
Molecular Subtypes and Targeted Therapeutic Strategies in Small Cell Lung Cancer: Advances, Challenges, and Future Perspectives

 , and
, and
Abstract
1. Introduction
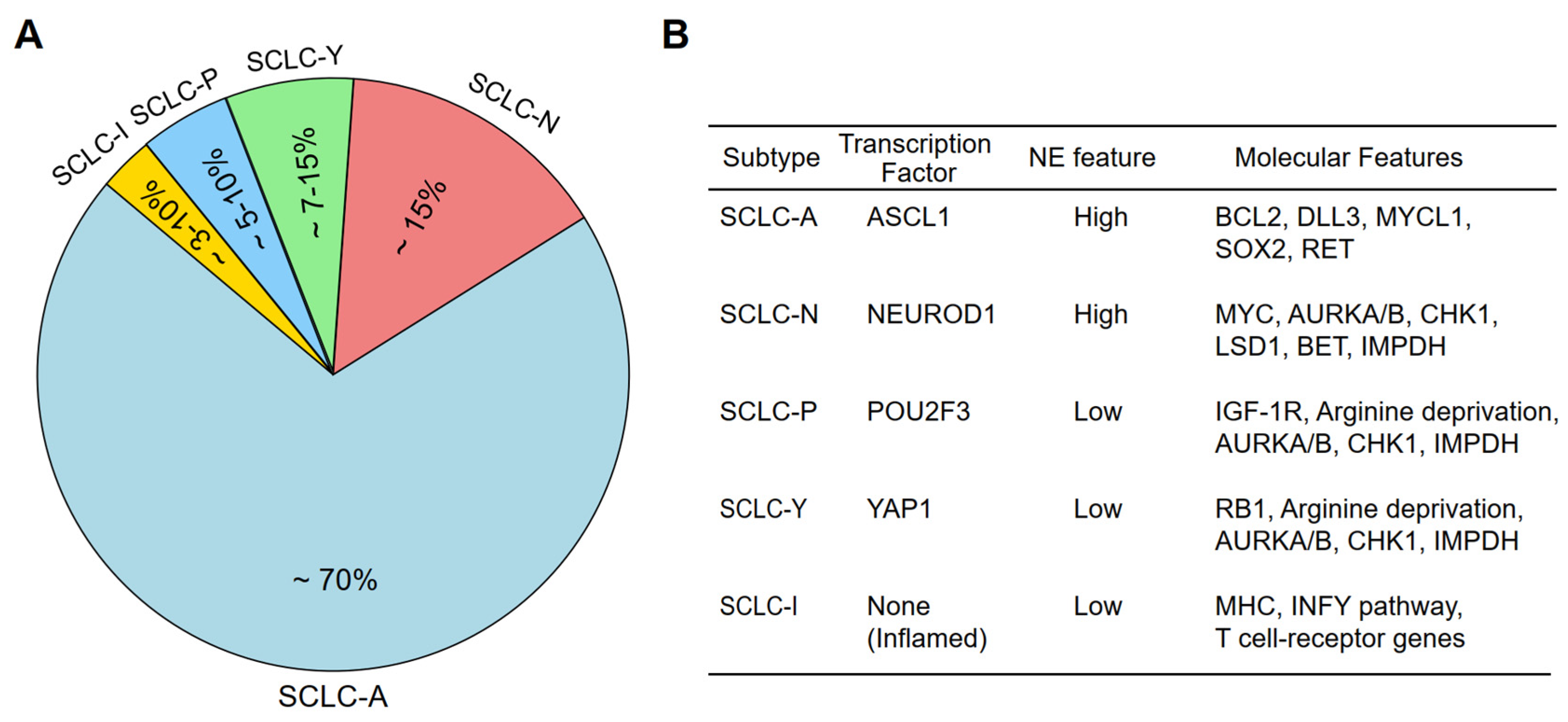
2. Molecular Subtypes of SCLC
2.1. Historical Perspective and Classification Evolution
2.2. Transcription Factor-Based Subtypes of SCLC
2.3. Emerging Subtypes and Multi-Omic Classifications
2.4. Challenges in SCLC Subtyping
3. Upstream Regulation Mechanism of Lineage-Defining Transcription Factors
3.1. Regulation of ASCL1
3.2. Regulation of NEUROD1
3.3. Regulation of POU2F3
4. Targeted Therapeutic Strategies for SCLC Subtypes
4.1. ASCL1-Driven SCLC (SCLC-A)
4.2. NEUROD1-Driven SCLC (SCLC-N)
4.3. POU2F3-Driven SCLC (SCLC-P)
4.4. Inflamed SCLC (SCLC-I) and Immunotherapy Approaches
4.5. Therapeutic Features Beyond Classical Subtypes
4.5.1. YAP1 Activation in Relapsed or Therapy-Resistant SCLC
4.5.2. RB1-Intact Phenotype in SCLC: Biology or Artifact?
4.5.3. Molecular Patterns Identified by NMF Clustering
5. Challenges in Targeting SCLC Subtypes
5.1. Heterogeneity and Tumor Evolution
5.2. Challenges in Biomarker-Driven Therapy
5.3. Resistance Mechanisms and Combination Approaches
5.4. Mixed Histology and Transformed SCLC: Distinct Clinical Entities
6. Conclusions and Future Perspectives
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- van Meerbeeck, J.P.; Fennell, D.A.; De Ruysscher, D.K. Small-cell lung cancer. Lancet 2011, 378, 1741–1755. [Google Scholar] [CrossRef]
- George, J.; Lim, J.S.; Jang, S.J.; Cun, Y.; Ozretić, L.; Kong, G.; Leenders, F.; Lu, X.; Fernández-Cuesta, L.; Bosco, G.; et al. Comprehensive genomic profiles of small cell lung cancer. Nature 2015, 524, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Detterbeck, F.C.; Nicholson, A.G.; Franklin, W.A.; Marom, E.M.; Travis, W.D.; Girard, N.; Arenberg, D.A.; Bolejack, V.; Donington, J.S.; Mazzone, P.J.; et al. The IASLC Lung Cancer Staging Project: Summary of Proposals for Revisions of the Classification of Lung Cancers with Multiple Pulmonary Sites of Involvement in the Forthcoming Eighth Edition of the TNM Classification. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2016, 11, 639–650. [Google Scholar] [CrossRef] [PubMed]
- Hecht, S.S. Lung carcinogenesis by tobacco smoke. Int. J. Cancer 2012, 131, 2724–2732. [Google Scholar] [CrossRef]
- Siegel, D.A.; Fedewa, S.A.; Henley, S.J.; Pollack, L.A.; Jemal, A. Proportion of Never Smokers Among Men and Women with Lung Cancer in 7 US States. JAMA Oncol. 2021, 7, 302–304. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Gümüş, Z.H.; Colarossi, C.; Memeo, L.; Wang, X.; Kong, C.Y.; Boffetta, P. SCLC: Epidemiology, Risk Factors, Genetic Susceptibility, Molecular Pathology, Screening, and Early Detection. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2023, 18, 31–46. [Google Scholar] [CrossRef] [PubMed]
- Ortega-Franco, A.; Ackermann, C.; Paz-Ares, L.; Califano, R. First-line immune checkpoint inhibitors for extensive stage small-cell lung cancer: Clinical developments and future directions. ESMO Open 2021, 6, 100003. [Google Scholar] [CrossRef] [PubMed]
- Früh, M.; De Ruysscher, D.; Popat, S.; Crinò, L.; Peters, S.; Felip, E. Small-cell lung cancer (SCLC): ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2013, 24 (Suppl. 6), VI99–VI105. [Google Scholar] [CrossRef]
- Lally, B.E.; Urbanic, J.J.; Blackstock, A.W.; Miller, A.A.; Perry, M.C. Small cell lung cancer: Have we made any progress over the last 25 years? Oncologist 2007, 12, 1096–1104. [Google Scholar] [CrossRef]
- Wang, S.; Tang, J.; Sun, T.; Zheng, X.; Li, J.; Sun, H.; Zhou, X.; Zhou, C.; Zhang, H.; Cheng, Z.; et al. Survival changes in patients with small cell lung cancer and disparities between different sexes, socioeconomic statuses and ages. Sci. Rep. 2017, 7, 1339. [Google Scholar] [CrossRef]
- Horn, L.; Mansfield, A.S.; Szczęsna, A.; Havel, L.; Krzakowski, M.; Hochmair, M.J.; Huemer, F.; Losonczy, G.; Johnson, M.L.; Nishio, M.; et al. First-Line Atezolizumab plus Chemotherapy in Extensive-Stage Small-Cell Lung Cancer. N. Engl. J. Med. 2018, 379, 2220–2229. [Google Scholar] [CrossRef] [PubMed]
- Ragavan, M.; Das, M. Systemic Therapy of Extensive Stage Small Cell Lung Cancer in the Era of Immunotherapy. Curr. Treat. Options Oncol. 2020, 21, 64. [Google Scholar] [CrossRef]
- Jordan, E.J.; Kim, H.R.; Arcila, M.E.; Barron, D.; Chakravarty, D.; Gao, J.; Chang, M.T.; Ni, A.; Kundra, R.; Jonsson, P.; et al. Prospective Comprehensive Molecular Characterization of Lung Adenocarcinomas for Efficient Patient Matching to Approved and Emerging Therapies. Cancer Discov. 2017, 7, 596–609. [Google Scholar] [CrossRef]
- Rudin, C.M.; Poirier, J.T.; Byers, L.A.; Dive, C.; Dowlati, A.; George, J.; Heymach, J.V.; Johnson, J.E.; Lehman, J.M.; MacPherson, D.; et al. Molecular subtypes of small cell lung cancer: A synthesis of human and mouse model data. Nat. Rev. Cancer 2019, 19, 289–297. [Google Scholar] [CrossRef] [PubMed]
- Caeser, R.; Egger, J.V.; Chavan, S.; Socci, N.D.; Jones, C.B.; Kombak, F.E.; Asher, M.; Roehrl, M.H.; Shah, N.S.; Allaj, V.; et al. Genomic and transcriptomic analysis of a library of small cell lung cancer patient-derived xenografts. Nat. Commun. 2022, 13, 2144. [Google Scholar] [CrossRef]
- Simpson, K.L.; Stoney, R.; Frese, K.K.; Simms, N.; Rowe, W.; Pearce, S.P.; Humphrey, S.; Booth, L.; Morgan, D.; Dynowski, M.; et al. A biobank of small cell lung cancer CDX models elucidates inter- and intratumoral phenotypic heterogeneity. Nat. Cancer 2020, 1, 437–451. [Google Scholar] [CrossRef]
- Liu, Q.; Zhang, J.; Guo, C.; Wang, M.; Wang, C.; Yan, Y.; Sun, L.; Wang, D.; Zhang, L.; Yu, H.; et al. Proteogenomic characterization of small cell lung cancer identifies biological insights and subtype-specific therapeutic strategies. Cell 2024, 187, 184–203.e128. [Google Scholar] [CrossRef] [PubMed]
- Gay, C.M.; Stewart, C.A.; Park, E.M.; Diao, L.; Groves, S.M.; Heeke, S.; Nabet, B.Y.; Fujimoto, J.; Solis, L.M.; Lu, W.; et al. Patterns of transcription factor programs and immune pathway activation define four major subtypes of SCLC with distinct therapeutic vulnerabilities. Cancer Cell 2021, 39, 346–360.e347. [Google Scholar] [CrossRef]
- Carney, D.N.; Gazdar, A.F.; Bepler, G.; Guccion, J.G.; Marangos, P.J.; Moody, T.W.; Zweig, M.H.; Minna, J.D. Establishment and identification of small cell lung cancer cell lines having classic and variant features. Cancer Res. 1985, 45, 2913–2923. [Google Scholar]
- Yang, Y.; Valdés-Rives, S.A.; Liu, Q.; Gao, T.; Burudpakdee, C.; Li, Y.; Tan, J.; Tan, Y.; Koch, C.A.; Rong, Y.; et al. Thyroid hormone suppresses medulloblastoma progression through promoting terminal differentiation of tumor cells. Cancer Cell 2024, 42, 1434–1449.e1435. [Google Scholar] [CrossRef]
- Kong, R.; Patel, A.S.; Sato, T.; Jiang, F.; Yoo, S.; Bao, L.; Sinha, A.; Tian, Y.; Fridrikh, M.; Liu, S.; et al. Transcriptional Circuitry of NKX2-1 and SOX1 Defines an Unrecognized Lineage Subtype of Small-Cell Lung Cancer. Am. J. Respir. Crit. Care Med. 2022, 206, 1480–1494. [Google Scholar] [CrossRef] [PubMed]
- Mollaoglu, G.; Guthrie, M.R.; Böhm, S.; Brägelmann, J.; Can, I.; Ballieu, P.M.; Marx, A.; George, J.; Heinen, C.; Chalishazar, M.D.; et al. MYC Drives Progression of Small Cell Lung Cancer to a Variant Neuroendocrine Subtype with Vulnerability to Aurora Kinase Inhibition. Cancer Cell 2017, 31, 270–285. [Google Scholar] [CrossRef]
- Borromeo, M.D.; Savage, T.K.; Kollipara, R.K.; He, M.; Augustyn, A.; Osborne, J.K.; Girard, L.; Minna, J.D.; Gazdar, A.F.; Cobb, M.H.; et al. ASCL1 and NEUROD1 Reveal Heterogeneity in Pulmonary Neuroendocrine Tumors and Regulate Distinct Genetic Programs. Cell Rep. 2016, 16, 1259–1272. [Google Scholar] [CrossRef]
- Poirier, J.T.; Gardner, E.E.; Connis, N.; Moreira, A.L.; de Stanchina, E.; Hann, C.L.; Rudin, C.M. DNA methylation in small cell lung cancer defines distinct disease subtypes and correlates with high expression of EZH2. Oncogene 2015, 34, 5869–5878. [Google Scholar] [CrossRef] [PubMed]
- McColl, K.; Wildey, G.; Sakre, N.; Lipka, M.B.; Behtaj, M.; Kresak, A.; Chen, Y.; Yang, M.; Velcheti, V.; Fu, P.; et al. Reciprocal expression of INSM1 and YAP1 defines subgroups in small cell lung cancer. Oncotarget 2017, 8, 73745–73756. [Google Scholar] [CrossRef]
- Baine, M.K.; Hsieh, M.S.; Lai, W.V.; Egger, J.V.; Jungbluth, A.A.; Daneshbod, Y.; Beras, A.; Spencer, R.; Lopardo, J.; Bodd, F.; et al. SCLC Subtypes Defined by ASCL1, NEUROD1, POU2F3, and YAP1: A Comprehensive Immunohistochemical and Histopathologic Characterization. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2020, 15, 1823–1835. [Google Scholar] [CrossRef]
- Zhang, X.; Wang, H.; Liu, W.; Xiao, Z.; Ma, Z.; Zhang, Z.; Gong, W.; Chen, J.; Liu, Z. Molecular features and evolutionary trajectory of ASCL1(+) and NEUROD1(+) SCLC cells. Br. J. Cancer 2023, 128, 748–759. [Google Scholar] [CrossRef]
- Wooten, D.J.; Groves, S.M.; Tyson, D.R.; Liu, Q.; Lim, J.S.; Albert, R.; Lopez, C.F.; Sage, J.; Quaranta, V. Systems-level network modeling of Small Cell Lung Cancer subtypes identifies master regulators and destabilizers. PLoS Comput. Biol. 2019, 15, e1007343. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Klingbeil, O.; He, X.Y.; Wu, X.S.; Arun, G.; Lu, B.; Somerville, T.D.D.; Milazzo, J.P.; Wilkinson, J.E.; Demerdash, O.E.; et al. POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer. Genes Dev. 2018, 32, 915–928. [Google Scholar] [CrossRef]
- Wu, X.S.; He, X.Y.; Ipsaro, J.J.; Huang, Y.H.; Preall, J.B.; Ng, D.; Shue, Y.T.; Sage, J.; Egeblad, M.; Joshua-Tor, L.; et al. OCA-T1 and OCA-T2 are coactivators of POU2F3 in the tuft cell lineage. Nature 2022, 607, 169–175. [Google Scholar] [CrossRef]
- George, J.; Maas, L.; Abedpour, N.; Cartolano, M.; Kaiser, L.; Fischer, R.N.; Scheel, A.H.; Weber, J.P.; Hellmich, M.; Bosco, G.; et al. Evolutionary trajectories of small cell lung cancer under therapy. Nature 2024, 627, 880–889. [Google Scholar] [CrossRef]
- Park, S.; Hong, T.H.; Hwang, S.; Heeke, S.; Gay, C.M.; Kim, J.; Jung, H.A.; Sun, J.M.; Ahn, J.S.; Ahn, M.J.; et al. Comprehensive analysis of transcription factor-based molecular subtypes and their correlation to clinical outcomes in small-cell lung cancer. EBioMedicine 2024, 102, 105062. [Google Scholar] [CrossRef]
- Ireland, A.S.; Micinski, A.M.; Kastner, D.W.; Guo, B.; Wait, S.J.; Spainhower, K.B.; Conley, C.C.; Chen, O.S.; Guthrie, M.R.; Soltero, D.; et al. MYC Drives Temporal Evolution of Small Cell Lung Cancer Subtypes by Reprogramming Neuroendocrine Fate. Cancer Cell 2020, 38, 60–78.e12. [Google Scholar] [CrossRef]
- Owonikoko, T.K.; Dwivedi, B.; Chen, Z.; Zhang, C.; Barwick, B.; Ernani, V.; Zhang, G.; Gilbert-Ross, M.; Carlisle, J.; Khuri, F.R.; et al. YAP1 Expression in SCLC Defines a Distinct Subtype With T-cell-Inflamed Phenotype. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2021, 16, 464–476. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z.; Liu, C.; Zheng, S.; Yao, Y.; Wang, S.; Wang, X.; Yin, E.; Zeng, Q.; Zhang, C.; Zhang, G.; et al. Molecular subtypes of neuroendocrine carcinomas: A cross-tissue classification framework based on five transcriptional regulators. Cancer Cell 2024, 42, 1106–1125.e1108. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; Su, J.; Li, F.L.; Chen, T.; Mayner, J.; Engler, A.; Ma, S.; Li, Q.; Guan, K.L. YAP silencing by RB1 mutation is essential for small-cell lung cancer metastasis. Nat. Commun. 2023, 14, 5916. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.; Cai, L.; Girard, L.; Prall, O.W.J.; Rajan, N.; Khoo, C.; Batrouney, A.; Byrne, D.J.; Boyd, D.K.; Kersbergen, A.J.; et al. Molecular and Pathologic Characterization of YAP1-Expressing Small Cell Lung Cancer Cell Lines Leads to Reclassification as SMARCA4-Deficient Malignancies. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2024, 30, 1846–1858. [Google Scholar] [CrossRef]
- Febres-Aldana, C.A.; Chang, J.C.; Ptashkin, R.; Wang, Y.; Gedvilaite, E.; Baine, M.K.; Travis, W.D.; Ventura, K.; Bodd, F.; Yu, H.A.; et al. Rb Tumor Suppressor in Small Cell Lung Cancer: Combined Genomic and IHC Analysis with a Description of a Distinct Rb-Proficient Subset. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2022, 28, 4702–4713. [Google Scholar] [CrossRef]
- Rekhtman, N. All That Is Small Is Not a Small-Cell Carcinoma: Thoracic SMARCA4-Deficient Undifferentiated Tumors Masquerading as SCLC. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2024, 30, 1708–1711. [Google Scholar] [CrossRef]
- Rekhtman, N.; Montecalvo, J.; Chang, J.C.; Alex, D.; Ptashkin, R.N.; Ai, N.; Sauter, J.L.; Kezlarian, B.; Jungbluth, A.; Desmeules, P.; et al. SMARCA4-Deficient Thoracic Sarcomatoid Tumors Represent Primarily Smoking-Related Undifferentiated Carcinomas Rather Than Primary Thoracic Sarcomas. J. Thorac. Oncol. 2020, 15, 231–247. [Google Scholar] [CrossRef]
- Jia, D.; Augert, A.; Kim, D.W.; Eastwood, E.; Wu, N.; Ibrahim, A.H.; Kim, K.B.; Dunn, C.T.; Pillai, S.P.S.; Gazdar, A.F.; et al. Crebbp Loss Drives Small Cell Lung Cancer and Increases Sensitivity to HDAC Inhibition. Cancer Discov. 2018, 8, 1422–1437. [Google Scholar] [CrossRef] [PubMed]
- Schaffer, B.E.; Park, K.S.; Yiu, G.; Conklin, J.F.; Lin, C.; Burkhart, D.L.; Karnezis, A.N.; Sweet-Cordero, E.A.; Sage, J. Loss of p130 accelerates tumor development in a mouse model for human small-cell lung carcinoma. Cancer Res. 2010, 70, 3877–3883. [Google Scholar] [CrossRef]
- Wu, Q.; Guo, J.; Liu, Y.; Zheng, Q.; Li, X.; Wu, C.; Fang, D.; Chen, X.; Ma, L.; Xu, P.; et al. YAP drives fate conversion and chemoresistance of small cell lung cancer. Sci. Adv. 2021, 7, eabg1850. [Google Scholar] [CrossRef]
- Pearsall, S.M.; Humphrey, S.; Revill, M.; Morgan, D.; Frese, K.K.; Galvin, M.; Kerr, A.; Carter, M.; Priest, L.; Blackhall, F.; et al. The Rare YAP1 Subtype of SCLC Revisited in a Biobank of 39 Circulating Tumor Cell Patient Derived Explant Models: A Brief Report. J. Thorac. Oncol. Off. Publ. Int. Assoc. Study Lung Cancer 2020, 15, 1836–1843. [Google Scholar] [CrossRef]
- Catozzi, A.; Peiris-Pagès, M.; Humphrey, S.; Revill, M.; Morgan, D.; Roebuck, J.; Chen, Y.; Davies-Williams, B.; Lallo, A.; Galvin, M.; et al. Functional Characterisation of the ATOH1 Molecular Subtype Indicates a Pro-Metastatic Role in Small Cell Lung Cancer. bioRxiv 2024. [Google Scholar] [CrossRef]
- Chen, Y.; Li, H.; Fan, Y. Shaping the tumor immune microenvironment of SCLC: Mechanisms, and opportunities for immunotherapy. Cancer Treat. Rev. 2023, 120, 102606. [Google Scholar] [CrossRef]
- Shirasawa, M.; Yoshida, T.; Shiraishi, K.; Takigami, A.; Takayanagi, D.; Imabayashi, T.; Matsumoto, Y.; Masuda, K.; Shinno, Y.; Okuma, Y.; et al. Identification of inflamed-phenotype of small cell lung cancer leading to the efficacy of anti-PD-L1 antibody and chemotherapy. Lung Cancer 2023, 179, 107183. [Google Scholar] [CrossRef] [PubMed]
- Dora, D.; Rivard, C.; Yu, H.; Bunn, P.; Suda, K.; Ren, S.; Lueke Pickard, S.; Laszlo, V.; Harko, T.; Megyesfalvi, Z.; et al. Neuroendocrine subtypes of small cell lung cancer differ in terms of immune microenvironment and checkpoint molecule distribution. Mol. Oncol. 2020, 14, 1947–1965. [Google Scholar] [CrossRef]
- Nabet, B.Y.; Hamidi, H.; Lee, M.C.; Banchereau, R.; Morris, S.; Adler, L.; Gayevskiy, V.; Elhossiny, A.M.; Srivastava, M.K.; Patil, N.S.; et al. Immune heterogeneity in small-cell lung cancer and vulnerability to immune checkpoint blockade. Cancer Cell 2024, 42, 429–443.e424. [Google Scholar] [CrossRef]
- Heeke, S.; Gay, C.M.; Estecio, M.R.; Tran, H.; Morris, B.B.; Zhang, B.; Tang, X.; Raso, M.G.; Rocha, P.; Lai, S.; et al. Tumor- and circulating-free DNA methylation identifies clinically relevant small cell lung cancer subtypes. Cancer Cell 2024, 42, 225–237.e225. [Google Scholar] [CrossRef]
- Chemi, F.; Pearce, S.P.; Clipson, A.; Hill, S.M.; Conway, A.M.; Richardson, S.A.; Kamieniecka, K.; Caeser, R.; White, D.J.; Mohan, S.; et al. cfDNA methylome profiling for detection and subtyping of small cell lung cancers. Nat. Cancer 2022, 3, 1260–1270. [Google Scholar] [CrossRef] [PubMed]
- Gillotin, S.; Davies, J.D.; Philpott, A. Subcellular localisation modulates ubiquitylation and degradation of Ascl1. Sci. Rep. 2018, 8, 4625. [Google Scholar] [CrossRef]
- Liu, Y.; Wu, Q.; Jiang, B.; Hou, T.; Wu, C.; Wu, M.; Song, H. Distinct Regulation of ASCL1 by the Cell Cycle and Chemotherapy in Small Cell Lung Cancer. Mol. Cancer Res. MCR 2024, 22, 613–624. [Google Scholar] [CrossRef]
- Zhang, J.; Zhu, L.; Zhou, J.; Yu, Q.; Yang, G.; Zhao, K.; Luo, C.; Meng, J.; Liu, J.; Yang, X. Ubiquitination of ASCL1 mediates CD47 transcriptional activation of the AKT signaling pathway, and glycolysis promotes osteogenic differentiation of hBMSCs. In vitro cellular & developmental biology. Animal 2023, 59, 636–648. [Google Scholar] [CrossRef]
- Redin, E.; Sridhar, H.; Zhan, Y.A.; Pereira Mello, B.; Zhong, H.; Durani, V.; Sabet, A.; Manoj, P.; Linkov, I.; Qiu, J.; et al. SMARCA4 controls state plasticity in small cell lung cancer through regulation of neuroendocrine transcription factors and REST splicing. J. Hematol. Oncol. 2024, 17, 58. [Google Scholar] [CrossRef] [PubMed]
- Cheng, Y.; Liao, S.; Xu, G.; Hu, J.; Guo, D.; Du, F.; Contreras, A.; Cai, K.Q.; Peri, S.; Wang, Y.; et al. NeuroD1 Dictates Tumor Cell Differentiation in Medulloblastoma. Cell Rep. 2020, 31, 107782. [Google Scholar] [CrossRef]
- Leiendecker, L.; Jung, P.S.; Krecioch, I.; Neumann, T.; Schleiffer, A.; Mechtler, K.; Wiesner, T.; Obenauf, A.C. LSD1 inhibition induces differentiation and cell death in Merkel cell carcinoma. EMBO Mol. Med. 2020, 12, e12525. [Google Scholar] [CrossRef]
- Jeon, S.J.; Kim, J.W.; Kim, K.C.; Han, S.M.; Go, H.S.; Seo, J.E.; Choi, C.S.; Ryu, J.H.; Shin, C.Y.; Song, M.R. Translational regulation of NeuroD1 expression by FMRP: Involvement in glutamatergic neuronal differentiation of cultured rat primary neural progenitor cells. Cell. Mol. Neurobiol. 2014, 34, 297–305. [Google Scholar] [CrossRef]
- Duplaquet, L.; So, K.; Ying, A.W.; Pal Choudhuri, S.; Li, X.; Xu, G.D.; Li, Y.; Qiu, X.; Li, R.; Singh, S.; et al. Mammalian SWI/SNF complex activity regulates POU2F3 and constitutes a targetable dependency in small cell lung cancer. Cancer Cell 2024, 42, 1352–1369.e1313. [Google Scholar] [CrossRef]
- He, T.; Xiao, L.; Qiao, Y.; Klingbeil, O.; Young, E.; Wu, X.S.; Mannan, R.; Mahapatra, S.; Redin, E.; Cho, H.; et al. Targeting the mSWI/SNF complex in POU2F-POU2AF transcription factor-driven malignancies. Cancer Cell 2024, 42, 1336–1351.e1339. [Google Scholar] [CrossRef]
- Szczepanski, A.; Tsuboyama, N.; Lyu, H.; Wang, P.; Beytullahoglu, O.; Zhang, T.; Singer, B.D.; Yue, F.; Zhao, Z.; Wang, L. A SWI/SNF-dependent transcriptional regulation mediated by POU2AF2/C11orf53 at enhancer. Nat. Commun. 2024, 15, 2067. [Google Scholar] [CrossRef] [PubMed]
- Murai, F.; Koinuma, D.; Shinozaki-Ushiku, A.; Fukayama, M.; Miyaozono, K.; Ehata, S. EZH2 promotes progression of small cell lung cancer by suppressing the TGF-β-Smad-ASCL1 pathway. Cell Discov. 2015, 1, 15026. [Google Scholar] [CrossRef] [PubMed]
- Lochmann, T.L.; Floros, K.V.; Naseri, M.; Powell, K.M.; Cook, W.; March, R.J.; Stein, G.T.; Greninger, P.; Maves, Y.K.; Saunders, L.R.; et al. Venetoclax Is Effective in Small-Cell Lung Cancers with High BCL-2 Expression. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2018, 24, 360–369. [Google Scholar] [CrossRef]
- Valko, Z.; Megyesfalvi, Z.; Schwendenwein, A.; Lang, C.; Paku, S.; Barany, N.; Ferencz, B.; Horvath-Rozsas, A.; Kovacs, I.; Schlegl, E.; et al. Dual targeting of BCL-2 and MCL-1 in the presence of BAX breaks venetoclax resistance in human small cell lung cancer. Br. J. Cancer 2023, 128, 1850–1861. [Google Scholar] [CrossRef] [PubMed]
- Oser, M.G.; Sabet, A.H.; Gao, W.; Chakraborty, A.A.; Schinzel, A.C.; Jennings, R.B.; Fonseca, R.; Bonal, D.M.; Booker, M.A.; Flaifel, A.; et al. The KDM5A/RBP2 histone demethylase represses NOTCH signaling to sustain neuroendocrine differentiation and promote small cell lung cancer tumorigenesis. Genes Dev. 2019, 33, 1718–1738. [Google Scholar] [CrossRef]
- Ramkumar, K.; Tanimoto, A.; Della Corte, C.M.; Stewart, C.A.; Wang, Q.; Shen, L.; Cardnell, R.J.; Wang, J.; Polanska, U.M.; Andersen, C.; et al. Targeting BCL2 Overcomes Resistance and Augments Response to Aurora Kinase B Inhibition by AZD2811 in Small Cell Lung Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2023, 29, 3237–3249. [Google Scholar] [CrossRef]
- Rudin, C.M.; Pietanza, M.C.; Bauer, T.M.; Ready, N.; Morgensztern, D.; Glisson, B.S.; Byers, L.A.; Johnson, M.L.; Burris, H.A., 3rd; Robert, F.; et al. Rovalpituzumab tesirine, a DLL3-targeted antibody-drug conjugate, in recurrent small-cell lung cancer: A first-in-human, first-in-class, open-label, phase 1 study. Lancet Oncol. 2017, 18, 42–51. [Google Scholar] [CrossRef]
- Morgensztern, D.; Besse, B.; Greillier, L.; Santana-Davila, R.; Ready, N.; Hann, C.L.; Glisson, B.S.; Farago, A.F.; Dowlati, A.; Rudin, C.M.; et al. Efficacy and Safety of Rovalpituzumab Tesirine in Third-Line and Beyond Patients with DLL3-Expressing, Relapsed/Refractory Small-Cell Lung Cancer: Results from the Phase II TRINITY Study. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 6958–6966. [Google Scholar] [CrossRef]
- Augert, A.; Eastwood, E.; Ibrahim, A.H.; Wu, N.; Grunblatt, E.; Basom, R.; Liggitt, D.; Eaton, K.D.; Martins, R.; Poirier, J.T.; et al. Targeting NOTCH activation in small cell lung cancer through LSD1 inhibition. Sci. Signal. 2019, 12, eaau2922. [Google Scholar] [CrossRef]
- Takagi, S.; Ishikawa, Y.; Mizutani, A.; Iwasaki, S.; Matsumoto, S.; Kamada, Y.; Nomura, T.; Nakamura, K. LSD1 Inhibitor T-3775440 Inhibits SCLC Cell Proliferation by Disrupting LSD1 Interactions with SNAG Domain Proteins INSM1 and GFI1B. Cancer Res. 2017, 77, 4652–4662. [Google Scholar] [CrossRef]
- Kim, J.; Sage, J. Taking SCLC on a Bad LSD(1) Trip One NOTCH Further. Trends Mol. Med. 2019, 25, 261–264. [Google Scholar] [CrossRef] [PubMed]
- Taniguchi, H.; Chakraborty, S.; Takahashi, N.; Banerjee, A.; Caeser, R.; Zhan, Y.A.; Tischfield, S.E.; Chow, A.; Nguyen, E.M.; Villalonga, Á.Q.; et al. ATR inhibition activates cancer cell cGAS/STING-interferon signaling and promotes antitumor immunity in small-cell lung cancer. Sci. Adv. 2024, 10, eado4618. [Google Scholar] [CrossRef] [PubMed]
- Scattolin, D.; Maso, A.D.; Ferro, A.; Frega, S.; Bonanno, L.; Guarneri, V.; Pasello, G. The emerging role of Schlafen-11 (SLFN11) in predicting response to anticancer treatments: Focus on small cell lung cancer. Cancer Treat. Rev. 2024, 128, 102768. [Google Scholar] [CrossRef]
- Costanzo, F.; Martínez Diez, M.; Santamaría Nuñez, G.; Díaz-Hernandéz, J.I.; Genes Robles, C.M.; Díez Pérez, J.; Compe, E.; Ricci, R.; Li, T.K.; Coin, F.; et al. Promoters of ASCL1- and NEUROD1-dependent genes are specific targets of lurbinectedin in SCLC cells. EMBO Mol. Med. 2022, 14, e14841. [Google Scholar] [CrossRef] [PubMed]
- Chakraborty, S.; Sen, U.; Ventura, K.; Jethalia, V.; Coleman, C.; Sridhar, S.; Banerjee, A.; Ozakinci, H.; Mahendravarman, Y.; Snioch, K.; et al. Lurbinectedin sensitizes PD-L1 blockade therapy by activating STING-IFN signaling in small-cell lung cancer. Cell Rep. Med. 2024, 5, 101852. [Google Scholar] [CrossRef]
- Garralda, E.; Beaulieu, M.E.; Moreno, V.; Casacuberta-Serra, S.; Martínez-Martín, S.; Foradada, L.; Alonso, G.; Massó-Vallés, D.; López-Estévez, S.; Jauset, T.; et al. MYC targeting by OMO-103 in solid tumors: A phase 1 trial. Nat. Med. 2024, 30, 762–771. [Google Scholar] [CrossRef]
- Chalishazar, M.D.; Wait, S.J.; Huang, F.; Ireland, A.S.; Mukhopadhyay, A.; Lee, Y.; Schuman, S.S.; Guthrie, M.R.; Berrett, K.C.; Vahrenkamp, J.M.; et al. MYC-Driven Small-Cell Lung Cancer is Metabolically Distinct and Vulnerable to Arginine Depletion. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2019, 25, 5107–5121. [Google Scholar] [CrossRef]
- Helfrich, B.A.; Kim, J.; Gao, D.; Chan, D.C.; Zhang, Z.; Tan, A.C.; Bunn, P.A., Jr. Barasertib (AZD1152), a Small Molecule Aurora B Inhibitor, Inhibits the Growth of SCLC Cell Lines In Vitro and In Vivo. Mol. Cancer Ther. 2016, 15, 2314–2322. [Google Scholar] [CrossRef]
- Huang, F.; Ni, M.; Chalishazar, M.D.; Huffman, K.E.; Kim, J.; Cai, L.; Shi, X.; Cai, F.; Zacharias, L.G.; Ireland, A.S.; et al. Inosine Monophosphate Dehydrogenase Dependence in a Subset of Small Cell Lung Cancers. Cell Metab. 2018, 28, 369–382.e365. [Google Scholar] [CrossRef]
- Chatterjee, D.; Svoboda, R.A.; Huisman, D.H.; Drapkin, B.J.; Vieira, H.M.; Rao, C.; Askew, J.W.; Fisher, K.W.; Lewis, R.E. KSR1 mediates small-cell lung carcinoma tumor initiation and cisplatin resistance. Mol. Cancer Res. MCR 2025, 24-0652. [Google Scholar] [CrossRef]
- Chen, H.; Gesumaria, L.; Park, Y.K.; Oliver, T.G.; Singer, D.S.; Ge, K.; Schrump, D.S. BET Inhibitors Target the SCLC-N Subtype of Small-Cell Lung Cancer by Blocking NEUROD1 Transactivation. Mol. Cancer Res. MCR 2023, 21, 91–101. [Google Scholar] [CrossRef]
- Lehman, J.M.; Hoeksema, M.D.; Staub, J.; Qian, J.; Harris, B.; Callison, J.C.; Miao, J.; Shi, C.; Eisenberg, R.; Chen, H.; et al. Somatostatin receptor 2 signaling promotes growth and tumor survival in small-cell lung cancer. Int. J. Cancer 2019, 144, 1104–1114. [Google Scholar] [CrossRef] [PubMed]
- Rudin, C.M.; Balli, D.; Lai, W.V.; Richards, A.L.; Nguyen, E.; Egger, J.V.; Choudhury, N.J.; Sen, T.; Chow, A.; Poirier, J.T.; et al. Clinical Benefit from Immunotherapy in Patients with SCLC Is Associated with Tumor Capacity for Antigen Presentation. J. Thorac. Oncol. 2023, 18, 1222–1232. [Google Scholar] [CrossRef]
- Xie, M.; Vuko, M.; Rodriguez-Canales, J.; Zimmermann, J.; Schick, M.; O’Brien, C.; Paz-Ares, L.; Goldman, J.W.; Garassino, M.C.; Gay, C.M.; et al. Molecular classification and biomarkers of outcome with immunotherapy in extensive-stage small-cell lung cancer: Analyses of the CASPIAN phase 3 study. Mol. Cancer 2024, 23, 115. [Google Scholar] [CrossRef]
- Meka, S.T.; Bojja, S.L.; Kumar, G.; Birangal, S.R.; Rao, C.M. Novel HDAC inhibitors provide neuroprotection in MPTP-induced Parkinson’s disease model of rats. Eur. J. Pharmacol. 2023, 959, 176067. [Google Scholar] [CrossRef] [PubMed]
- O’Mahony, A.G.; Mazzocchi, M.; Morris, A.; Morales-Prieto, N.; Guinane, C.; Wyatt, S.L.; Collins, L.M.; Sullivan, A.M.; O’Keeffe, G.W. The class-IIa HDAC inhibitor TMP269 promotes BMP-Smad signalling and is neuroprotective in in vitro and in vivo 6-hydroxydopamine models of Parkinson’s disease. Neuropharmacology 2025, 268, 110319. [Google Scholar] [CrossRef]
- Mazzocchi, M.; Goulding, S.R.; Morales-Prieto, N.; Foley, T.; Collins, L.M.; Sullivan, A.M.; O’Keeffe, G.W. Peripheral administration of the Class-IIa HDAC inhibitor MC1568 partially protects against nigrostriatal neurodegeneration in the striatal 6-OHDA rat model of Parkinson’s disease. Brain Behav. Immun. 2022, 102, 151–160. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Li, Z.; Yang, Y.; Ji, W.; Yu, Y.; Niu, X.; Zeng, Q.; Xia, W.; Lu, S. The Hippo/YAP1 pathway interacts with FGFR1 signaling to maintain stemness in lung cancer. Cancer Lett. 2018, 423, 36–46. [Google Scholar] [CrossRef]
- Szulzewsky, F.; Holland, E.C.; Vasioukhin, V. YAP1 and its fusion proteins in cancer initiation, progression and therapeutic resistance. Dev. Biol. 2021, 475, 205–221. [Google Scholar] [CrossRef]
- Zhang, J.; Zeng, X.; Guo, Q.; Sheng, Z.; Chen, Y.; Wan, S.; Zhang, L.; Zhang, P. Small cell lung cancer: Emerging subtypes, signaling pathways, and therapeutic vulnerabilities. Exp. Hematol. Oncol. 2024, 13, 78. [Google Scholar] [CrossRef]
- Rozkiewicz, D.; Hermanowicz, J.M.; Kwiatkowska, I.; Krupa, A.; Pawlak, D. Bruton’s Tyrosine Kinase Inhibitors (BTKIs): Review of Preclinical Studies and Evaluation of Clinical Trials. Molecules 2023, 28, 2400. [Google Scholar] [CrossRef]
- Zhao, Y.; Sun, B.; Fu, X.; Zuo, Z.; Qin, H.; Yao, K. YAP in development and disease: Navigating the regulatory landscape from retina to brain. Biomed. Pharmacother. 2024, 175, 116703. [Google Scholar] [CrossRef]
- Meuwissen, R.; Linn, S.C.; Linnoila, R.I.; Zevenhoven, J.; Mooi, W.J.; Berns, A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003, 4, 181–189. [Google Scholar] [CrossRef]
- Wildey, G.; Shay, A.M.; McColl, K.S.; Yoon, S.; Shatat, M.A.; Perwez, A.; Spainhower, K.B.; Kresak, A.M.; Lipka, M.; Yang, M.; et al. Retinoblastoma Expression and Targeting by CDK4/6 Inhibitors in Small Cell Lung Cancer. Mol. Cancer Ther. 2023, 22, 264–273. [Google Scholar] [CrossRef] [PubMed]
- Stewart, C.A.; Gay, C.M.; Xi, Y.; Sivajothi, S.; Sivakamasundari, V.; Fujimoto, J.; Bolisetty, M.; Hartsfield, P.M.; Balasubramaniyan, V.; Chalishazar, M.D.; et al. Single-cell analyses reveal increased intratumoral heterogeneity after the onset of therapy resistance in small-cell lung cancer. Nat. Cancer 2020, 1, 423–436. [Google Scholar] [CrossRef]
- Jin, Y.; Wu, Y.; Reuben, A.; Zhu, L.; Gay, C.M.; Wu, Q.; Zhou, X.; Mo, H.; Zheng, Q.; Ren, J.; et al. Single-cell and spatial proteo-transcriptomic profiling reveals immune infiltration heterogeneity associated with neuroendocrine features in small cell lung cancer. Cell Discov. 2024, 10, 93. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Sun, X.; Liu, Y.; Zhang, Y.; Yang, Z.; Dong, J.; Wang, N.; Ying, J.; Zhou, M.; Yang, L. Spatial Transcriptome-Wide Profiling of Small Cell Lung Cancer Reveals Intra-Tumoral Molecular and Subtype Heterogeneity. Adv. Sci. 2024, 11, e2402716. [Google Scholar] [CrossRef] [PubMed]
- Barrett, M.T.; Lenkiewicz, E.; Evers, L.; Holley, T.; Ruiz, C.; Bubendorf, L.; Sekulic, A.; Ramanathan, R.K.; Von Hoff, D.D. Clonal evolution and therapeutic resistance in solid tumors. Front. Pharmacol. 2013, 4, 2. [Google Scholar] [CrossRef]
- Laplane, L.; Maley, C.C. The evolutionary theory of cancer: Challenges and potential solutions. Nat. Rev. Cancer 2024, 24, 718–733. [Google Scholar] [CrossRef]
- Kim, D.H.; Park, H.; Choi, Y.J.; Im, K.; Lee, C.W.; Kim, D.S.; Pack, C.G.; Kim, H.Y.; Choi, C.M.; Lee, J.C.; et al. Identification of exosomal microRNA panel as diagnostic and prognostic biomarker for small cell lung cancer. Biomark. Res. 2023, 11, 80. [Google Scholar] [CrossRef]
- Weber, M.C.; Izzo, L.T.; Oliver, T.G. Epigenetic Regulators Open the Door to SCLC Plasticity. Cancer Res. 2023, 83, 3495–3497. [Google Scholar] [CrossRef]
- Rubin, M.A.; Bristow, R.G.; Thienger, P.D.; Dive, C.; Imielinski, M. Impact of Lineage Plasticity to and from a Neuroendocrine Phenotype on Progression and Response in Prostate and Lung Cancers. Mol. Cell 2020, 80, 562–577. [Google Scholar] [CrossRef]
- Shi, Z.-D.; Pang, K.; Wu, Z.-X.; Dong, Y.; Hao, L.; Qin, J.-X.; Wang, W.; Chen, Z.-S.; Han, C.-H. Tumor cell plasticity in targeted therapy-induced resistance: Mechanisms and new strategies. Signal Transduct. Target. Ther. 2023, 8, 113. [Google Scholar] [CrossRef]
- Khan, T.; Nagarajan, M.; Kang, I.; Wu, C.; Wangpaichitr, M. Targeting Metabolic Vulnerabilities to Combat Drug Resistance in Cancer Therapy. J. Pers. Med. 2025, 15, 50. [Google Scholar] [CrossRef]
- Leung, A.W.; de Silva, T.; Bally, M.B.; Lockwood, W.W. Synthetic lethality in lung cancer and translation to clinical therapies. Mol. Cancer 2016, 15, 61. [Google Scholar] [CrossRef]
- Yang, H.; Cui, W.; Wang, L. Epigenetic synthetic lethality approaches in cancer therapy. Clin. Epigenetics 2019, 11, 136. [Google Scholar] [CrossRef]
- Vyse, S.; Howitt, A.; Huang, P.H. Exploiting Synthetic Lethality and Network Biology to Overcome EGFR Inhibitor Resistance in Lung Cancer. J. Mol. Biol. 2017, 429, 1767–1786. [Google Scholar] [CrossRef]
- Thomas, A.; Takahashi, N.; Rajapakse, V.N.; Zhang, X.; Sun, Y.; Ceribelli, M.; Wilson, K.M.; Zhang, Y.; Beck, E.; Sciuto, L.; et al. Therapeutic targeting of ATR yields durable regressions in small cell lung cancers with high replication stress. Cancer Cell 2021, 39, 566–579.e567. [Google Scholar] [CrossRef]
- Raso, M.G.; Bota-Rabassedas, N.; Wistuba, I.I. Pathology and Classification of SCLC. Cancers 2021, 13, 820. [Google Scholar] [CrossRef]
- Andrini, E.; Marchese, P.V.; De Biase, D.; Mosconi, C.; Siepe, G.; Panzuto, F.; Ardizzoni, A.; Campana, D.; Lamberti, G. Large Cell Neuroendocrine Carcinoma of the Lung: Current Understanding and Challenges. J. Clin. Med. 2022, 11, 1461. [Google Scholar] [CrossRef]
- Lantuejoul, S.; Fernandez-Cuesta, L.; Damiola, F.; Girard, N.; McLeer, A. New molecular classification of large cell neuroendocrine carcinoma and small cell lung carcinoma with potential therapeutic impacts. Transl. Lung Cancer Res. 2020, 9, 2233–2244. [Google Scholar] [CrossRef]
- Ruffini, E.; Rena, O.; Oliaro, A.; Filosso, P.L.; Bongiovanni, M.; Arslanian, A.; Papalia, E.; Maggi, G. Lung tumors with mixed histologic pattern. Clinico-pathologic characteristics and prognostic significance. Eur. J. Cardio-Thorac. Surg. 2002, 22, 701–707. [Google Scholar] [CrossRef]
- Zullo, L.; Dall’Olio, F.G.; Rossi, G.; Dellepiane, C.; Barletta, G.; Bennicelli, E.; Ingaliso, M.; Tagliamento, M.; Genova, C. Molecular and Genetic Advances in Small Cell Lung Cancer Landscape: From Homogeneity to Diversity. Int. J. Mol. Sci. 2023, 25, 224. [Google Scholar] [CrossRef]
- Zhai, X.; Zhang, Z.; Chen, Y.; Wu, Y.; Zhen, C.; Liu, Y.; Lin, Y.; Chen, C. Current and future therapies for small cell lung carcinoma. J. Hematol. Oncol. 2025, 18, 37. [Google Scholar] [CrossRef]
- Yang, Y.; Fan, S. Small cell lung cancer transformations from non-small cell lung cancer: Biological mechanism and clinical relevance. Chin. Med. J. Pulm. Crit. Care Med. 2024, 2, 42–47. [Google Scholar] [CrossRef]
- Marcoux, N.; Gettinger, S.N.; O’Kane, G.; Arbour, K.C.; Neal, J.W.; Husain, H.; Evans, T.L.; Brahmer, J.R.; Muzikansky, A.; Bonomi, P.D.; et al. EGFR-Mutant Adenocarcinomas That Transform to Small-Cell Lung Cancer and Other Neuroendocrine Carcinomas: Clinical Outcomes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2019, 37, 278–285. [Google Scholar] [CrossRef]
- Wang, S.; Wang, Y.; Wu, X.; Yang, L.; Zhang, X. Patients outcomes in lung adenocarcinoma transforming to small-cell lung cancer after tyrosine kinase inhibitor therapy. World J. Surg. Oncol. 2025, 23, 34. [Google Scholar] [CrossRef]
- Mambetsariev, I.; Arvanitis, L.; Fricke, J.; Pharaon, R.; Baroz, A.R.; Afkhami, M.; Koczywas, M.; Massarelli, E.; Salgia, R. Small Cell Lung Cancer Transformation following Treatment in EGFR-Mutated Non-Small Cell Lung Cancer. J. Clin. Med. 2022, 11, 1429. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, K.; Wang, H. The emerging landscape and future perspective of SCLC transformation: From molecular mechanisms to therapeutic strategies. Crit. Rev. Oncol. Hematol. 2025, 207, 104616. [Google Scholar] [CrossRef]
- Herzog, B.H.; Devarakonda, S.; Govindan, R. Overcoming Chemotherapy Resistance in SCLC. J. Thorac. Oncol. 2021, 16, 2002–2015. [Google Scholar] [CrossRef]
- Pacesa, M.; Pelea, O.; Jinek, M. Past, present, and future of CRISPR genome editing technologies. Cell 2024, 187, 1076–1100. [Google Scholar] [CrossRef]
- Modell, A.E.; Lim, D.; Nguyen, T.M.; Sreekanth, V.; Choudhary, A. CRISPR-based therapeutics: Current challenges and future applications. Trends Pharmacol. Sci. 2022, 43, 151–161. [Google Scholar] [CrossRef]
- Henning, N.J.; Boike, L.; Spradlin, J.N.; Ward, C.C.; Liu, G.; Zhang, E.; Belcher, B.P.; Brittain, S.M.; Hesse, M.J.; Dovala, D.; et al. Deubiquitinase-targeting chimeras for targeted protein stabilization. Nat. Chem. Biol. 2022, 18, 412–421. [Google Scholar] [CrossRef]
- Liu, J.; Yu, X.; Chen, H.; Kaniskan, H.; Xie, L.; Chen, X.; Jin, J.; Wei, W. TF-DUBTACs Stabilize Tumor Suppressor Transcription Factors. J. Am. Chem. Soc. 2022, 144, 12934–12941. [Google Scholar] [CrossRef]
- Leong, Y.Q.; Koh, R.Y.; Chye, S.M.; Ng, K.Y. Unravelling the genetic links between Parkinson’s disease and lung cancer. Biol. Chem. 2023, 404, 551–567. [Google Scholar] [CrossRef]
- Georgiannakis, E.; Zougou, T.; Mavrommatis, E. Paraneoplastic Syndromes of the Nervous System in Patients Suffering from SCLC. A Review of the Recent Literature. Acta Medica Acad. 2024, 53, 176–182. [Google Scholar] [CrossRef]
- Lu, X.; Liu, Q.X.; Zhang, J.; Zhou, D.; Yang, G.X.; Li, M.Y.; Qiu, Y.; Chen, Q.; Zheng, H.; Dai, J.G. PINK1 Overexpression Promotes Cell Migration and Proliferation via Regulation of Autophagy and Predicts a Poor Prognosis in Lung Cancer Cases. Cancer Manag. Res. 2020, 12, 7703–7714. [Google Scholar] [CrossRef]
- Chanda, S.; Ang, C.E.; Davila, J.; Pak, C.; Mall, M.; Lee, Q.Y.; Ahlenius, H.; Jung, S.W.; Südhof, T.C.; Wernig, M. Generation of induced neuronal cells by the single reprogramming factor ASCL1. Stem Cell Rep. 2014, 3, 282–296. [Google Scholar] [CrossRef]
- Tutukova, S.; Tarabykin, V.; Hernandez-Miranda, L.R. The Role of Neurod Genes in Brain Development, Function, and Disease. Front. Mol. Neurosci. 2021, 14, 662774. [Google Scholar] [CrossRef]
- Pavlinkova, G.; Smolik, O. NEUROD1: Transcriptional and epigenetic regulator of human and mouse neuronal and endocrine cell lineage programs. Front. Cell Dev. Biol. 2024, 12, 1435546. [Google Scholar] [CrossRef]
- Schagen, S.B.; Wefel, J.S. Chemotherapy-related changes in cognitive functioning. Eur. J. Cancer Suppl. 2013, 11, 225–232. [Google Scholar] [CrossRef]
- Miyashita, M. Chemotherapy-related cognitive impairment: What we need to know and what we can do. Asia-Pac. J. Oncol. Nurs. 2024, 11, 100334. [Google Scholar] [CrossRef]
- Vega, J.N.; Dumas, J.; Newhouse, P.A. Cognitive Effects of Chemotherapy and Cancer-Related Treatments in Older Adults. Am. J. Geriatr. Psychiatry Off. J. Am. Assoc. Geriatr. Psychiatry 2017, 25, 1415–1426. [Google Scholar] [CrossRef]
- Trecarichi, A.; Flatters, S.J.L. Mitochondrial dysfunction in the pathogenesis of chemotherapy-induced peripheral neuropathy. Int. Rev. Neurobiol. 2019, 145, 83–126. [Google Scholar] [CrossRef]

{kind=link}
| Transcription Factor | Regulator | Outcomes | Potential Treatments | Reference |
|---|---|---|---|---|
| ASCL1 | TCF3/CDK2/Cyclin A2 | Stabilization | CDK2 inhibitors | [53] |
| HUWE1 | Degradation | None | [52] | |
| USP8 | Stabilization | USP8 inhibitors | [54] | |
| NEUROD1 | SMARCA4 | Promotes its transcription | SMARCA4 inhibitors | [55] |
| EZH2 | Suppresses its expression | None | [62] | |
| LSD1 | Enhances transcriptional activity | LSD1 inhibitor | [57] | |
| FMRP | Inhibits its translation | None | [58] | |
| POU2F3 | IGF1R | Promotes its expression | IGF1R inhibitors | [29] |
| OCA-T1/ OCA-T2 | Activation | None | [30] | |
| mSWI/SNF complex | Promotes its transcription | SMARCA2/4 inhibitor; BRD9 degrader | [59,60,61] |
| SCLC Subtype | Targeted Therapies |
|---|---|
| SCLC-A | BCL2 inhibitors (venetoclax), DLL3-targeting agents (tarlatamab), LSD1 inhibitors (ORY-1001), HDAC inhibitors (pracinostat), ATR inhibitors, PARP inhibitors (olaparib), CEACAM5-targeting agent (labetuzumab govitecan), Lurbinectedin |
| SCLC-N | MYC-targeting agents (OMO-103), AURKA/B inhibitors (alisertib, barasertib), IMPDH inhibitors, mTOR inhibitors, CHK1 inhibitors, BET inhibitors (CPI-0610, JQ1), SSTR2-targeting agents |
| SCLC-P | IGF1R inhibitors (linsitinib), PARP inhibitors, antifolate agents, SWI/SNF ATPase inhibitors (SMARCA4/2 inhibitors, BRD9 degraders) |
| SCLC-I | Immune checkpoint inhibitors (ICIs), BTK inhibitors (ibrutinib), HDAC inhibitors (pracinostat) |
| YAP1 Activation | RTK inhibitors (EGFR, FGFR), BTK inhibitors (ibrutinib) |
| RB1-Intact | CDK4/6 inhibitors (palbociclib, abemaciclib) |
| NMF Clusters | NMF1: PARP/ATR inhibitors; NMF2: DLL3 ADCs, ICIs; NMF3: RTK inhibitors; NMF4: AURKA inhibitors |
| Challenge | Description | Potential Solutions |
|---|---|---|
| Tumor Heterogeneity | Coexistence of multiple subtypes within the same tumor | Single-cell RNA sequencing, spatial transcriptomics |
| Subtype Plasticity | Dynamic transition between subtypes under therapy pressure | Real-time monitoring with liquid biopsy |
| Resistance Mechanisms | Epigenetic reprogramming, clonal evolution, drug resistance | Combination therapy, synthetic lethality approaches |
| Limited Biomarkers | Lack of standardized predictive biomarkers for treatment selection | Integration of ctDNA, RNA-seq, and proteomics |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Huang, D.; Wang, J.; Chen, L.; Jiang, W.; Inuzuka, H.; Simon, D.K.; Wei, W. Molecular Subtypes and Targeted Therapeutic Strategies in Small Cell Lung Cancer: Advances, Challenges, and Future Perspectives. Molecules 2025, 30, 1731. https://doi.org/10.3390/molecules30081731
Huang D, Wang J, Chen L, Jiang W, Inuzuka H, Simon DK, Wei W. Molecular Subtypes and Targeted Therapeutic Strategies in Small Cell Lung Cancer: Advances, Challenges, and Future Perspectives. Molecules. 2025; 30(8):1731. https://doi.org/10.3390/molecules30081731
Chicago/Turabian StyleHuang, Daoyuan, Jingchao Wang, Li Chen, Weiwei Jiang, Hiroyuki Inuzuka, David K. Simon, and Wenyi Wei. 2025. "Molecular Subtypes and Targeted Therapeutic Strategies in Small Cell Lung Cancer: Advances, Challenges, and Future Perspectives" Molecules 30, no. 8: 1731. https://doi.org/10.3390/molecules30081731
APA StyleHuang, D., Wang, J., Chen, L., Jiang, W., Inuzuka, H., Simon, D. K., & Wei, W. (2025). Molecular Subtypes and Targeted Therapeutic Strategies in Small Cell Lung Cancer: Advances, Challenges, and Future Perspectives. Molecules, 30(8), 1731. https://doi.org/10.3390/molecules30081731