al. Anticancer activity of CX-3543: A direct inhibitor of rRNA biogenesis. Cancer Res. 2009, 69, 7653–7661. [Google Scholar] [CrossRef]
- Xu, H.; Di Antonio, M.; McKinney, S.; Mathew, V.; Ho, B.; O’Neil, N.J.; Santos, N.D.; Silvester, J.; Wei, V.; Garcia, J.; et al. CX-5461 is a DNA G-quadruplex stabilizer with selective lethality in BRCA1/2 deficient tumours. Nat. Commun. 2017, 8, 14432. [Google Scholar] [CrossRef]
- Huesca, M.; Lock, L.S.; Khine, A.A.; Viau, S.; Peralta, R.; Cukier, I.H.; Jin, H.; Al-Qawasmeh, R.A.; Lee, Y.; Wright, J.; et al. A novel small molecule with potent anticancer activity inhibits cell growth by modulating intracellular labile zinc homeostasis. Mol. Cancer Ther. 2009, 8, 2586–2596. [Google Scholar] [CrossRef]
- Local, A.; Zhang, H.; Benbatoul, K.D.; Folger, P.; Sheng, X.; Tsai, C.Y.; Howell, S.B.; Rice, W.G. APTO-253 Stabilizes G-quadruplex DNA, Inhibits MYC Expression, and Induces DNA Damage in Acute Myeloid Leukemia Cells. Mol. Cancer Ther. 2018, 17, 1177–1186. [Google Scholar] [CrossRef]
- Ahmed, A.A.; Angell, R.; Oxenford, S.; Worthington, J.; Williams, N.; Barton, N.; Fowler, T.G.; O’Flynn, D.E.; Sunose, M.; McConville, M.; et al. Asymmetrically Substituted Quadruplex-Binding Naphthalene Diimide Showing Potent Activity in Pancreatic Cancer Models. ACS Med. Chem. Lett. 2020, 11, 1634–1644. [Google Scholar] [CrossRef]
- Read, M.; Harrison, R.J.; Romagnoli, B.; Tanious, F.A.; Gowan, S.H.; Reszka, A.P.; Wilson, W.D.; Kelland, L.R.; Neidle, S. Structure-based design of selective and potent G quadruplex-mediated telomerase inhibitors. Proc. Natl. Acad. Sci. USA 2001, 98, 4844–4849. [Google Scholar] [CrossRef] [PubMed]
- Burger, A.M.; Dai, F.; Schultes, C.M.; Reszka, A.P.; Moore, M.J.; Double, J.A.; Neidle, S. The G-quadruplex-interactive molecule BRACO-19 inhibits tumor growth, consistent with telomere targeting and interference with telomerase function. Cancer Res. 2005, 65, 1489–1496. [Google Scholar] [CrossRef] [PubMed]
- Gowan, S.M.; Harrison, J.R.; Patterson, L.; Valenti, M.; Read, M.A.; Neidle, S.; Kelland, L.R. A G-quadruplex-interactive potent small-molecule inhibitor of telomerase exhibiting in vitro and in vivo antitumor activity. Mol. Pharmacol. 2002, 61, 1154–1162. [Google Scholar] [CrossRef] [PubMed]
- Gowan, S.M.; Heald, R.; Stevens, M.F.; Kelland, L.R. Potent inhibition of telomerase by small-molecule pentacyclic acridines capable of interacting with G-quadruplexes. Mol. Pharmacol. 2001, 60, 981–988. [Google Scholar] [CrossRef]
- Berardinelli, F.; Siteni, S.; Tanzarella, C.; Stevens, M.F.; Sgura, A.; Antoccia, A. The G-quadruplex-stabilising agent RHPS4 induces telomeric dysfunction and enhances radiosensitivity in glioblastoma cells. DNA Repair 2015, 25, 104–115. [Google Scholar] [CrossRef]
- Falabella, M.; Kolesar, J.E.; Wallace, C.; de Jesus, D.; Sun, L.; Taguchi, Y.V.; Wang, C.; Wang, T.; Xiang, I.M.; Alder, J.K.; et al. G-quadruplex dynamics contribute to regulation of mitochondrial gene expression. Sci. Rep. 2019, 9, 5605. [Google Scholar] [CrossRef]
- Wheelhouse, R.T.; Sun, D.; Han, H.; Han, F.X.; Hurley, L.H. Cationic Porphyrins as Telomerase Inhibitors: the Interaction of Tetra-(N-methyl-4-pyridyl)porphine with Quadruplex DNA. J. Am. Chem. Soc. 1998, 120, 3261–3262. [Google Scholar] [CrossRef]
- Bhattacharjee, A.J.; Ahluwalia, K.; Taylor, S.; Jin, O.; Nicoludis, J.M.; Buscaglia, R.; Brad Chaires, J.; Kornfilt, D.J.; Marquardt, D.G.; Yatsunyk, L.A. Induction of G-quadruplex DNA structure by Zn(II) 5,10,15,20-tetrakis(N-methyl-4-pyridyl)porphyrin. Biochimie 2011, 93, 1297–1309. [Google Scholar] [CrossRef]
- Rosu, F.; Gabelica, V.; Shin-ya, K.; De Pauw, E. Telomestatin-induced stabilization of the human telomeric DNA quadruplex monitored by electrospray mass spectrometry. Chem. Commun. 2003, 21, 2702–2703. [Google Scholar] [CrossRef]
- Rodriguez, R.; Müller, S.; Yeoman, J.A.; Trentesaux, C.; Riou, J.F.; Balasubramanian, S. A novel small molecule that alters shelterin integrity and triggers a DNA-damage response at telomeres. J. Am. Chem. Soc. 2008, 130, 15758–15759. [Google Scholar] [CrossRef]
- Liu, L.Y.; Ma, T.Z.; Zeng, Y.L.; Liu, W.; Mao, Z.W. Structural Basis of Pyridostatin and Its Derivatives Specifically Binding to G-Quadruplexes. J. Am. Chem. Soc. 2022, 144, 11878–11887. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.C.; Tang, G.X.; Luo, W.H.; Shao, W.; Dai, J.; Zeng, S.T.; Huang, Z.S.; Chen, S.B.; Tan, J.H. Monitoring and Modulating mtDNA G-Quadruplex Dynamics Reveal Its Close Relationship to Cell Glycolysis. J. Am. Chem. Soc. 2021, 143, 20779–20791. [Google Scholar] [CrossRef] [PubMed]
- Li, M.L.; Dai, L.T.; Gao, Z.Y.; Yan, J.T.; Xu, S.M.; Tan, J.H.; Huang, Z.S.; Chen, S.B.; Chen, X.C. Discovery of Novel Coumarin-quinolinium Derivatives as Pan-KRAS Translation Inhibitors by Targeting 5′-UTR RNA G-Quadruplexes. J. Med. Chem. 2024, 67, 1961–1981. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.Y.; Chen, A.C.; Yin, Q.K.; Li, Z.; Huang, S.M.; Du, G.; He, J.H.; Zan, L.P.; Wang, S.K.; Xu, Y.H.; et al. New Disubstituted Quindoline Derivatives Inhibiting Burkitt’s Lymphoma Cell Proliferation by Impeding c-MYC Transcription. J. Med. Chem. 2017, 60, 5438–5454. [Google Scholar] [CrossRef]
- Chen, S.B.; Tan, J.H.; Ou, T.M.; Huang, S.L.; An, L.K.; Luo, H.B.; Li, D.; Gu, L.Q.; Huang, Z.S. Pharmacophore-based discovery of triaryl-substituted imidazole as new telomeric G-quadruplex ligand. Bioorganic Med. Chem. Lett. 2011, 21, 1004–1009. [Google Scholar] [CrossRef]
- Hu, M.H.; Chen, S.B.; Wang, B.; Ou, T.M.; Gu, L.Q.; Tan, J.H.; Huang, Z.S. Specific targeting of telomeric multimeric G-quadruplexes by a new triaryl-substituted imidazole. Nucleic Acids Res. 2017, 45, 1606–1618. [Google Scholar] [CrossRef]
- Hu, M.H.; Wang, Y.Q.; Yu, Z.Y.; Hu, L.N.; Ou, T.M.; Chen, S.B.; Huang, Z.S.; Tan, J.H. Discovery of a New Four-Leaf Clover-Like Ligand as a Potent c-MYC Transcription Inhibitor Specifically Targeting the Promoter G-Quadruplex. J. Med. Chem. 2018, 61, 2447–2459. [Google Scholar] [CrossRef]
- Lemarteleur, T.; Gomez, D.; Paterski, R.; Mandine, E.; Mailliet, P.; Riou, J.F. Stabilization of the c-myc gene promoter quadruplex by specific ligands’ inhibitors of telomerase. Biochem. Biophys. Res. Commun. 2004, 323, 802–808. [Google Scholar] [CrossRef]
- Bandeira, S.; Gonzalez-Garcia, J.; Pensa, E.; Albrecht, T.; Vilar, R. A Redox-Activated G-Quadruplex DNA Binder Based on a Platinum(IV)-Salphen Complex. Angew. Chem. Int. Ed. Engl. 2018, 57, 310–313. [Google Scholar] [CrossRef]
- Viglasky, V. Platination of telomeric sequences and nuclease hypersensitive elements of human c-myc and PDGF-A promoters and their ability to form G-quadruplexes. FEBS J. 2009, 276, 401–409. [Google Scholar] [CrossRef]
- Liu, L.Y.; Ma, T.Z.; Zeng, Y.L.; Liu, W.; Zhang, H.; Mao, Z.W. Organic-Platinum Hybrids for Covalent Binding of G-Quadruplexes: Structural Basis and Application to Cancer Immunotherapy. Angew. Chem. Int. Ed. Engl. 2023, 62, e202305645. [Google Scholar] [CrossRef] [PubMed]
- Long, W.; Zheng, B.X.; Li, Y.; Huang, X.H.; Lin, D.M.; Chen, C.C.; Hou, J.Q.; Ou, T.M.; Wong, W.L.; Zhang, K.; et al. Rational design of small-molecules to recognize G-quadruplexes of c-MYC promoter and telomere and the evaluation of their in vivo antitumor activity against breast cancer. Nucleic Acids Res. 2022, 50, 1829–1848. [Google Scholar] [CrossRef] [PubMed]
- Lauer, N.K.; Maier, S.M.; Oberringer, M.; Schulte, M.; Mutschler, W.; Hanselmann, R.G.; Schartl, M.; Scherer, S.J.; Wirbel, R.J. Absence of telomerase activity in malignant bone tumors and soft-tissue sarcomas. Sarcoma 2002, 6, 43–46. [Google Scholar] [CrossRef] [PubMed]
- Odago, F.O.; Gerson, S.L. Telomerase inhibition and telomere erosion: A two-pronged strategy in cancer therapy. Trends Pharmacol. Sci. 2003, 24, 328–331. [Google Scholar] [CrossRef]
- Mo, Y.; Gan, Y.; Song, S.; Johnston, J.; Xiao, X.; Wientjes, M.G.; Au, J.L. Simultaneous targeting of telomeres and telomerase as a cancer therapeutic approach. Cancer Res. 2003, 63, 579–585. [Google Scholar]
- Neidle, S.; Read, M.A. G-quadruplexes as therapeutic targets. Biopolymers 2000, 56, 195–208. [Google Scholar] [CrossRef]
- Campbell, N.H.; Parkinson, G.N.; Reszka, A.P.; Neidle, S. Structural basis of DNA quadruplex recognition by an acridine drug. J. Am. Chem. Soc. 2008, 130, 6722–6724. [Google Scholar] [CrossRef]
- Incles, C.M.; Schultes, C.M.; Kempski, H.; Koehler, H.; Kelland, L.R.; Neidle, S. A G-quadruplex telomere targeting agent produces p16-associated senescence and chromosomal fusions in human prostate cancer cells. Mol. Cancer Ther. 2004, 3, 1201–1206. [Google Scholar] [CrossRef]
- Gunaratnam, M.; Greciano, O.; Martins, C.; Reszka, A.P.; Schultes, C.M.; Morjani, H.; Riou, J.F.; Neidle, S. Mechanism of acridine-based telomerase inhibition and telomere shortening. Biochem. Pharmacol. 2007, 74, 679–689. [Google Scholar] [CrossRef]
- Zhou, G.; Liu, X.; Li, Y.; Xu, S.; Ma, C.; Wu, X.; Cheng, Y.; Yu, Z.; Zhao, G.; Chen, Y. Telomere targeting with a novel G-quadruplex-interactive ligand BRACO-19 induces T-loop disassembly and telomerase displacement in human glioblastoma cells. Oncotarget 2016, 7, 14925–14939. [Google Scholar] [CrossRef]
- Heald, R.A.; Modi, C.; Cookson, J.C.; Hutchinson, I.; Laughton, C.A.; Gowan, S.M.; Kelland, L.R.; Stevens, M.F. Antitumor polycyclic acridines. 8.1 Synthesis and telomerase-inhibitory activity of methylated pentacyclic acridinium salts. J. Med. Chem. 2002, 45, 590–597. [Google Scholar] [CrossRef] [PubMed]
- Leonetti, C.; Amodei, S.; D’Angelo, C.; Rizzo, A.; Benassi, B.; Antonelli, A.; Elli, R.; Stevens, M.F.; D’Incalci, M.; Zupi, G.; et al. Biological activity of the G-quadruplex ligand RHPS4 (3,11-difluoro-6,8,13-trimethyl-8H-quino[4,3,2-kl]acridinium methosulfate) is associated with telomere capping alteration. Mol. Pharmacol. 2004, 66, 1138–1146. [Google Scholar] [CrossRef] [PubMed]
- Salvati, E.; Leonetti, C.; Rizzo, A.; Scarsella, M.; Mottolese, M.; Galati, R.; Sperduti, I.; Stevens, M.F.; D’Incalci, M.; Blasco, M.; et al. Telomere damage induced by the G-quadruplex ligand RHPS4 has an antitumor effect. J. Clin. Investig. 2007, 117, 3236–3247. [Google Scholar] [CrossRef] [PubMed]
- Phatak, P.; Cookson, J.C.; Dai, F.; Smith, V.; Gartenhaus, R.B.; Stevens, M.F.; Burger, A.M. Telomere uncapping by the G-quadruplex ligand RHPS4 inhibits clonogenic tumour cell growth in vitro and in vivo consistent with a cancer stem cell targeting mechanism. Br. J. Cancer 2007, 96, 1223–1233. [Google Scholar] [CrossRef]
- Rizzo, A.; Salvati, E.; Porru, M.; D’Angelo, C.; Stevens, M.F.; D’Incalci, M.; Leonetti, C.; Gilson, E.; Zupi, G.; Biroccio, A. Stabilization of quadruplex DNA perturbs telomere replication leading to the activation of an ATR-dependent ATM signaling pathway. Nucleic Acids Res. 2009, 37, 5353–5364. [Google Scholar] [CrossRef]
- Lipps, H.J.; Rhodes, D. G-quadruplex structures: In vivo evidence and function. Trends Cell Biol. 2009, 19, 414–422. [Google Scholar] [CrossRef]
- Jackson, S.P.; Bartek, J. The DNA-damage response in human biology and disease. Nature 2009, 461, 1071–1078. [Google Scholar] [CrossRef]
- Hänsel-Hertsch, R.; Di Antonio, M.; Balasubramanian, S. DNA G-quadruplexes in the human genome: Detection, functions and therapeutic potential. Nat. Rev. Mol. Cell Biol. 2017, 18, 279–284. [Google Scholar] [CrossRef]
- Rodriguez, R.; Miller, K.M.; Forment, J.V.; Bradshaw, C.R.; Nikan, M.; Britton, S.; Oelschlaegel, T.; Xhemalce, B.; Balasubramanian, S.; Jackson, S.P. Small-molecule-induced DNA damage identifies alternative DNA structures in human genes. Nat. Chem. Biol. 2012, 8, 301–310. [Google Scholar] [CrossRef]
- Beauvarlet, J.; Bensadoun, P.; Darbo, E.; Labrunie, G.; Rousseau, B.; Richard, E.; Draskovic, I.; Londono-Vallejo, A.; Dupuy, J.W.; Nath Das, R.; et al. Modulation of the ATM/autophagy pathway by a G-quadruplex ligand tips the balance between senescence and apoptosis in cancer cells. Nucleic Acids Res. 2019, 47, 2739–2756. [Google Scholar] [CrossRef]
- De Magis, A.; Manzo, S.G.; Russo, M.; Marinello, J.; Morigi, R.; Sordet, O.; Capranico, G. DNA damage and genome instability by G-quadruplex ligands are mediated by R loops in human cancer cells. Proc. Natl. Acad. Sci. USA 2019, 116, 816–825. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Zhou, J.; Lin, L.; Hu, M.H. Discovery of a far-red carbazole-benzoindolium fluorescent ligand that selectively targets mitochondrial DNA and suppresses breast cancer growth. Eur. J. Med. Chem. 2024, 264, 116046. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, J.; Djavaheri-Mergny, M.; Ferret, L.; Mergny, J.L.; Cruz, C. Harnessing G-quadruplex ligands for lung cancer treatment: A comprehensive overview. Drug Discov. Today 2023, 28, 103808. [Google Scholar] [CrossRef]
- Prior, I.A.; Hood, F.E.; Hartley, J.L. The Frequency of Ras Mutations in Cancer. Cancer Res. 2020, 80, 2969–2974. [Google Scholar] [CrossRef]
- Wang, K.B.; Liu, Y.; Li, J.; Xiao, C.; Wang, Y.; Gu, W.; Li, Y.; Xia, Y.Z.; Yan, T.; Yang, M.H.; et al. Structural insight into the bulge-containing KRAS oncogene promoter G-quadruplex bound to berberine and coptisine. Nat. Commun. 2022, 13, 6016. [Google Scholar] [CrossRef]
- Lejault, P.; Mitteaux, J.; Sperti, F.R.; Monchaud, D. How to untie G-quadruplex knots and why? Cell Chem. Biol. 2021, 28, 436–455. [Google Scholar] [CrossRef]
- Mitteaux, J.; Lejault, P.; Wojciechowski, F.; Joubert, A.; Boudon, J.; Desbois, N.; Gros, C.P.; Hudson, R.H.E.; Boulé, J.B.; Granzhan, A.; et al. Identifying G-Quadruplex-DNA-Disrupting Small Molecules. J. Am. Chem. Soc. 2021, 143, 12567–12577. [Google Scholar] [CrossRef]
- Grabosch, S.; Bulatovic, M.; Zeng, F.; Ma, T.; Zhang, L.; Ross, M.; Brozick, J.; Fang, Y.; Tseng, G.; Kim, E.; et al. Cisplatin-induced immune modulation in ovarian cancer mouse models with distinct inflammation profiles. Oncogene 2019, 38, 2380–2393. [Google Scholar] [CrossRef]
- Ohyanagi, F.; Yamamoto, N.; Horiike, A.; Harada, H.; Kozuka, T.; Murakami, H.; Gomi, K.; Takahashi, T.; Morota, M.; Nishimura, T.; et al. Phase II trial of S-1 and cisplatin with concurrent radiotherapy for locally advanced non-small-cell lung cancer. Br. J. Cancer 2009, 101, 225–231. [Google Scholar] [CrossRef]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Abe, T.; Barber, G.N. Cytosolic-DNA-mediated, STING-dependent proinflammatory gene induction necessitates canonical NF-κB activation through TBK1. J. Virol. 2014, 88, 5328–5341. [Google Scholar] [CrossRef] [PubMed]
- Chen, Q.; Sun, L.; Chen, Z.J. Regulation and function of the cGAS-STING pathway of cytosolic DNA sensing. Nat. Immunol. 2016, 17, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Liu, P. Cytosolic DNA sensing by cGAS: Regulation, function, and human diseases. Signal Transduct. Target. Ther. 2021, 6, 170. [Google Scholar] [CrossRef]
- Diamond, M.S.; Kinder, M.; Matsushita, H.; Mashayekhi, M.; Dunn, G.P.; Archambault, J.M.; Lee, H.; Arthur, C.D.; White, J.M.; Kalinke, U.; et al. Type I interferon is selectively required by dendritic cells for immune rejection of tumors. J. Exp. Med. 2011, 208, 1989–2003. [Google Scholar] [CrossRef]
- Wang-Bishop, L.; Wehbe, M.; Shae, D.; James, J.; Hacker, B.C.; Garland, K.; Chistov, P.P.; Rafat, M.; Balko, J.M.; Wilson, J.T. Potent STING activation stimulates immunogenic cell death to enhance antitumor immunity in neuroblastoma. J. Immunother. Cancer 2020, 8, e000282. [Google Scholar] [CrossRef]
- Gu, Z.; Hao, Y.; Schomann, T.; Ossendorp, F.; Ten Dijke, P.; Cruz, L.J. Enhancing anti-tumor immunity through liposomal oxaliplatin and localized immunotherapy via STING activation. J. Control. Release 2023, 357, 531–544. [Google Scholar] [CrossRef]
- Miglietta, G.; Russo, M.; Duardo, R.C.; Capranico, G. G-quadruplex binders as cytostatic modulators of innate immune genes in cancer cells. Nucleic Acids Res. 2021, 49, 6673–6686. [Google Scholar] [CrossRef]
- Ling, Y.Y.; Xia, X.Y.; Hao, L.; Wang, W.J.; Zhang, H.; Liu, L.Y.; Liu, W.; Li, Z.Y.; Tan, C.P.; Mao, Z.W. Simultaneous Photoactivation of cGAS-STING Pathway and Pyroptosis by Platinum(II) Triphenylamine Complexes for Cancer Immunotherapy. Angew. Chem. Int. Ed. Engl. 2022, 61, e202210988. [Google Scholar] [CrossRef]
- Ma, T.Z.; Liu, L.Y.; Zeng, Y.L.; Ding, K.; Zhang, H.; Liu, W.; Cao, Q.; Xia, W.; Xiong, X.; Wu, C.; et al. G-quadruplex-guided cisplatin triggers multiple pathways in targeted chemotherapy and immunotherapy. Chem. Sci. 2024, 15, 9756–9774. [Google Scholar] [CrossRef]
- Wang, X.D.; Liu, Y.S.; Chen, M.D.; Hu, M.H. Discovery of a triphenylamine-based ligand that targets mitochondrial DNA G-quadruplexes and activates the cGAS-STING immunomodulatory pathway. Eur. J. Med. Chem. 2024, 269, 116361. [Google Scholar] [CrossRef]
- Mender, I.; Zhang, A.; Ren, Z.; Han, C.; Deng, Y.; Siteni, S.; Li, H.; Zhu, J.; Vemula, A.; Shay, J.W.; et al. Telomere Stress Potentiates STING-Dependent Anti-tumor Immunity. Cancer Cell 2020, 38, 400–411.e406. [Google Scholar] [CrossRef]
- Von Zglinicki, T. Oxidative stress shortens telomeres. Trends Biochem. Sci. 2002, 27, 339–344. [Google Scholar] [CrossRef]
- Jurk, D.; Wilson, C.; Passos, J.F.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2014, 2, 4172. [Google Scholar] [CrossRef]
- Chabanon, R.M.; Rouanne, M.; Lord, C.J.; Soria, J.C.; Pasero, P.; Postel-Vinay, S. Targeting the DNA damage response in immuno-oncology: Developments and opportunities. Nat. Rev. Cancer 2021, 21, 701–717. [Google Scholar] [CrossRef]
- Nihira, N.T.; Kudo, R.; Ohta, T. Inflammation and tumor immune escape in response to DNA damage. Semin. Cancer Biol. 2025, 110, 36–45. [Google Scholar] [CrossRef]
- Wang, X.D.; Wang, J.X.; Hu, M.H. Novel phenanthrene imidazoles as telomeric G-quadruplex ligands trigger potent immunogenic cell death in triple-negative breast cancer. Int. J. Biol. Macromol. 2023, 249, 126068. [Google Scholar] [CrossRef]
- Nassour, J.; Aguiar, L.G.; Correia, A.; Schmidt, T.T.; Mainz, L.; Przetocka, S.; Haggblom, C.; Tadepalle, N.; Williams, A.; Shokhirev, M.N.; et al. Telomere-to-mitochondria signalling by ZBP1 mediates replicative crisis. Nature 2023, 614, 767–773. [Google Scholar] [CrossRef]
- Scionti, F.; Juli, G.; Rocca, R.; Polerà, N.; Nadai, M.; Grillone, K.; Caracciolo, D.; Riillo, C.; Altomare, E.; Ascrizzi, S.; et al. TERRA G-quadruplex stabilization as a new therapeutic strategy for multiple myeloma. J. Exp. Clin. Cancer Res. 2023, 42, 71. [Google Scholar] [CrossRef]
- Mao, J.; Zhang, Q.; Wang, Y.; Zhuang, Y.; Xu, L.; Ma, X.; Guan, D.; Zhou, J.; Liu, J.; Wu, X.; et al. TERT activates endogenous retroviruses to promote an immunosuppressive tumour microenvironment. EMBO Rep. 2022, 23, e52984. [Google Scholar] [CrossRef]
- Leão, R.; Apolónio, J.D.; Lee, D.; Figueiredo, A.; Tabori, U.; Castelo-Branco, P. Mechanisms of human telomerase reverse transcriptase (hTERT) regulation: Clinical impacts in cancer. J. Biomed. Sci. 2018, 25, 22. [Google Scholar] [CrossRef]
- Liu, Z.; Li, Q.; Li, K.; Chen, L.; Li, W.; Hou, M.; Liu, T.; Yang, J.; Lindvall, C.; Björkholm, M.; et al. Telomerase reverse transcriptase promotes epithelial-mesenchymal transition and stem cell-like traits in cancer cells. Oncogene 2013, 32, 4203–4213. [Google Scholar] [CrossRef]
- Hannen, R.; Bartsch, J.W. Essential roles of telomerase reverse transcriptase hTERT in cancer stemness and metastasis. FEBS Lett. 2018, 592, 2023–2031. [Google Scholar] [CrossRef]
- Zahler, A.M.; Williamson, J.R.; Cech, T.R.; Prescott, D.M. Inhibition of telomerase by G-quartet DNA structures. Nature 1991, 350, 718–720. [Google Scholar] [CrossRef]
- Mohammed, A.A.; Rashed, H.E.; Abdelrahman, A.E.; Obaya, A.A.; Toam, M.; Abdel Nour, H.M.; Abdelhamid, M.I.; Elsayed, F.M. C-MYC and BCL2: Correlation between Protein Over-Expression and Gene Translocation and Impact on Outcome in Diffuse Large B Cell Lymphoma. Asian Pac. J. Cancer Prev. 2019, 20, 1463–1470. [Google Scholar] [CrossRef]
- Casacuberta-Serra, S.; Soucek, L. Myc and Ras, the Bonnie and Clyde of immune evasion. Transl. Cancer Res. 2018, 7, S457–S459. [Google Scholar] [CrossRef]
- Dhanasekaran, R.; Deutzmann, A.; Mahauad-Fernandez, W.D.; Hansen, A.S.; Gouw, A.M.; Felsher, D.W. The MYC oncogene—The grand orchestrator of cancer growth and immune evasion. Nat. Rev. Clin. Oncol. 2022, 19, 23–36. [Google Scholar] [CrossRef]
- Pello, O.M.; Andrés, V. Role of c-MYC in tumor-associated macrophages and cancer progression. Oncoimmunology 2013, 2, e22984. [Google Scholar] [CrossRef]
- Rakhra, K.; Bachireddy, P.; Zabuawala, T.; Zeiser, R.; Xu, L.; Kopelman, A.; Fan, A.C.; Yang, Q.; Braunstein, L.; Crosby, E.; et al. CD4+ T cells contribute to the remodeling of the microenvironment required for sustained tumor regression upon oncogene inactivation. Cancer Cell 2010, 18, 485–498. [Google Scholar] [CrossRef]
- Casey, S.C.; Tong, L.; Li, Y.; Do, R.; Walz, S.; Fitzgerald, K.N.; Gouw, A.M.; Baylot, V.; Gütgemann, I.; Eilers, M.; et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352, 227–231. [Google Scholar] [CrossRef]
- Swaminathan, S.; Hansen, A.S.; Heftdal, L.D.; Dhanasekaran, R.; Deutzmann, A.; Fernandez, W.D.M.; Liefwalker, D.F.; Horton, C.; Mosley, A.; Liebersbach, M.; et al. MYC functions as a switch for natural killer cell-mediated immune surveillance of lymphoid malignancies. Nat. Commun. 2020, 11, 2860. [Google Scholar] [CrossRef]
- Nanbakhsh, A.; Pochon, C.; Mallavialle, A.; Amsellem, S.; Bourhis, J.H.; Chouaib, S. c-Myc regulates expression of NKG2D ligands ULBP1/2/3 in AML and modulates their susceptibility to NK-mediated lysis. Blood 2014, 123, 3585–3595. [Google Scholar] [CrossRef]
- Zimmerli, D.; Brambillasca, C.S.; Talens, F.; Bhin, J.; Linstra, R.; Romanens, L.; Bhattacharya, A.; Joosten, S.E.P.; Da Silva, A.M.; Padrao, N.; et al. MYC promotes immune-suppression in triple-negative breast cancer via inhibition of interferon signaling. Nat. Commun. 2022, 13, 6579. [Google Scholar] [CrossRef]
- Markovits, E.; Harush, O.; Baruch, E.N.; Shulman, E.D.; Debby, A.; Itzhaki, O.; Anafi, L.; Danilevsky, A.; Shomron, N.; Ben-Betzalel, G.; et al. MYC Induces Immunotherapy and IFNγ Resistance Through Downregulation of JAK2. Cancer Immunol. Res. 2023, 11, 909–924. [Google Scholar] [CrossRef]
- Yang, C.; Liu, Y.; Hu, Y.; Fang, L.; Huang, Z.; Cui, H.; Xie, J.; Hong, Y.; Chen, W.; Xiao, N.; et al. Myc inhibition tips the immune balance to promote antitumor immunity. Cell. Mol. Immunol. 2022, 19, 1030–1041. [Google Scholar] [CrossRef]
- Downward, J. Targeting RAS signalling pathways in cancer therapy. Nat. Rev. Cancer 2003, 3, 11–22. [Google Scholar] [CrossRef]
- Punekar, S.R.; Velcheti, V.; Neel, B.G.; Wong, K.K. The current state of the art and future trends in RAS-targeted cancer therapies. Nat. Rev. Clin. Oncol. 2022, 19, 637–655. [Google Scholar] [CrossRef]
- Wellenstein, M.D.; de Visser, K.E. Cancer-Cell-Intrinsic Mechanisms Shaping the Tumor Immune Landscape. Immunity 2018, 48, 399–416. [Google Scholar] [CrossRef]
- Zdanov, S.; Mandapathil, M.; Abu Eid, R.; Adamson-Fadeyi, S.; Wilson, W.; Qian, J.; Carnie, A.; Tarasova, N.; Mkrtichyan, M.; Berzofsky, J.A.; et al. Mutant KRAS Conversion of Conventional T Cells into Regulatory T Cells. Cancer Immunol. Res. 2016, 4, 354–365. [Google Scholar] [CrossRef]
- Coelho, M.A.; de Carné Trécesson, S.; Rana, S.; Zecchin, D.; Moore, C.; Molina-Arcas, M.; East, P.; Spencer-Dene, B.; Nye, E.; Barnouin, K.; et al. Oncogenic RAS Signaling Promotes Tumor Immunoresistance by Stabilizing PD-L1 mRNA. Immunity 2017, 47, 1083–1099.e1086. [Google Scholar] [CrossRef]
- Liao, W.; Overman, M.J.; Boutin, A.T.; Shang, X.; Zhao, D.; Dey, P.; Li, J.; Wang, G.; Lan, Z.; Li, J.; et al. KRAS-IRF2 Axis Drives Immune Suppression and Immune Therapy Resistance in Colorectal Cancer. Cancer Cell 2019, 35, 559–572.e557. [Google Scholar] [CrossRef]
- Ji, H.; Houghton, A.M.; Mariani, T.J.; Perera, S.; Kim, C.B.; Padera, R.; Tonon, G.; McNamara, K.; Marconcini, L.A.; Hezel, A.; et al. K-ras activation generates an inflammatory response in lung tumors. Oncogene 2006, 25, 2105–2112. [Google Scholar] [CrossRef]
- Pylayeva-Gupta, Y.; Lee, K.E.; Hajdu, C.H.; Miller, G.; Bar-Sagi, D. Oncogenic Kras-induced GM-CSF production promotes the development of pancreatic neoplasia. Cancer Cell 2012, 21, 836–847. [Google Scholar] [CrossRef]
- Ischenko, I.; Zhi, J.; Hayman, M.J.; Petrenko, O. KRAS-dependent suppression of MYC enhances the sensitivity of cancer cells to cytotoxic agents. Oncotarget 2017, 8, 17995–18009. [Google Scholar] [CrossRef]
- Casacuberta-Serra, S.; González-Larreategui, Í.; Capitán-Leo, D.; Soucek, L. MYC and KRAS cooperation: From historical challenges to therapeutic opportunities in cancer. Signal Transduct. Target. Ther. 2024, 9, 205. [Google Scholar] [CrossRef]
- Hu, H.; Cheng, R.; Wang, Y.; Wang, X.; Wu, J.; Kong, Y.; Zhan, S.; Zhou, Z.; Zhu, H.; Yu, R.; et al. Oncogenic KRAS signaling drives evasion of innate immune surveillance in lung adenocarcinoma by activating CD47. J. Clin. Investig. 2023, 133, e153470. [Google Scholar] [CrossRef]
- Zhu, Z.; Aref, A.R.; Cohoon, T.J.; Barbie, T.U.; Imamura, Y.; Yang, S.; Moody, S.E.; Shen, R.R.; Schinzel, A.C.; Thai, T.C.; et al. Inhibition of KRAS-driven tumorigenicity by interruption of an autocrine cytokine circuit. Cancer Discov. 2014, 4, 452–465. [Google Scholar] [CrossRef]
- Mugarza, E.; van Maldegem, F.; Boumelha, J.; Moore, C.; Rana, S.; Llorian Sopena, M.; East, P.; Ambler, R.; Anastasiou, P.; Romero-Clavijo, P.; et al. Therapeutic KRAS(G12C) inhibition drives effective interferon-mediated antitumor immunity in immunogenic lung cancers. Sci. Adv. 2022, 8, eabm8780. [Google Scholar] [CrossRef]
- Muthalagu, N.; Monteverde, T.; Raffo-Iraolagoitia, X.; Wiesheu, R.; Whyte, D.; Hedley, A.; Laing, S.; Kruspig, B.; Upstill-Goddard, R.; Shaw, R.; et al. Repression of the Type I Interferon Pathway Underlies MYC- and KRAS-Dependent Evasion of NK and B Cells in Pancreatic Ductal Adenocarcinoma. Cancer Discov. 2020, 10, 872–887. [Google Scholar] [CrossRef]
- Molina-Arcas, M.; Downward, J. Exploiting the therapeutic implications of KRAS inhibition on tumor immunity. Cancer Cell 2024, 42, 338–357. [Google Scholar] [CrossRef]
- Matsumoto, K.; Okamoto, K.; Okabe, S.; Fujii, R.; Ueda, K.; Ohashi, K.; Seimiya, H. G-quadruplex-forming nucleic acids interact with splicing factor 3B subunit 2 and suppress innate immune gene expression. Genes Cells 2021, 26, 65–82. [Google Scholar] [CrossRef] [PubMed]
- Iachettini, S.; Stevens, M.F.; Frigerio, M.; Hummersone, M.G.; Hutchinson, I.; Garner, T.P.; Searle, M.S.; Wilson, D.W.; Munde, M.; Nanjunda, R.; et al. On and off-target effects of telomere uncapping G-quadruplex selective ligands based on pentacyclic acridinium salts. J. Exp. Clin. Cancer Res. 2013, 32, 68. [Google Scholar] [CrossRef] [PubMed]
- Iachettini, S.; Biroccio, A.; Zizza, P. Therapeutic Use of G4-Ligands in Cancer: State-of-the-Art and Future Perspectives. Pharmaceuticals 2024, 17, 771. [Google Scholar] [CrossRef] [PubMed]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lin, J.; Gong, Z.; Lu, Y.; Cai, J.; Zhang, J.; Tan, J.; Huang, Z.; Chen, S. Recent Progress and Potential of G4 Ligands in Cancer Immunotherapy. Molecules 2025, 30, 1805. https://doi.org/10.3390/molecules30081805
Lin J, Gong Z, Lu Y, Cai J, Zhang J, Tan J, Huang Z, Chen S. Recent Progress and Potential of G4 Ligands in Cancer Immunotherapy. Molecules. 2025; 30(8):1805. https://doi.org/10.3390/molecules30081805
Chicago/Turabian StyleLin, Jiahui, Zhu Gong, Yingyue Lu, Jiongheng Cai, Junjie Zhang, Jiaheng Tan, Zhishu Huang, and Shuobin Chen. 2025. "Recent Progress and Potential of G4 Ligands in Cancer Immunotherapy" Molecules 30, no. 8: 1805. https://doi.org/10.3390/molecules30081805
APA StyleLin, J., Gong, Z., Lu, Y., Cai, J., Zhang, J., Tan, J., Huang, Z., & Chen, S. (2025). Recent Progress and Potential of G4 Ligands in Cancer Immunotherapy. Molecules, 30(8), 1805. https://doi.org/10.3390/molecules30081805