Metastable LaOClx Phase Stabilization as an Effective Strategy for Controllable Chlorination of Ethane into 1,2-Dichloroethane


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
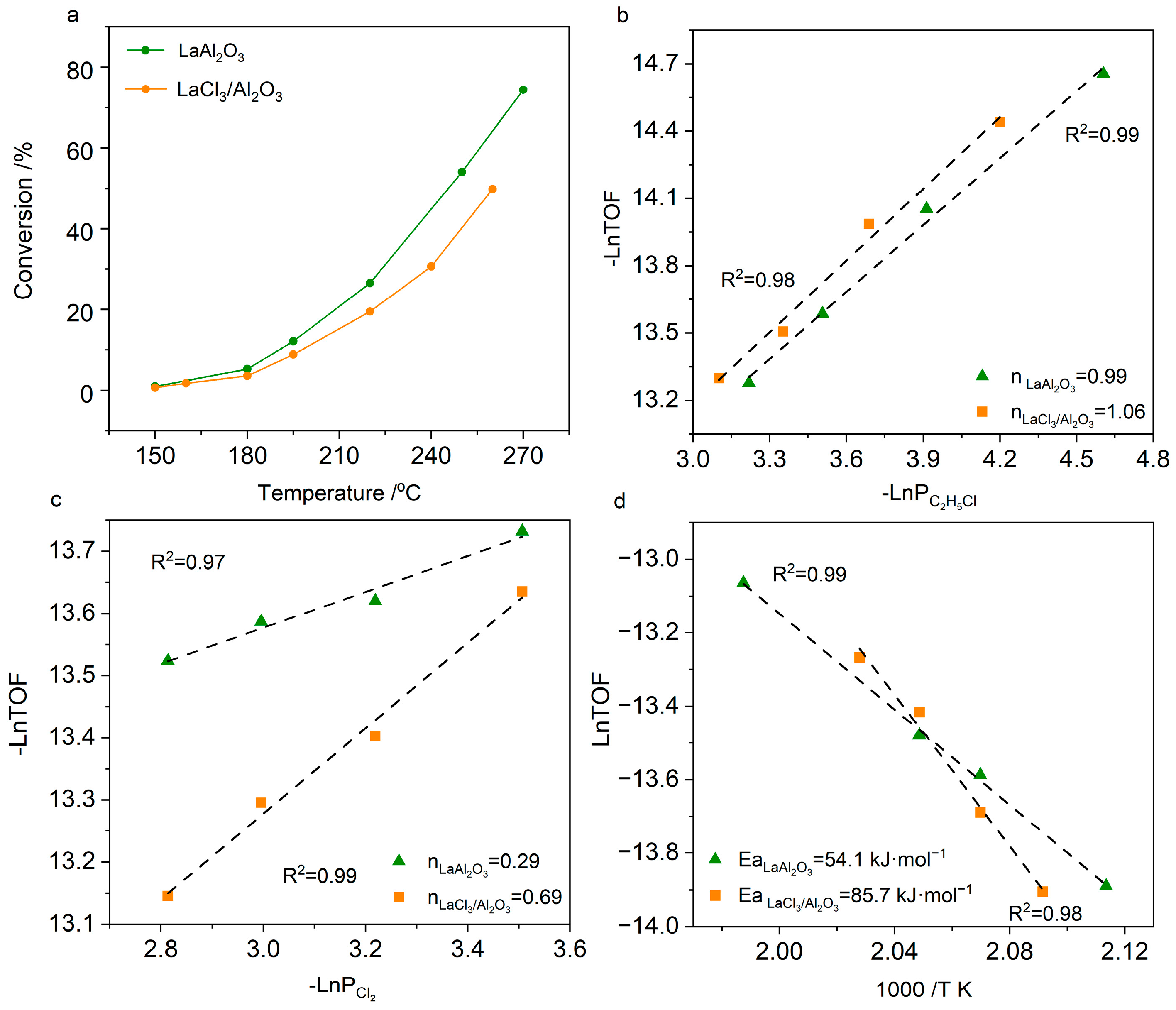
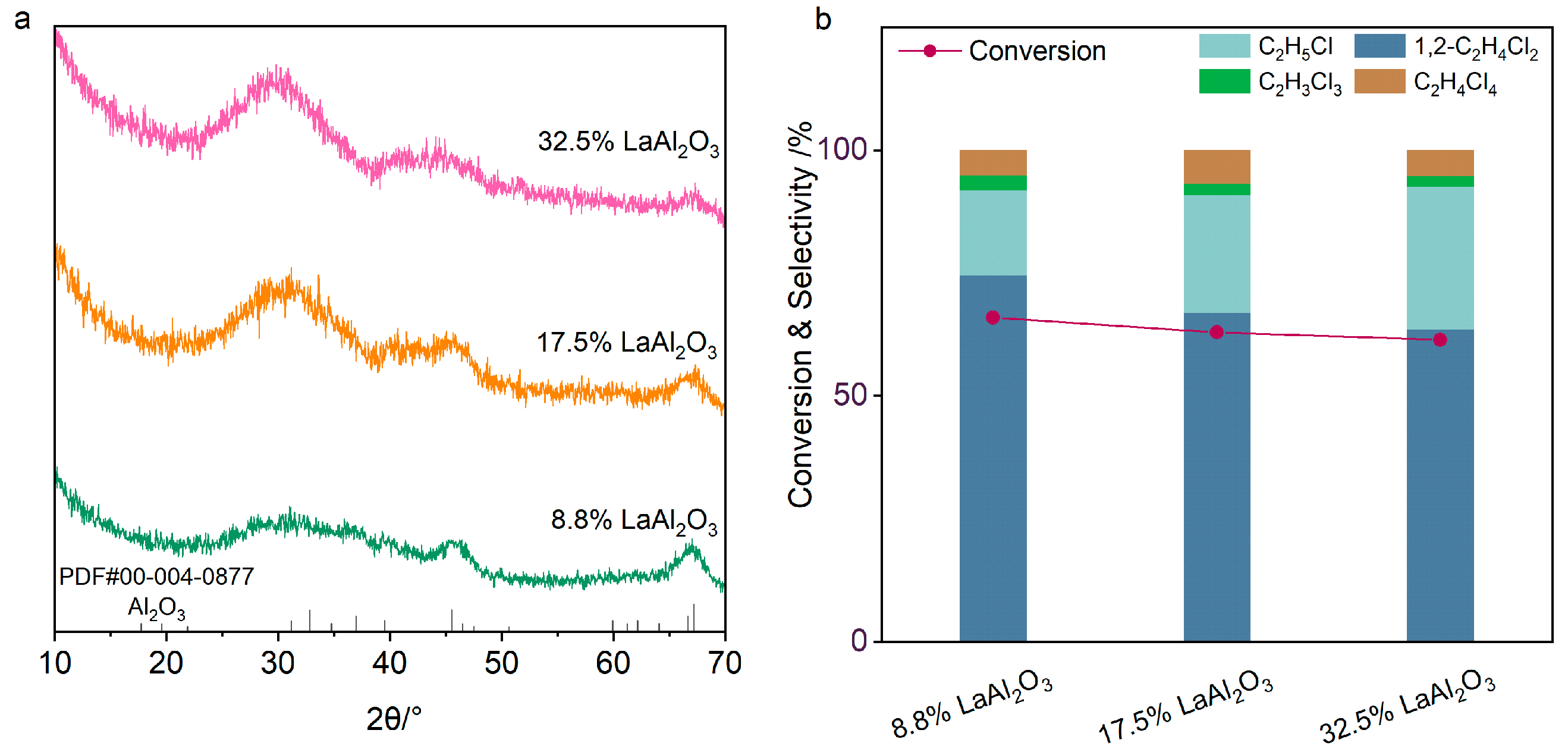
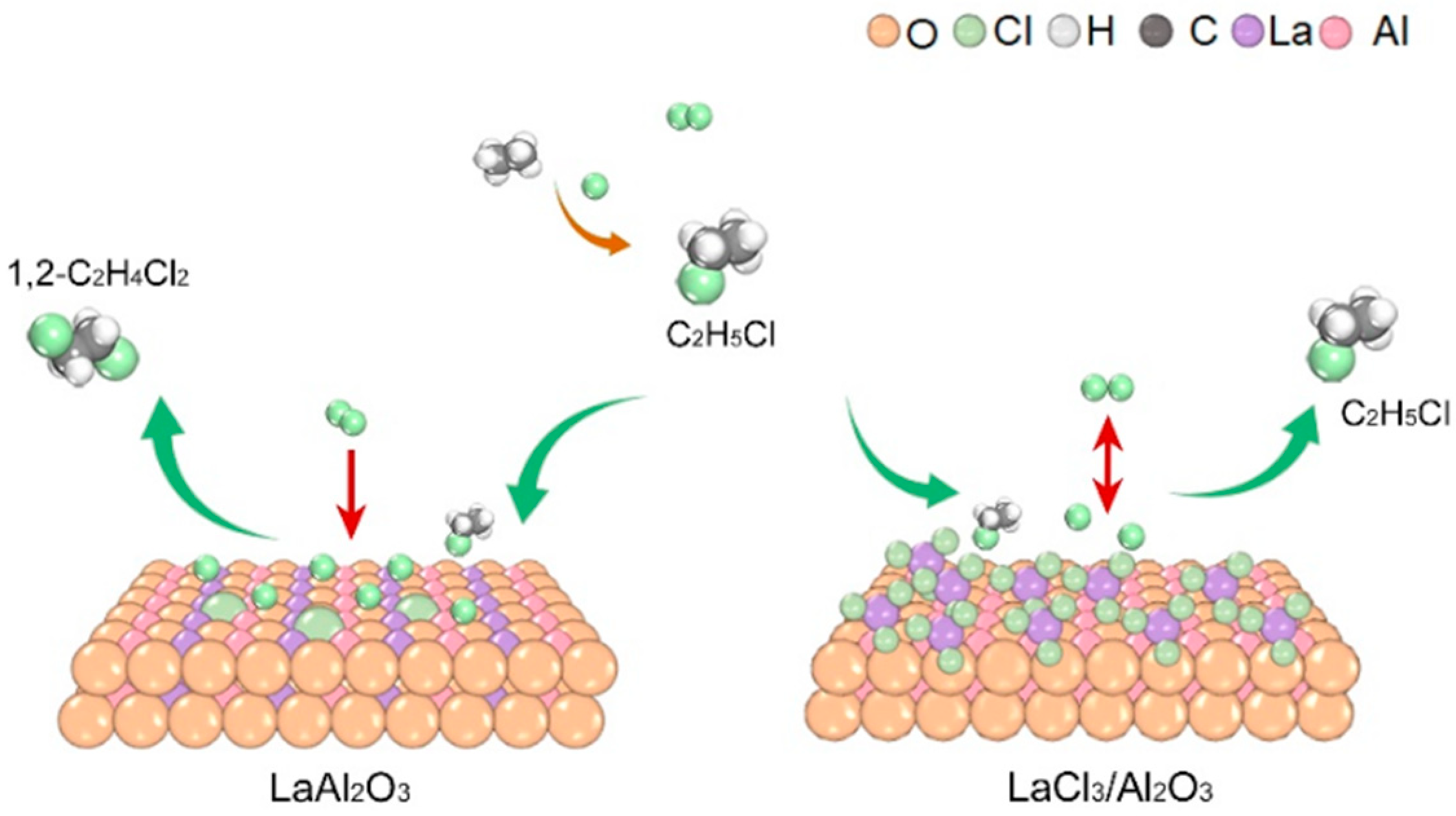
2. Results and Discussion
3. Experimental Section
3.1. Catalyst Preparation
3.2. Characterization
3.3. Catalyst Evaluation
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Skouteris, A.; Giannikopoulos, I.; Edgar, T.F.; Baldea, M.; Allen, D.T.; Stadtherr, M.A. Systems Analysis of Natural Gas Liquid Resources for Chemical Manufacturing: Strategic Utilization of Ethane. Ind. Eng. Chem. Res. 2021, 60, 12377–12389. [Google Scholar] [CrossRef]
- Jing, H.; Tao, L.; Jiesheng, L.; Jie, W. Utilization of Natural Gases as Inexpensive Feedstocks for Fine Chemical Synthesis Through Photocatalysis. In Springer Handbook of Inorganic Photochemistry; Springer International Publishing: Cham, Switzerland, 2022; pp. 1627–1659. [Google Scholar]
- McFarland, E. Unconventional Chemistry for Unconventional Natural Gas. Science 2012, 338, 340–342. [Google Scholar] [CrossRef] [PubMed]
- Medrano-García, J.D.; Giulimondi, V.; Ceruti, A.; Zichittella, G.; Pérez-Ramírez, J.; Guillén-Gosálbez, G. Economic and Environmental Competitiveness of Ethane-Based Technologies for Vinyl Chloride Synthesis. ACS Sustain. Chem. Eng. 2023, 11, 13062–13069. [Google Scholar] [CrossRef]
- He, X.; Li, Z.; Hu, H.; Chen, J.; Zeng, L.; Zhang, J.; Lin, W.; Wang, C. Chemical looping conversion of ethane to ethanol via photo-assisted nitration of ethane. Cell Rep. Phys. Sci. 2021, 2, 100481. [Google Scholar] [CrossRef]
- Wang, M.; Ma, D. Reaction: Direct chlorination of ethane to dichloroethane. Chem 2022, 8, 886–887. [Google Scholar] [CrossRef]
- Zhu, Y.; Shi, S.; Wang, C.; Hu, Y.H. Photocatalytic conversion of ethane: Status and perspective. Int. J. Energy Res. 2020, 44, 708–717. [Google Scholar] [CrossRef]
- De Rosa, S.E.; Allen, D.T. Impact of Natural Gas and Natural Gas Liquids Supplies on the United States Chemical Manufacturing Industry: Production Cost Effects and Identification of Bottleneck Intermediates. ACS Sustain. Chem. Eng. 2015, 3, 451–459. [Google Scholar] [CrossRef]
- Wernet, G.; Bauer, C.; Steubing, B.; Reinhard, J.; Moreno-Ruiz, E.; Weidema, B. The ecoinvent database version 3 (part I): Overview and methodology. Int. J. Life Cycle Assess. 2016, 21, 1218–1230. [Google Scholar] [CrossRef]
- Jin, R.; Peng, M.; Li, A.; Deng, Y.; Jia, Z.; Huang, F.; Ling, Y.; Yang, F.; Fu, H.; Xie, J.; et al. Low Temperature Oxidation of Ethane to Oxygenates by Oxygen over Iridium-Cluster Catalysts. J. Am. Chem. Soc. 2019, 141, 18921–18925. [Google Scholar] [CrossRef]
- Yilang, L.; Charles, J.M.; William, H.G.; Prashant, D. Effects of surface species and homogeneous reactions on rates and selectivity in ethane oxidation on oxide catalysts. AlChE J. 2021, 67, 17483. [Google Scholar]
- Abdulrhman, S.A.-A.; Ahmed Mohamed, E.-T.; Joselito, P.L.; Aslam, K.; Hamid, G.; Attiyah, A.A.-Z.; Ahmed, E.A.; Saeed, M.A.-Z. Mesoporous Organo-Silica Supported Chromium Oxide Catalyst for Oxidative Dehydrogenation of Ethane to Ethylene with CO2. Catalysts 2021, 5, 642. [Google Scholar]
- Grabowski, R. Kinetics of Oxidative Dehydrogenation of C2–C3 Alkanes on Oxide Catalysts. Catal. Rev. 2006, 48, 199–268. [Google Scholar] [CrossRef]
- Duanxing, L.; Zenghao, W.; Kazuhiro, T. Oxidative Dehydrogenation of Ethane to Ethylene over Potassium Tungstate Catalysts. Ind. Eng. Chem. Res. 2024, 63, 5646–5654. [Google Scholar]
- Aaron, S.; Michele, P.; Elaine, G.; Randall, J.M.; Sara, Y.; Henry, K.; Michael, C.; Yuan, G. Catalytic limitations on alkane dehydrogenation under H2 deficient conditions relevant to membrane reactors. Energy Environ. Sci. 2022, 15, 2120–2129. [Google Scholar]
- Novotný, P.; Yusuf, S.; Li, F.; Lamb, H.H. Oxidative dehydrogenation of ethane using MoO3/Fe2O3 catalysts in a cyclic redox mode. Catal. Today 2018, 317, 50–55. [Google Scholar] [CrossRef]
- Henning, D.A.; Schmidt, L.D. Oxidative dehydrogenation of ethane at short contact times: Species and temperature profiles within and after the catalyst. Chem. Eng. Sci. 2002, 57, 2615–2625. [Google Scholar] [CrossRef]
- Zhang, H.-M.; Fan, Q.-Y.; Zhang, Q.-H.; Kang, J.-C.; Wang, Y.; Cheng, J. Understanding Catalytic Mechanisms of Alkane Oxychlorination from the Perspective of Energy Levels. J. Phys. Chem. C 2020, 124, 6070–6077. [Google Scholar] [CrossRef]
- Zichittella, G.; Pérez-Ramírez, J. Ethane-Based Catalytic Process for Vinyl Chloride Manufacture. Angew. Chem. Int. Ed. 2021, 60, 24089–24095. [Google Scholar] [CrossRef]
- Ma, H.; Ma, G.; Qi, Y.; Wang, Y.; Chen, Q.; Rout, K.R.; Fuglerud, T.; Chen, D. Nitrogen-Doped Carbon-Assisted One-pot Tandem Reaction for Vinyl Chloride Production via Ethylene Oxychlorination. Angew. Chem. Int. Ed. 2020, 59, 22080–22085. [Google Scholar] [CrossRef]
- Mehrpooya, M.; Ghorbani, B.; Khalili, M. A new developed integrated process configuration for production of hydrogen chloride using geothermal and wind energy resources. Sustain. Energy Technol. Assess. 2021, 45, 101173. [Google Scholar] [CrossRef]
- Scharfe, M.; Lira-Parada, P.A.; Paunović, V.; Moser, M.; Amrute, A.P.; Pérez-Ramírez, J. Oxychlorination–Dehydrochlorination Chemistry on Bifunctional Ceria Catalysts for Intensified Vinyl Chloride Production. Angew. Chem. Int. Ed. 2016, 55, 3068–3072. [Google Scholar] [CrossRef]
- Li, C.; Hu, G.; Zhong, W.; He, W.; Du, W.; Qian, F. Coke Deposition Influence Based on a Run Length Simulation of a 1,2-Dichloroethane Cracker. Ind. Eng. Chem. Res. 2013, 52, 17501–17516. [Google Scholar] [CrossRef]
- Scharfe, M.; Zichittella, G.; Kondratenko, V.A.; Kondratenko, E.V.; López, N.; Pérez-Ramírez, J. Mechanistic origin of the diverging selectivity patterns in catalyzed ethane and ethene oxychlorination. J. Catal. 2019, 377, 233–244. [Google Scholar] [CrossRef]
- Lin, R.; Amrute, A.P.; Pérez-Ramírez, J. Halogen-Mediated Conversion of Hydrocarbons to Commodities. Chem. Rev. 2017, 117, 4182–4247. [Google Scholar] [CrossRef]
- Johnston, P.; Carthey, N.; Hutchings, G.J. Discovery, Development, and Commercialization of Gold Catalysts for Acetylene Hydrochlorination. J. Am. Chem. Soc. 2015, 137, 14548–14557. [Google Scholar] [CrossRef] [PubMed]
- Malta, G.; Kondrat, S.A.; Freakley, S.J.; Davies, C.J.; Lu, L.; Dawson, S.; Thetford, A.; Gibson, E.K.; Morgan, D.J.; Jones, W.; et al. Identification of single-site gold catalysis in acetylene hydrochlorination. Science 2017, 355, 1399–1402. [Google Scholar] [CrossRef]
- Muddada, N.B.; Olsbye, U.; Caccialupi, L.; Cavani, F.; Leofanti, G.; Gianolio, D.; Bordiga, S.; Lamberti, C. Influence of additives in defining the active phase of the ethylene oxychlorination catalyst. Phys. Chem. Chem. Phys. 2010, 12, 5605–5618. [Google Scholar] [CrossRef]
- Conte, M.; Carley, A.F.; Hutchings, G.J. Reactivation of a Carbon-supported Gold Catalyst for the Hydrochlorination of Acetylene. Catal. Lett. 2008, 124, 165–167. [Google Scholar] [CrossRef]
- Li, C.; Zhou, G.; Wang, L.; Li, Z.; Xue, Y.; Cheng, T. Effect of impregnation procedure of La2O3 precursor on copper-based catalysts for ethane oxychlorination. Catal. Commun. 2011, 13, 22–25. [Google Scholar] [CrossRef]
- He, J.; Xu, T.; Wang, Z.; Zhang, Q.; Deng, W.; Wang, Y. Transformation of Methane to Propylene: A Two-Step Reaction Route Catalyzed by Modified CeO2 Nanocrystals and Zeolites. Angew. Chem. Int. Ed. Engl. 2012, 51, 2438–2442. [Google Scholar] [CrossRef]
- Xie, Q.; Zhang, H.; Kang, J.; Cheng, J.; Zhang, Q.; Wang, Y. Oxidative Dehydrogenation of Propane to Propylene in the Presence of HCl Catalyzed by CeO2 and NiO-Modified CeO2 Nanocrystals. ACS Catal. 2018, 8, 4902–4916. [Google Scholar] [CrossRef]
- Akintola, J.; Patinvoh, R.; Moradeyo, O.; Akpan, J.; Umoh, G.; Wilson, E.; Moses, Q.; Udom, P.; Osagie, E. Optimization and techno-economic evaluation of an integrated process route for the synthesis of vinyl chloride monomer. RSC Sustain. 2025, 3, 526–539. [Google Scholar] [CrossRef]
- Zichittella, G.; Aellen, N.; Paunović, V.; Amrute, A.P.; Pérez-Ramírez, J. Olefins from Natural Gas by Oxychlorination. Angew. Chem. 2017, 56, 13670–13674. [Google Scholar] [CrossRef] [PubMed]
- Hickman, D.A.; Jones, M.E.; Jovanovic, Z.R.; Olken, M.M.; Podkolzin, S.G.; Stangland, E.E.; Thompson, R.K. Reactor Scale-up for Fluidized Bed Conversion of Ethane to Vinyl Chloride. Ind. Eng. Chem. Res. 2010, 49, 10674–10681. [Google Scholar] [CrossRef]
- Scharfe, M.; Lira-Parada, P.A.; Amrute, A.P.; Mitchell, S.; Pérez-Ramírez, J. Lanthanide compounds as catalysts for the one-step synthesis of vinyl chloride from ethylene. J. Catal. 2016, 344, 524–534. [Google Scholar] [CrossRef]
- Podkolzin, S.G.; Stangland, E.E.; Jones, M.E.; Peringer, E.; Lercher, J.A. Methyl Chloride Production from Methane over Lanthanum-Based Catalysts. J. Am. Chem. Soc. 2007, 129, 2569–2576. [Google Scholar] [CrossRef]
- Starchevskyy, V.; Shparij, M.; Hrynchuk, Y.; Reutskyy, V.; Kurta, S.; Khatsevich, O. Modification of the Catalytic System for the Industrial Chlorine Processing of Ethylene in 1,2-Dichloroethane. Chem. Chem. Technol. 2020, 14, 394–402. [Google Scholar] [CrossRef]
- Kim, S.W.; Jyoko, K.; Masui, T.; Imanaka, N. Green-emitting (La,M,Tb)OCl (M=Mg, Ca, and Sr) phosphors. Opt. Mater. 2012, 35, 280–284. [Google Scholar] [CrossRef]
- Zhang, A.; Jiang, H.; Wu, Z.; Wu, C.; Qian, B. Internal stress, lattice deformation, and modulus of polymers. J. Appl. Polym. Sci. 1991, 42, 1779–1791. [Google Scholar] [CrossRef]
- Lin, L.; Zhang, L.; Fu, Z.; Lou, J.; Gao, Z.; Wu, J.; Li, C.; Han, C.; Zhou, D.; Wang, Z. Unraveling Mechanism for Microstructure Engineering toward High-Capacity Nickel-Rich Cathode Materials. Adv. Mater. 2024, 36, 2406175. [Google Scholar] [CrossRef]
- Jaffar, B.M.; Swart, H.C.; Seed Ahmed, H.A.A.; Yousif, A.; Kroon, R.E. Comparative study of the luminescence of Bi doped LaOCl and LaOBr phosphor powders. J. Lumin. 2022, 250, 119050. [Google Scholar] [CrossRef]
- Uwamino, Y.; Tsuge, A.; Ishizuka, T.; Yamatera, H. X-Ray Photoelectron Spectroscopy of Rare Earth Halides. Bull. Chem. Soc. Jpn. 2006, 59, 2263–2267. [Google Scholar] [CrossRef]
- Terlingen, B.; Oord, R.; Ahr, M.; Hutter, E.M.; van Lare, C.; Weckhuysen, B.M. Favoring the Methane Oxychlorination Reaction over EuOCl by Synergistic Effects with Lanthanum. ACS Catal. 2022, 12, 5698–5710. [Google Scholar] [CrossRef]
- Ma, H.; Wang, Y.; Qi, Y.; Rout, K.R.; Chen, D. Critical Review of Catalysis for Ethylene Oxychlorination. ACS Catal. 2020, 10, 9299–9319. [Google Scholar] [CrossRef]
- Dahl, I.M.; Myhrvold, E.M.; Olsbye, U.; Rohr, F.; Rokstad, O.A.; Swang, O. On the gas-phase chlorination of ethane. Ind. Eng. Chem. Res. 2001, 40, 2226–2235. [Google Scholar] [CrossRef]
- Zaidman, O.A.; Kurlyandskaya, I.I.; Sonin, E.V.; Treger, Y.A.; Pashcenko, L.E.; Verkhutova, E.I.; Kutcher, V.L. Kinetics and Mechanism of Ethane Chlorination in Volume Reaction Inhibited by Oxygen. Khim. Promst. 1990, 271–274. [Google Scholar]

Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, Y.; Zhu, Z.; Wu, X.; Ma, L.; Sun, X.; Liu, Q. Metastable LaOClx Phase Stabilization as an Effective Strategy for Controllable Chlorination of Ethane into 1,2-Dichloroethane. Molecules 2025, 30, 1746. https://doi.org/10.3390/molecules30081746
Li Y, Zhu Z, Wu X, Ma L, Sun X, Liu Q. Metastable LaOClx Phase Stabilization as an Effective Strategy for Controllable Chlorination of Ethane into 1,2-Dichloroethane. Molecules. 2025; 30(8):1746. https://doi.org/10.3390/molecules30081746
Chicago/Turabian StyleLi, Yuting, Zihan Zhu, Xia Wu, Lei Ma, Xiaohui Sun, and Qinggang Liu. 2025. "Metastable LaOClx Phase Stabilization as an Effective Strategy for Controllable Chlorination of Ethane into 1,2-Dichloroethane" Molecules 30, no. 8: 1746. https://doi.org/10.3390/molecules30081746
APA StyleLi, Y., Zhu, Z., Wu, X., Ma, L., Sun, X., & Liu, Q. (2025). Metastable LaOClx Phase Stabilization as an Effective Strategy for Controllable Chlorination of Ethane into 1,2-Dichloroethane. Molecules, 30(8), 1746. https://doi.org/10.3390/molecules30081746