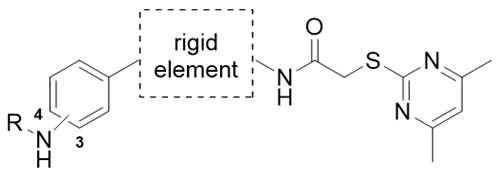
4.1. Chemical Synthesis
Solvents and reagents were purchased commercially and used without further purification. Reaction control was performed via thin-layer chromatography (TLC) using 0.2 mm silica-gel-coated POLYGRAM
® SIL G/UV254 polyester sheets (Macherey-Nagel, Düren, Germany) by UV-light visualization (254 nm). Flash column chromatography was performed using SiO
2 60 (0.040–0.063 mm, 230–400 mesh ASTM) by Merk (Darmstadt, Germany). Melting points were determined using a Büchi B-540 melting point meter (Fawil, Switzerland). The Perkin Elmer FT-IR BXII/1000 spectrometer (Waltham, MA, USA) in combination with DuraSamp IR II Diamond ATR sensor (Smiths Detection, London, UK) was used to perform infrared spectroscopy (IR). The IR spectra was recorded from wavenumbers 4000 to 650 cm
−1, and crucial absorption bands are given as wavenumber (
) in cm
−1. High-resolution mass spectroscopy (HR-MS) was performed using a Jeol Mstation 700 (Akishima, Japan) or JMS GCmate II Jeol (Akishima, Japan) instrument for electron impact ionization (EI). Electrospray ionization (ESI) HR-MS was performed using a Thermo Finnigan LTQ (Thermo Fisher Scientific, Waltham, MA, USA) instrument. NMR spectra were recorded with Avance III HD 400 MHz Bruker BioSpin (Bruker Corporation, Billerica, MA, USA) for
1H NMR: 400 MHz and for
13C NMR: 101 MHz or Avance III HD 500 MHz Bruker BioSpin for
1H NMR: 500 MHz and for
13C NMR: 126 MHz. MestreNova 14.3.0 (Mestrelab Research S.L., Santiago de Compostela, Spain) was used as analysis program, and the obtained chemical shifts δ (ppm, parts per million) were referenced to the deuterated solvent peak (DMSO-
d6: δH = 2.50 ppm, δC = 39.52 ppm; CD
2Cl
2: δH = 5.32 ppm, δC = 53.84 ppm; CDCl
3: δH = 7.26 ppm, δC = 77.16 ppm). Coupling constants J are assigned in Hertz (Hz) and multiplicities are reported as follows: s (singlett), d (doublet), t (triplet), and q (quartet); derivatives of that or m (multiplet). The compound numbering follows IUPAC conventions, but, for clarity purposes, the assignment of the hierarchy levels of NMR signals (indicated by apostrophes) was carried out based on the order of synthesis. HPLC purity was determined at 210 nm and 254 nm with 1100/1200 diode array detector using HP Agilent 1100 HPLC device by Agilent (Santa Clara, CA, USA) with Zorbax Eclipse Plus
® C18 5 µm (4.6 × 150 mm) column by Agilent and acetonitrile/water as eluent and Chromeleon 7.2.9 Software by Thermo Fisher Scientific (Waltham, MA, USA).
1H and
13C NMR spectra of synthesized compounds and HPLC chromatograms of final test substances are provided in
Supplementary Materials.
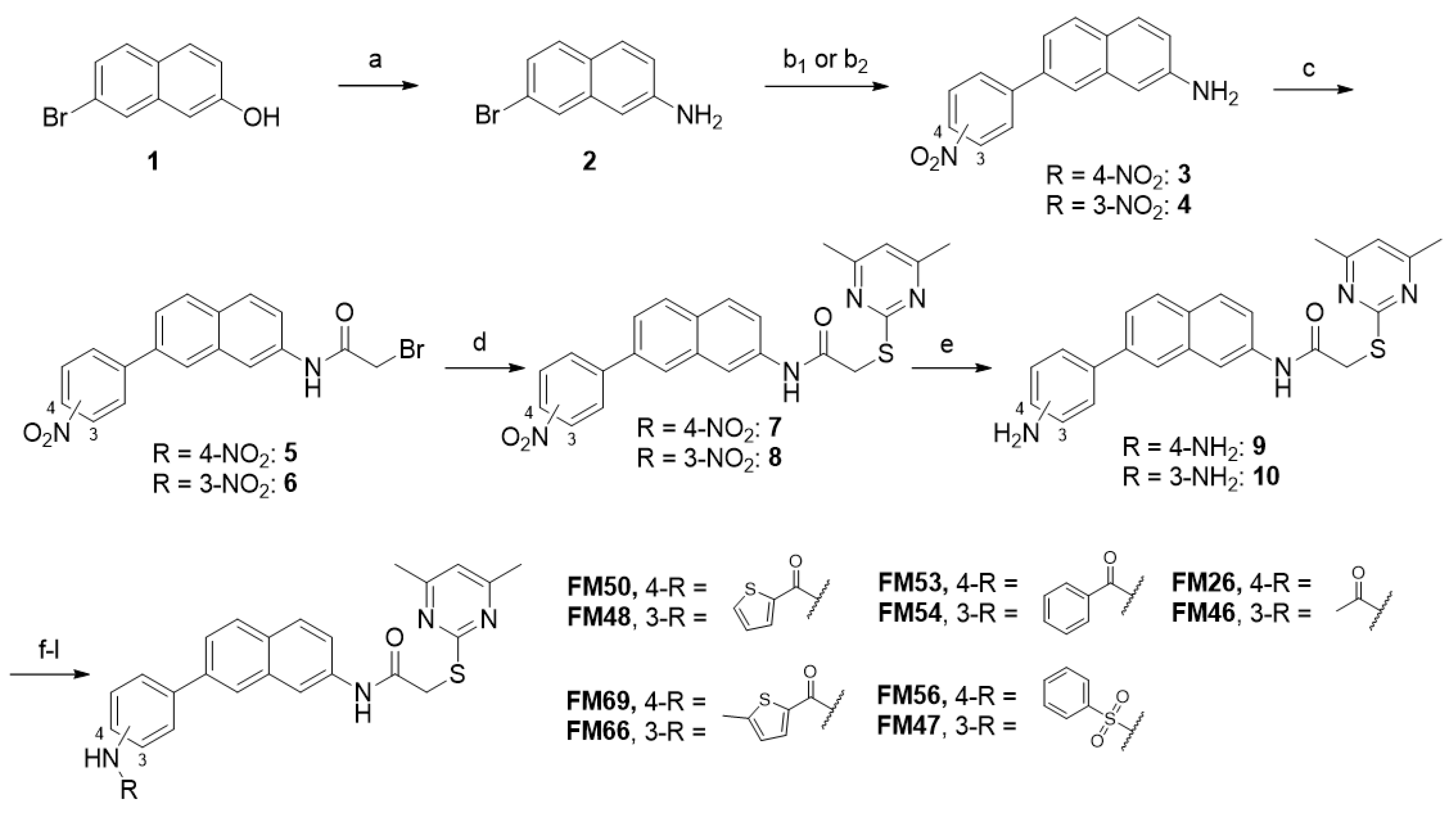
7-(4-Nitrophenyl)naphthalen-2-amine (3). At room temperature, 7-bromonaphthalene-2-amine (2, 3.40 g, 15.3 mmol, 1.00 eq), 4-nitrophenyl boronic acid (3.06 g, 18.4 mmol, 1.20 eq), K2CO3 (6.32 g, 38.3 mmol, 2.50 eq), and Pd(PPh3)4 (0.884 g, 0.765 mmol, 0.0500 eq) were weighed out into a flask, which was put under nitrogen afterwards. Degassed anhydrous DMF (15 mL) was added via syringe, and the reaction mixture was stirred for 23 h at 105 °C and then for another 2 h at room temperature under nitrogen atmosphere. Subsequently, the mixture was diluted with water (100 mL) and brine (200 mL) and extracted with EtOAc (4 × 100 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 7:3 → 3:7) to yield the title compound as a red-orange solid (1.70 g, 6.44 mmol, 42%). m.p.: 233 °C, Rf: 0.16 (hexanes/EtOAc 7:3). 1H NMR (400 MHz, DMSO-d6) δ [ppm] = 8.34–8.29 (m, 2H, 3′-H, 5′-H), 8.08–8.02 (m, 2H, 2′-H, 6′-H), 7.95 (d, J = 1.9 Hz, 1H, 8-H), 7.77 (d, J = 8.5 Hz, 1H, 5-H), 7.65 (d, J = 8.7 Hz, 1H, 4-H), 7.48 (dd, J = 8.4, 1.9 Hz, 1H, 6-H), 7.00 (dd, J = 8.7, 2.2 Hz, 1H, 3-H), 6.93 (d, J = 2.2 Hz, 1H, 1-H), 5.52 (s, 2H, NH2). 13C NMR (101 MHz, DMSO-d6) δ [ppm] = 147.4 (C-2), 147.2 (C-1′), 146.4 (C-4′), 135.2 (C-7), 135.0 (C-8a), 128.6 (C-5), 128.3 (C-4), 127.9 (C-2′, C-6′), 126.2 (C-4a), 124.04 (C-3′, C-5′), 123.97 (C-8), 119.6 (C-6), 119.5 (C-3), 106.3 (C-1). IR (ATR) ν῀ [cm−1] = 3442, 3355, 1630, 1588, 1501, 1462, 1392, 1340, 1279, 1248, 1108, 895, 855, 835, 750, 696. HRMS (EI): m/z = [M]•+ calculated for C16H12N2O2•+: 264.0893; found: 264.0899.
7-(3-Nitrophenyl)naphthalen-2-amine (4). At room temperature, 7-bromonaphthalene-2-amine (2, 3.40 g, 15.8 mmol, 1.00 eq), 3-nitrophenyl boronic acid (3.16 g, 18.9 mmol, 1.20 eq), K2CO3 (6.32 g, 38.3 mmol, 2.50 eq), and Pd(PPh3)4 (0.991 g, 0.788 mmol, 0.0500 eq) were weighed out into a flask, which was put under nitrogen afterwards. Degassed anhydrous DMF (15 mL) was added via syringe, and the reaction mixture was stirred for 23 h at 105 °C and then for another 2 h at room temperature under nitrogen atmosphere. Subsequently, the mixture was diluted with water (100 mL) and brine (200 mL) and extracted with EtOAc (4 × 100 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 7:3 → 3:7) to yield the title compound as a pink-red solid (1.61 g, 6.07 mmol, 38%). m.p.: 160 °C, Rf: 0.23 (hexanes/EtOAc 7:3). 1H NMR (400 MHz, DMSO-d6) δ [ppm] = 8.52 (t, J = 2.1 Hz, 1H, 2′-H), 8.26–8.18 (m, 2H, 4′-H, 6′-H), 7.94 (d, J = 1.9 Hz, 1H, 8-H), 7.82–7.73 (m, 2H, 5-H, 5′-H), 7.65 (d, J = 8.7 Hz, 1H, 4-H), 7.48 (dd, J = 8.4, 1.9 Hz, 1H, 6-H), 6.98 (dd, J = 8.7, 2.2 Hz, 1H, 3-H), 6.94 (d, J = 2.2 Hz, 1H, 1-H), 5.50 (s, 2H, NH2). 13C NMR (101 MHz, DMSO-d6) δ [ppm] = 148.5 (C-3′), 147.3 (C-2), 142.3 (C-1′), 135.3 (C-7), 135.1 (C-8a), 133.4 (C-6′), 130.4 (C-5′), 128.6 (C-5), 128.3 (C-4), 126.0 (C-4a), 123.4 (C-8), 121.9 (C-4′), 121.1 (C-2′), 119.6 (C-6), 119.2 (C-3), 106.2 (C-1). IR (ATR) ν῀ [cm−1] = 3450, 3372, 1631, 1526, 1513, 1485, 1460, 1388, 1340, 1308, 1269, 1224, 1190, 1138, 1101, 1082, 890, 870, 838, 814, 776, 738, 716, 682, 659. HRMS (EI): m/z = [M]•+ calculated for C16H12N2O2•+: 264.0893; found: 264.0897.
2-Bromo-N-(7-(4-nitrophenyl)naphthalen-2-yl)acetamide (5). At 0 °C, bromoacetyl bromide (1.38 mL, 15.9 mmol, 8.00 eq) was added dropwise to a stirred suspension of amine 3 (525 mg, 1.99 mmol, 1.00 eq) in anhydrous DCM (100 mL). After 2 h, water (50 mL) and brine (150 mL) were added, and the reaction mixture was extracted with DCM (4 × 100 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 7:3 → 1:9) to yield the title compound as a yellow-orange solid (684 mg, 1.78 mmol, 89%). m.p.: 189 °C, Rf: 0.20 (hexanes/EtOAc 7:3). 1H NMR (400 MHz, CD2Cl2) δ [ppm] = 8.35 (d, J = 1.9 Hz, 1H, 1-H), 8.35–8.31 (m, 2H, 3′-H, 5′-H), 8.26 (bs, 1H, NHCO), 8.12 (d, J = 2.3 Hz, 1H, 8-H), 7.95 (d, J = 8.5 Hz, 1H, 4-H), 7.93–7.87 (m, 3H, 5-H, 2′-H, 6′-H), 7.74 (dd, J = 8.5, 1.9 Hz, 1H, 6-H), 7.55 (dd, J = 8.8, 2.2 Hz, 1H, 3-H), 4.09 (s, 2H, CH2). 13C NMR (101 MHz, CD2Cl2) δ [ppm] = 164.3 (NHCO), 147.83 (C-1′), 147.80 (C-4′), 137.4 (C-2), 136.1 (C-7), 134.4 (C-8a), 131.3 (C-4a), 129.3 (C-4), 129.2 (C-5), 128.6 (C-2′, C-6′), 127.2 (C-8), 125.0 (C-6), 124.7 (C-3′, C-5′), 121.2 (C-3), 117.7 (C-1), 30.2 (CH2). IR (ATR) ν῀ [cm−1] = 3302, 2926, 1670, 1590, 1561, 1548, 1506, 142, 1397, 1334, 1287, 1214, 1108, 903, 850, 833, 750, 716, 690, 657. HRMS (ESI): m/z = [M-H]− calculated for C18H1279BrN2O3−: 383.0037; found: 383.0041.
2-Bromo-N-(7-(3-nitrophenyl)naphthalen-2-yl)acetamide (6). At 0 °C, bromoacetyl bromide (736 µL, 8.45 mmol, 3.00 eq) was added dropwise to a stirred suspension of amine 4 (745 mg, 2.82 mmol, 1.00 eq) in anhydrous DCM (100 mL). After 2 h, water (50 mL) and brine (150 mL) were added, and the reaction mixture was extracted with DCM (4 × 100 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 7:3 → 1:9) to yield the title compound as a dark-yellow solid (981 mg, 2.55 mmol, 90%). m.p.: 200 °C, Rf: 0.20 (hexanes/EtOAc 7:3). 1H NMR (500 MHz, CD2Cl2) δ [ppm] = 8.58 (t, J = 2.0 Hz, 1H, 2′-H), 8.34 (d, J = 2.2 Hz, 1H, 1-H), 8.27 (s, 1H, NHCO), 8.23 (ddd, J = 8.2, 2.3, 1.0 Hz, 1H, 4′-H), 8.11 (dd, J = 1.8, 0.8 Hz, 1H, 8-H), 8.08 (ddd, J = 7.7, 1.9, 1.0 Hz, 1H, 6′-H), 7.96 (d, J = 8.5 Hz, 1H, 5-H), 7.90 (d, J = 8.7 Hz, 1H, 4-H), 7.74 (dd, J = 8.5, 1.9 Hz, 1H, 6-H), 7.68 (t, J = 8.0 Hz, 1H, 5′-H), 7.55 (dd, J = 8.8, 2.2 Hz, 1H, 3-H), 4.09 (s, 2H, CH2). 13C NMR (126 MHz, CD2Cl2) δ [ppm] = 164.3 (NHCO), 149.4 (C-3′), 143.1 (C-1′), 137.4 (C-7), 136.1 (C-8a), 134.4 (C-2), 133.9 (C-6′), 131.1 (C-4a), 130.5 (C-5′), 129.3 (C-4), 129.2 (C-5), 126.7 (C-8), 125.0 (C-6), 122.7 (C-4′), 122.6 (C-2′), 121.0 (C-3), 117.6 (C-1), 30.2 (CH2). IR (ATR) ν῀ [cm−1] = 3266, 3078, 1687, 1662, 1632, 1611, 1528, 1516, 1463, 1398, 1334, 1271, 1243, 1227, 1187, 1172, 1098, 984, 953, 885, 866, 842, 804, 741, 830, 680. HRMS (ESI): m/z = [M-H]− calculated for C18H1279BrN2O3−: 383.0037; found: 383.0035.
2-((4,6-Dimethylpyrimidin-2-yl)thio)-N-(7-(4-nitrophenyl)naphthalen-2-yl)acetamide (7). At room temperature, potassium tert-butoxide (346 mg, 3.08 mmol, 2.00 eq) was added to a stirred solution of 2-bromoacetamide 5 (594 mg, 1.54 mmol, 1.00 eq) and 4,6-dimethylpyrimidine-2-thiol (260 mg, 1.85 mmol, 1.20 eq) in anhydrous DMF (10 mL). After 3 h, water (150 mL) and brine (100 mL) were added, and the reaction mixture was extracted with EtOAc (4 × 100 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 6:4) to yield the title compound as a yellow-orange solid (684 mg, 1.78 mmol, 89%). m.p.: 185 °C, Rf: 0.18 (hexanes/EtOAc 6:4). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 10.53 (s, 1H, NHCO), 8.42 (d, J = 2.2 Hz, 1H, 1-H), 8.37–8.30 (m, 2H, 3′-H, 5′-H), 8.29 (d, J = 1.9 Hz, 1H, 8-H), 8.15–8.10 (m, 2H, 2′-H, 6′-H), 7.99 (d, J = 8.5 Hz, 1H, 5-H), 7.94 (d, J = 8.8 Hz, 1H, 4-H), 7.82 (dd, J = 8.5, 1.9 Hz, 1H, 6-H), 7.67 (dd, J = 8.8, 2.1 Hz, 1H, 3-H), 6.96 (s, 1H, 5″-H), 4.12 (s, 2H, CH2), 2.33 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 169.3 (C-2″), 166.98 (C-4″, C-6″), 166.94 (NHCO), 146.7 (C-4′), 146.5 (C-1′), 137.3 (C-2), 135.6 (C-7), 133.6 (C-8a), 129.6 (C-4a), 128.6 (C-5), 128.3 (C-4), 128.1 (C-2′, C-6′), 126.2 (C-8), 124.1 (C-3′, C-5′), 123.5 (C-6), 120.9 (C-3), 116.1 (C-5″), 115.8 (C-1), 35.6 (CH2), 23.3 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 3240, 3078, 1667, 1584, 1560, 1512, 1428, 1393, 1344, 1315, 1268, 1221, 1175, 1152, 1107, 904, 884, 847, 752, 696. HRMS (ESI): m/z = [M+H]+ calculated for C24H21N4O3S+: 445.1329; found: 445.1329.
2-((4,6-Dimethylpyrimidin-2-yl)thio)-N-(7-(3-nitrophenyl)naphthalen-2-yl)acetamide (8). At room temperature, potassium tert-butoxide (522 mg, 4.65 mmol, 2.00 eq) was added to a stirred solution of 2-bromoacetamide 6 (896 mg, 2.32 mmol, 1.00 eq) and 4,6-dimethylpyrimidine-2-thiol (391 mg, 2.79 mmol, 1.20 eq) in anhydrous DMF (10 mL). After 3 h, water (150 mL) and brine (100 mL) were added, and the reaction mixture was extracted with EtOAc (4 × 100 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 6:4) to yield the title compound as a white-pink solid (895 mg, 2.01 mmol, 87%). m.p.: 94–98 °C, Rf: 0.17 (hexanes/EtOAc 6:4). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 10.51 (s, 1H, NHCO), 8.60 (t, J = 2.1 Hz, 1H, 2′-H), 8.42 (d, J = 2.1 Hz, 1H, 1-H), 8.34–8.28 (m, 2H, 8-H, 6′-H), 8.25 (ddd, J = 8.0, 2.3, 0.9 Hz, 1H, 4′-H), 7.99 (d, J = 8.5 Hz, 1H, 5-H), 7.93 (d, J = 8.8 Hz, 1H, 4-H), 7.86–7.77 (m, 2H, 6-H, 5′-H), 7.65 (dd, J = 8.8, 2.1 Hz, 1H, 3-H), 6.97 (s, 1H, 5″-H), 4.12 (s, 2H, CH2), 2.33 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 169.3 (C-2″), 167.0 (C-4″, C-6″), 166.9 (NHCO), 148.5 (C-3′), 141.7 (C-1′), 137.3 (C-2), 135.6 (C-7), 133.7 (C-8a), 133.6 (C-6′), 130.5 (C-5′), 129.4 (C-4a), 128.5 (C-5), 128.3 (C-4), 125.7 (C-8), 123.5 (C-6), 122.2 (C-4′), 121.4 (C-2′), 120.6 (C-3), 116.1 (C-5″), 115.7 (C-1), 35.6 (CH2), 23.3 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 3289, 3066, 1688, 1582, 1527, 1512, 1433, 1341, 1266, 1240, 893, 841, 806, 737, 685. HRMS (ESI): m/z = [M+H]+ calculated for C24H21N4O3S+: 445.1329; found: 445.1324.
N-(7-(4-Aminophenyl)naphthalen-2-yl)-2-((4,6-dimethylpyrimidin-2-yl)thio)acetamide (9). Under nitrogen atmosphere, nitro compound 7 (0.533 g, 1.20 mmol, 1.00 eq) was suspended in acetic acid (5 mL), and iron powder (2.34 g, 42.0 mmol, 35.0 eq) was added. After the reaction mixture was stirred at 50 °C for 3 h, it was filtered and then diluted with water (50 mL) and brine (100 mL). The mixture was extracted with EtOAc (4 × 50 mL), and the combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 1:1) to yield the title compound as a yellow solid (397 mg, 0.957 mmol, 80%). m.p.: 202 °C, Rf: 0.10 (hexanes/EtOAc 1:1). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 10.42 (s, 1H, NHCO), 8.25 (d, J = 2.0 Hz, 1H, 1-H), 7.91 (d, J = 1.9 Hz, 1H, 8-H), 7.84–7.79 (m, 2H, 4-H, 5-H), 7.64 (dd, J = 8.6, 1.8 Hz, 1H, 6-H), 7.53 (dd, J = 8.7, 2.1 Hz, 1H, 3-H), 7.52–7.49 (m, 2H, 2′-H, 6′-H), 6.97 (s, 1H, 5″-H), 6.70–6.64 (m, 2H, 3′-H, 5′-H), 5.27 (s, 2H, NH2), 4.10 (s, 2H, CH2), 2.34 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 169.4 (C-2″), 167.0 (C-4″, C-6″), 166.8 (NHCO), 148.6 (C-4′), 138.5 (C-7), 136.8 (C-2), 134.0 (C-8a), 128.2 (C-4a), 128.0 (C-4), 127.8 (C-5), 127.5 (C-2′, C-6′), 127.0 (C-1′), 123.3 (C-6), 122.4 (C-8), 119.1 (C-3), 116.1 (C-5″), 115.3 (C-1), 114.3 (C-3′, C-5′), 35.6 (CH2), 23.3 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 3439, 3355, 3248, 2968, 2911, 1660, 1618, 1606, 1577, 1532, 1506, 1338, 1264, 1248, 1228, 1190, 1168, 984, 952, 896, 851, 828, 802, 728, 706. HRMS (ESI): m/z = [M-H]− calculated for C24H21N4OS−: 413.1442; found: 413.1442.
N-(7-(3-Aminophenyl)naphthalen-2-yl)-2-((4,6-dimethylpyrimidin-2-yl)thio)acetamide (10). Under nitrogen atmosphere, nitro compound 8 (0.684 g, 1.54 mmol, 1.00 eq) was suspended in acetic acid (5 mL), and iron powder (3.01 g, 53.9 mmol, 35.0 eq) was added. After the reaction mixture was stirred at 50 °C for 3 h, it was filtered and then diluted with water (50 mL) and brine (100 mL). The mixture was extracted with EtOAc (4 × 50 mL), and the combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 1:1) to yield the title compound as a yellow solid (480 mg, 1.16 mmol, 75%). m.p.: 167 °C, Rf: 0.11 (hexanes/EtOAc 1:1). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 10.46 (s, 1H, NHCO), 8.30 (d, J = 2.1 Hz, 1H, 1-H), 7.95 (d, J = 1.8 Hz, 1H, 8-H), 7.92–7.85 (m, 2H, 4-H, 5-H), 7.66–7.58 (m, 2H, 3-H, 6-H), 7.14 (t, J = 7.8 Hz, 1H, 5′-H), 6.99 (t, J = 2.0 Hz, 1H, 2′-H), 6.98 (s, 1H, 5″-H), 6.92 (ddd, J = 7.6, 1.8, 1.0 Hz, 1H, 6′-H), 6.60 (ddd, J = 7.9, 2.2, 0.9 Hz, 1H, 4′-H), 5.17 (s, 2H, NH2), 4.11 (s, 2H, CH2), 2.34 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 169.3 (C-2″), 170.0 (C-4″, C-6″), 166.8 (NHCO), 149.2 (C-3′), 140.7 (C-1′), 138.9 (C-7), 137.0 (C-2), 133.7 (C-8a), 129.5 (C-5′), 128.9 (C-4a), 128.2 (C-5), 128.0 (C-4), 124.2 (C-8), 123.9 (C-6), 119.8 (C-3), 116.1 (C-5″), 115.4 (C-1), 114.6 (C-6′), 113.3 (C-4′), 112.4 (C-2′), 35.6 (CH2), 23.3 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 3430, 3224, 3097, 1656, 1631, 1611, 1584, 1546, 1516, 1493, 1404, 1341, 1322, 1272, 1236, 922, 940, 904, 886, 858, 840, 774, 749, 691. HRMS (ESI): m/z = [M-H]− calculated for C24H21N4OS−: 413.1442; found: 413.1440.
N-(7-(4-Acetamidophenyl)naphthalen-2-yl)-2-((4,6-dimethylpyrimidin-2-yl)thio)acetamide (FM26). At room temperature, acetic anhydride (9.03 µL, 0.0955 mmol, 2.00 eq) was added dropwise to a stirred solution of amine 9 (19.8 mg, 0.0478 mmol, 1.00 eq) and NEt3 (13.3 µL, 0.0955 mmol, 2.00 eq) in anhydrous DCM (5 mL). After 2 h, water (25 mL) and brine (25 mL) were added, and the reaction mixture was extracted with EtOAc (3 × 25 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 2:8) to yield the title compound as a white solid (15.6 mg, 0.0342 mmol, 72%). m.p.: 194 °C, Rf: 0.23 (hexanes/EtOAc 2:8). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 10.46 (s, 1H, 2′-NHCO), 10.05 (s, 1H, 4′-NHCO), 8.32 (d, J = 2.0 Hz, 1H, 8-H), 8.06 (d, J = 1.8 Hz, 1H, 1-H), 7.89 (d, J = 8.6 Hz, 1H, 5-H), 7.87 (d, J = 8.8 Hz, 1H, 4-H), 7.80–7.73 (m, 2H, 2′-H, 6′-H), 7.73–7.68 (m, 3H, 3-H, 3′-H, 5′-H), 7.59 (dd, J = 8.8, 2.1 Hz, 1H, 6-H), 6.97 (s, 1H, 5″-H), 4.11 (s, 2H, CH2), 2.33 (s, 6H, 4″-CH3, 6″-CH3), 2.08 (s, 3H, CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 169.4 (C-2″), 168.4 (4′-NHCO), 167.0 (C-4″, C-6″), 166.8 (2″-NHCO), 138.9 (C-4′), 137.5 (C-7), 137.0 (C-2), 134.4 (C-1′), 133.8 (C-8a), 128.8 (C-4a), 128.2 (C-4), 128.1 (C-5), 127.2 (C-2′, C-6′), 124.0 (C-8), 123.5 (C-6), 119.8 (C-3), 119.3 (C-3′, C-5′), 116.1 (C-5″), 115.5 (C-1), 35.6 (CH2), 24.1 (CH3), 23.3 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 3250, 3050, 2926, 1658, 1578, 1521, 1504, 1424, 1400, 1369, 1337, 1316, 1291, 1266, 1239, 1224, 1180, 1164, 1015, 984, 956, 892, 836, 826, 804, 713. HRMS (ESI): m/z = [M-H]− calculated for C26H23N4O2S−: 445.1547; found: 445.1546. Purity (HPLC): ≥98%.
N-(7-(3-Acetamidophenyl)naphthalen-2-yl)-2-((4,6-dimethylpyrimidin-2-yl)thio)acetamide (FM46). At room temperature, acetic anhydride (36.5 µL, 0.386 mmol, 2.00 eq) was added dropwise to a stirred solution of amine 10 (80.1 mg, 0.193 mmol, 1.00 eq) and NEt3 (53.9 µL, 0.386 mmol, 2.00 eq) in anhydrous DCM (5 mL). After 2 h, water (25 mL) and brine (25 mL) were added, and the reaction mixture was extracted with EtOAc (3 × 25 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 3:7) to yield the title compound as a white solid (80.2 mg, 0.176 mmol, 91%). m.p.: 213 °C, Rf: 0.12 (hexanes/EtOAc 3:7). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 10.48 (s, 1H, 2-NHCO), 10.05 (s, 1H, 3′-NHCO), 8.33 (d, J = 2.1 Hz, 1H, 1-H), 8.01 (d, J = 1.8 Hz, 1H, 8-H), 8.00 (t, J = 2.0 Hz, 1H, 2′-H), 7.93 (d, J = 8.6 Hz, 1H, 5-H), 7.90 (d, J = 8.8 Hz, 1H, 4-H), 7.66 (dd, J = 8.5, 1.8 Hz, 1H, 6-H), 7.64–7.59 (m, 2H, 3-H, 4′-H), 7.47 (dt, J = 7.7, 1.5 Hz, 1H, 6′-H), 7.41 (t, J = 7.8 Hz, 1H, 5′-H), 6.97 (s, 1H, 5″-H), 4.12 (s, 2H, CH2), 2.34 (s, 6H, 4″-CH3, 6″-CH3), 2.08 (s, 3H, CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 169.3 (C-2″), 168.4 (3′-NHCO), 167.0 (C-4″, C-6″), 166.9 (2-NHCO), 140.5 (C-1′), 139.9 (C-3′), 138.1 (C-7), 137.1 (C-2), 133.7 (C-8a), 129.4 (C-5′), 129.0 (C-4a), 128.3 (C-4), 128.2 (C-5), 124.7 (C-8), 123.7 (C-6), 121.7 (C-6′), 120.1 (C-3), 118.2 (C-4′), 117.6 (C-2′), 116.1 (C-5″), 115.4 (C-1), 35.6 (CH2), 24.1 (4″-CH3, 6″-CH3), 23.3 (CH3). IR (ATR) ν῀ [cm−1] = 3290, 2923, 2854, 1665, 1609, 1584, 1559, 1542, 1489, 1433, 1394, 1372, 1320, 1268, 1218, 1194, 1172, 1150, 1022, 970, 901, 885, 838, 679, 778, 720, 692. HRMS (ESI): m/z = [M-H]− calculated for C26H23N4O2S−: 445.1547; found: 445.1542. Purity (HPLC): >99%.
2-((4,6-Dimethylpyrimidin-2-yl)thio)-N-(7-(3-(phenylsulfonamido)phenyl)naphthalen-2-yl)acetamide (FM47). At room temperature, benzenesulfonyl chloride (49.3 µL, 0.386 mmol, 2.00 eq) was added dropwise to a stirred solution of amine 10 (80.1 mg, 0.193 mmol, 1.00 eq) and NEt3 (53.9 µL, 0.386 mmol, 2.00 eq) in anhydrous DCM (5 mL). After 4 h, water (25 mL) and brine (25 mL) were added, and the reaction mixture was extracted with EtOAc (4 × 25 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 1:1) to yield the title compound as a white solid (119 mg, 0.0899 mmol, 47%). m.p.: 176 °C (decomposition), Rf: 0.19 (hexanes/EtOAc 1:1). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 10.49 (s, 1H, NHCO), 10.42 (s, 1H, NHSO2), 8.32 (d, J = 2.1 Hz, 1H, 1-H), 7.93–7.87 (m, 3H, 4-H, 5-H, 8-H), 7.85–7.79 (m, 2H, 2‴-H, 6‴-H), 7.64–7.55 (m, 4H, 3-H, 3‴-H, 4‴-H, 5‴-H), 7.54 (dd, J = 8.4, 1.8 Hz, 1H, 6-H), 7.46 (dt, J = 8.0, 1.1 Hz, 1H, 6′-H), 7.44 (t, J = 2.0 Hz, 1H, 2′-H), 7.35 (t, J = 7.9 Hz, 1H, 5′-H), 7.12 (ddd, J = 8.1, 2.2, 1.0 Hz, 1H, 4′-H), 6.97 (s, 1H, 5″-H), 4.11 (s, 2H, CH2), 2.34 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 169.3 (C-2″), 167.0 (C-4″, C-6″), 166.9 (NHCO), 141.0 (C-1′), 139.5 (C-1‴), 138.3 (C-3′), 137.4 (C-7), 137.2 (C-2), 133.6 (C-8a), 133.0 (C-4‴), 129.8 (C-5′), 129.3 (C-3‴, C-5‴), 129.1 (C-4a), 128.3 (C-5), 128.2 (C-4), 126.8 (C-2‴, C-6‴), 124.7 (C-8), 123.5 (C-6), 122.9 (C-6′), 120.2 (C-3), 119.2 (C-4′), 118.6 (C-2′), 116.1 (C-5″), 115.4 (C-1), 35.6 (CH2), 23.3 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 3294, 3211, 2912, 1670, 1581, 1530, 1511, 1489, 1446, 1385, 1330, 1309, 1265, 1226, 1165, 1094, 944, 906, 893, 846, 839, 786, 758, 737, 685. HRMS (ESI): m/z = [M-H]− calculated for C30H25N4O3S2−: 553.1374; found: 553.1370. Purity (HPLC): >99%.
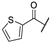
N-(3-(7-(2-((4,6-Dimethylpyrimidin-2-yl)thio)acetamido)naphthalen-2-yl)phenyl)thiophene-2-carboxamide (FM48). At room temperature, 2-thiophenecarbonyl chloride (21.9 µL, 0.205 mmol, 2.00 eq) was added dropwise to a stirred solution of amine 10 (42.4 mg, 0.102 mmol, 1.00 eq) and NEt3 (28.5 µL, 0.205 mmol, 2.00 eq) in anhydrous DCM (5 mL). After 2 h, water (25 mL) and brine (25 mL) were added, and the reaction mixture was extracted with DCM (3 × 25 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 1:1) to yield the title compound as a white solid (16.3 mg, 0.0313 mmol, 31%). m.p.: 180 °C, Rf: 0.21 (hexanes/EtOAc 1:1). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 10.49 (s, 1H, 2-NHCO), 10.33 (s, 1H, 3′-NHCO), 8.36 (d, J = 2.1 Hz, 1H, 1-H), 8.15 (t, J = 1.9 Hz, 1H, 2′-H), 8.09–8.05 (m, 2H, 8-H, 3‴-H), 7.96 (d, J = 8.5 Hz, 1H, 5-H), 7.91 (d, J = 8.9 Hz, 1H, 4-H), 7.88 (dd, J = 5.0, 1.2 Hz, 1H, 5‴-H), 7.81 (ddd, J = 8.0, 2.2, 1.1 Hz, 1H, 4-H), 7.72 (dd, J = 8.5, 1.8 Hz, 1H, 6-H), 7.63 (dd, J = 8.8, 2.1 Hz, 1H, 3-H), 7.56 (dt, J = 7.8, 1.4 Hz, 1H, 6′-H), 7.49 (t, J = 7.9 Hz, 1H, 5′-H), 7.25 (dd, J = 5.0, 3.7 Hz, 1H, 4‴-H), 6.97 (s, 1H, 5″-H), 4.12 (s, 2H, CH2), 2.34 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 169.3 (C-2″), 167.0 (C-4″, C-6″), 166.9 (2-NHCO), 160.0 (3′-NHCO), 140.5 (C-1′), 140.0 (C-2‴), 139.4 (C-3′), 137.9 (C-7), 137.2 (C-2), 133.8 (C-8a), 132.0 (C-5‴), 129.4 (C-5′), 129.2 (C-3‴), 129.1 (C-4a), 128.3 (C-5), 128.2 (C-4), 128.1 (C-4‴), 124.7 (C-8), 123.7 (C-6), 122.4 (C-6′), 120.1 (C-4), 119.4 (C-4′), 118.9 (C-2′), 116.1 (C-5″), 115.5 (C-1), 35.6 (CH2), 23.3 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 3299, 3078, 2957, 1660, 1634, 1608, 1583, 1538, 1487, 1428, 1335, 1306, 1265, 1227, 891, 841, 786, 718, 698. HRMS (ESI): m/z = [M-H]− calculated for C29H23N4O2S2−: 525.1268; found: 523.1263. Purity (HPLC): >99%.
N-(4-(7-(2-((4,6-Dimethylpyrimidin-2-yl)thio)acetamido)naphthalen-2-yl)phenyl)thiophene-2-carboxamide (FM50). At room temperature, 2-thiophenecarbonyl chloride (17.6 µL, 0.165 mmol, 2.00 eq) was added dropwise to a stirred solution of amine 9 (34.1 mg, 0.0823 mmol, 1.00 eq) and NEt3 (22.9 µL, 0.165 mmol, 2.00 eq) in anhydrous DCM (5 mL). After 2 h, water (25 mL) and brine (25 mL) were added, and the reaction mixture was extracted with EtOAc (3 × 25 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 1:1) to yield the title compound as a white solid (53.3 mg, 0.0438 mmol, 53%). m.p.: 195–198 °C, Rf: 0.25 (hexanes/EtOAc 1:1). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 10.47 (s, 1H, 2-NHCO), 10.34 (s, 1H, 4′-NHCO), 8.33 (d, J = 2.0 Hz, 1H, 1-H), 8.11 (d, J = 1.9 Hz, 1H, 8-H), 8.06 (dd, J = 3.8, 1.2 Hz, 1H, 3‴-H), 7.91 (d, J = 8.6 Hz, 1H, 5-H), 7.90–7.85 (m, 4H, 4-H, 3′-H, 5′-H, 5‴-H), 7.84 (d, J = 8.9 Hz, 2H, 2′-H, 6′-H), 7.76 (dd, J = 8.5, 1.8 Hz, 1H, 6-H), 7.61 (dd, J = 8.8, 2.1 Hz, 1H, 3-H), 7.25 (dd, J = 5.0, 3.7 Hz, 1H, 4‴-H), 6.97 (s, 1H, 5″-H), 4.12 (s, 2H, CH2), 2.34 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 169.4 (C-2″), 167.0 (C-4″, C-6″), 166.8 (2-NHCO), 159.9 (4′-NHCO), 140.0 (C-2‴), 138.4 (C-4′), 137.4 (C-7), 137.0 (C-2), 135.1 (C-1′), 133.9 (C-8a), 132.0 (C-5‴), 129.2 (C-3‴), 128.9 (C-4a), 128.2 (C-5), 128.14 (C-4), 128.11 (C-4‴), 127.2 (C-2′, C-6′), 124.1 (C-8), 123.5 (C-6), 120.6 (C-3′, C-5′), 119.9 (C-3), 116.1 (C-5″), 115.5 (C-1), 35.6 (CH2), 23.3 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 3509, 3242, 1665, 1632, 1584, 1529, 1507, 1424, 1355, 1339, 1264, 1250, 1229, 1191, 984, 952, 901, 865, 834, 801, 732, 718. HRMS (ESI): m/z = [M+H]+ calculated for C29H25N4O2S2+: 525.1413; found: 525.1408. Purity (HPLC): ≥98%.
N-(4-(7-(2-((4,6-Dimethylpyrimidin-2-yl)thio)acetamido)naphthalen-2-yl)phenyl)benzamide (FM53). At room temperature, benzoyl chloride (36.7 µL, 0.316 mmol, 2.00 eq) was added dropwise to a stirred solution of amine 9 (65.5 mg, 0.158 mmol, 1.00 eq) and NEt3 (44.0 µL, 0.316 mmol, 2.00 eq) in anhydrous DCM (5 mL). After 2 h, water (25 mL) and brine (25 mL) were added, and the reaction mixture was extracted with EtOAc (3 × 25 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (DCM/MeOH 100:2) to yield the title compound as a white solid (64.1 mg, 0.124 mmol, 78%). m.p.: 207 °C, Rf: 0.24 (DCM/MeOH 100:2). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 10.47 (s, 1H, 2-NHCO), 10.37 (s, 1H, 4′-NHCO), 8.34 (d, J = 2.0 Hz, 1H, 1-H), 8.11 (d, J = 1.9 Hz, 1H, 8-H), 8.01–7.97 (m, 2H, 2‴-H, 6‴-H), 7.96–7.90 (m, 3H, 5-H, 3′-H, 5′-H), 7.88 (d, J = 8.9 Hz, 1H, 4-H), 7.85–7.82 (m, 2H, 2′-H, 6′-H), 7.76 (dd, J = 8.5, 1.9 Hz, 1H, 6-H), 7.63–7.59 (m, 2H, 3-H, 4‴-H), 7.57–7.53 (m, 2H, 3‴-H, 5‴-H), 6.97 (s, 1H, 5″-H), 4.12 (s, 2H, CH2), 2.34 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 169.4 (C-2″), 167.0 (C-4″, C-6″), 166.8 (2-NHCO), 165.6 (4′-NHCO), 138.8 (C-4′), 137.5 (C-7), 137.0 (C-2), 135.0 (C-1′), 134.9 (C-1‴), 133.9 (C-8a), 131.6 (C-4‴), 128.8 (C-4a), 128.4 (C-3‴, C-5‴), 128.2 (C-4), 128.1 (C-5), 127.7 (C-2‴, C-6‴), 127.1 (C-2′, C-6′), 124.1 (C-8), 123.6 (C-6), 120.6 (C-3′, C-5′), 119.8 (C-3), 116.1 (C-5″), 115.5 (C-1), 35.6 (CH2), 23.3 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 3283, 3038, 1680, 1645, 1581, 1521, 1504, 1426, 1323, 1263, 892, 823, 795, 688. HRMS (ESI): m/z = [M-H]− calculated for C31H25N4O2S−: 517.1704; found: 517.1701. Purity (HPLC): >99%.
N-(3-(7-(2-((4,6-Dimethylpyrimidin-2-yl)thio)acetamido)naphthalen-2-yl)phenyl)benzamide (FM54). At room temperature, benzoyl chloride (34.7 µL, 0.299 mmol, 2.00 eq) was added dropwise to a stirred solution of amine 10 (62.0 mg, 0.150 mmol, 1.00 eq) and NEt3 (41.7 µL, 0.299 mmol, 2.00 eq) in anhydrous DCM (5 mL). After 2 h, water (25 mL) and brine (25 mL) were added, and the reaction mixture was extracted with DCM (3 × 25 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 1:1) to yield the title compound as a white solid (68.0 mg, 0.131 mmol, 87%). m.p.: 192 °C, Rf: 0.20 (hexanes/EtOAc 1:1). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 10.49 (s, 1H, 2-NHCO), 10.35 (s, 1H, 3′-NHCO), 8.36 (d, J = 2.1 Hz, 1H, 1-H), 8.23 (t, J = 1.9 Hz, 1H, 2′-H), 8.07 (d, J = 1.8 Hz, 1H, 8-H), 8.02–7.99 (m, 2H, 2‴-H, 6‴-H), 7.96 (d, J = 8.6 Hz, 1H, 5-H), 7.91 (d, J = 8.8 Hz, 1H, 4-H), 7.87 (ddd, J = 8.1, 2.2, 1.1 Hz, 1H, 4′-H), 7.72 (dd, J = 8.5, 1.8 Hz, 1H, 6-H), 7.65–7.59 (m, 2H, 3-H, 4‴-H), 7.58–7.54 (m, 3H, 6′-H, 3‴-H, 5‴-H), 7.49 (t, J = 7.9 Hz, 1H, 5′-H), 6.97 (s, 1H, 5″-H), 4.12 (s, 2H, CH2), 2.34 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 169.3 (C-2″), 167.0 (C-4″, C-6″), 166.9 (2-NHCO), 165.6 (3′-NHCO), 140.4 (C-1′), 139.7 (C-3′), 138.0 (C-7), 137.2 (C-2), 134.8 (C-1‴), 133.8 (C-8a), 131.7 (C-4‴), 129.3 (C-5′), 129.1 (C-4a), 128.4 (C-3‴, C-5‴), 128.3 (C-4), 128.2 (C-5), 127.7 (C-2‴, C-6‴), 124.7 (C-8), 123.7 (C-6), 122.3 (C-6′), 120.1 (C-3), 119.4 (C-4′), 118.9 (C-2′), 116.1 (C-5″), 115.5 (C-1), 35.6 (CH2), 23.3 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 3282, 3059, 1667, 1649, 1608, 1581, 1537, 1487, 1438, 1396, 1370, 1333, 1299, 1265, 1222, 1027, 889, 838, 787, 697. HRMS (ESI): m/z = [M-H]− calculated for C31H25N4O2S−: 517.1704; found: 517.1701. Purity (HPLC): >99%.
2-((4,6-Dimethylpyrimidin-2-yl)thio)-N-(7-(4-(phenylsulfonamido)phenyl)naphthalen-2-yl)acetamide (FM56). At room temperature, benzenesulfonyl chloride (19.9 µL, 0.156 mmol, 0.95 eq) was added dropwise to a stirred solution of amine 9 (67.9 mg, 0.164 mmol, 1.00 eq) and NEt3 (21.7 µL, 0.156 mmol, 0.95 eq) in anhydrous DCM (5 mL). After heating to reflux for 7h, water (25 mL) and brine (25 mL) were added, and the reaction mixture was extracted with DCM (3 × 25 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (DCM/MeOH 120:1) to yield the title compound as a white solid (39.5 mg, 0.0617 mmol, 40%). m.p.: 135 °C (decomposition), Rf: 0.04 (DCM/MeOH 120:1). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 10.45 (s, 2H, NHCO, NHSO2), 8.29 (d, J = 2.0 Hz, 1H, 1-H), 8.00 (d, J = 1.8 Hz, 1H, 8-H), 7.87 (d, J = 6.0 Hz, 1H, 5-H), 7.85 (d, J = 6.2 Hz, 1H, 4-H), 7.83–7.80 (m, 2H, 2‴-H, 6‴-H), 7.71–7.68 (m, 2H, 2′-H, 6′-H), 7.65 (dd, J = 8.6, 1.9 Hz, 1H, 6-H), 7.63–7.55 (m, 4H, 3-H, 3‴-H, 4‴-H, 5‴-H), 7.24–7.19 (m, 2H, 3′-H, 5′-H), 6.97 (s, 1H, 5″-H), 4.10 (s, 2H, CH2), 2.33 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 169.3 (C-2″), 167.0 (C-4″, C-6″), 166.8 (NHCO), 139.6 (C-1‴), 137.2 (C-4′), 137.1 (C-7), 137.0 (C-2), 135.5 (C-1′), 133.7 (C-8a), 133.0 (C-4‴), 129.4 (C-3‴, C-5‴), 128.8 (C-4a), 128.14 (C-5), 128.11 (C-4), 127.7 (C-2′, C-6′), 126.6 (C-2‴, C-6‴), 124.2 (C-8), 123.5 (C-6), 120.2 (C-3′, C-5′), 119.9 (C-3), 116.1 (C-5″), 115.4 (C-1), 35.6 (CH2), 23.3 (4″-CH3, 6″-CH3). IR (ATR) ν῀[cm−1] = 3056, 2923, 2854, 1668, 1583, 1532, 1502, 1329, 1265, 1158, 1091, 902, 830, 720, 687. HRMS (ESI): m/z = [M-H]- calculated for C30H25N4O3S2-: 553.1374; found: 553.1373. Purity (HPLC): >99%.
N-(3-(7-(2-((4,6-Dimethylpyrimidin-2-yl)thio)acetamido)naphthalen-2-yl)phenyl)-5-methylthiophene-2-carboxamide (FM66). At room temperature, previously prepared 5-methylthiophene-2-carbonyl chloride (27.7 µL, 0.228 mmol, 2.00 eq) was added dropwise to a stirred solution of amine 10 (47.2 mg, 0.114 mmol, 1.00 eq) and NEt3 (31.7 µL, 0.228 mmol, 2.00 eq) in anhydrous DCM (5 mL). After 5 h, water (25 mL) and brine (25 mL) were added, and the reaction mixture was extracted with DCM (3 × 25 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 1:1) to yield the title compound as a white solid (51.0 mg, 0.0947 mmol, 83%). m.p.: 122 °C (decomposition), Rf: 0.28 (hexanes/EtOAc 1:1). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 10.49 (s, 1H, 2-NHCO), 10.20 (s, 1H, 3′-NHCO), 8.35 (d, J = 2.0 Hz, 1H, 1-H), 8.14 (t, J = 2.0 Hz, 1H, 2′-H), 8.07 (d, J = 1.8 Hz, 1H, 8-H), 7.95 (d, J = 8.6 Hz, 1H, 5-H), 7.91 (d, J = 8.9 Hz, 1H, 4-H), 7.87 (d, J = 3.7 Hz, 1H, 3‴-H), 7.79 (ddd, J = 8.0, 2.1, 1.0 Hz, 1H, 4′-H), 7.71 (dd, J = 8.5, 1.8 Hz, 1H, 6-H), 7.63 (dd, J = 8.8, 2.0 Hz, 1H, 3-H), 7.54 (dt, J = 7.9, 1.4 Hz, 1H, 6′-H), 7.47 (t, J = 7.9 Hz, 1H, 5′-H), 6.97 (s, 1H, 5″-H), 6.95 (dd, J = 3.8, 1.2 Hz, 1H, 4‴-H), 4.12 (s, 2H, CH2), 2.51 (s, 3H, CH3), 2.34 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 169.3 (2″-H), 167.0 (C-4″, C-6″), 166.9 (2-NHCO), 159.9 (3′-NHCO), 146.0 (C-5‴), 140.5 (C-1′), 139.5 (C-3′), 137.9 (C-7), 137.4 (C-2‴), 137.1 (C-2), 133.8 (C-8a), 129.4 (C-3‴), 129.3 (C-5′), 129.1 (C-4a), 128.3 (C-5), 128.2 (C-4), 126.7 (C-4‴), 124.7 (C-8), 123.7 (C-6), 122.2 (C-6′), 120.1 (C-3), 119.3 (C-4′), 118.8 (C-2′), 116.1 (C-5″), 115.5 (C-1), 35.6 (CH2), 23.3 (4″-CH3, 6″-CH3), 15.3 (CH3). IR (ATR) ν῀ [cm−1] = 3287, 3059, 2920, 1633, 1607, 1582, 1531, 1510, 1486, 1459, 1428, 1398, 1336, 1303, 1263, 1171, 1085, 1032, 891, 839, 801, 785, 730, 697. HRMS (EI): m/z = [M]•+ calculated for C30H26N4O2S2•+: 538.1492; found: 538.1500. Purity (HPLC): >96%.
N-(4-(7-(2-((4,6-Dimethylpyrimidin-2-yl)thio)acetamido)naphthalen-2-yl)phenyl)-5-methylthiophene-2-carboxamide (FM69). At room temperature, previously prepared 5-methylthiophene-2-carbonyl chloride (24.5 µL, 0.201 mmol, 2.00 eq) was added dropwise to a stirred solution of amine 9 (41.7 mg, 0.101 mmol, 1.00 eq) and NEt3 (28.0 µL, 0.201 mmol, 2.00 eq) in anhydrous DCM (5 mL). After heating to reflux for 3 h, water (25 mL) and brine (25 mL) were added, and the reaction mixture was extracted with DCM (3 × 25 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (DCM/MeOH 120:1) to yield the title compound as a white solid (40.2 mg, 0.0746 mmol, 74%). m.p.: 191–193 °C, Rf: 0.07 (DCM/MeOH 120:1). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 10.47 (s, 1H, 2-NHCO), 10.22 (s, 1H, 4′-NHCO), 8.33 (d, J = 2.0 Hz, 1H, 1-H), 8.10 (d, J = 1.8 Hz, 1H, 8-H), 7.91 (d, J = 8.6 Hz, 1H, 5-H), 7.89–7.79 (m, 6H, 4-H, 2′-H, 3′-H, 5′-H, 6′-H, 3‴-H), 7.75 (dd, J = 8.5, 1.8 Hz, 1H, 6-H), 7.60 (dd, J = 8.8, 2.1 Hz, 1H, 3-H), 6.97 (s, 1H, 5″-H), 6.95–6.93 (m, 1H, 4‴-H), 4.11 (s, 2H, CH2), 2.51 (s, 3H, CH3), 2.34 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 169.4 (C-2″), 167.0 (C-4″, C-6″), 166.8 (2-NHCO), 159.8 (4′-NHCO), 146.0 (C-5‴), 138.5 (C-4′), 137.43 (C-7, C-2‴), 137.0 (C-2), 134.9 (C-1′), 133.9 (C-8a), 129.5 (C-3‴), 128.8 (C-4a), 128.2 (C-5), 128.1 (C-4), 127.1 (C-2′, C-6′), 126.7 (C-4‴), 124.1 (C-8), 123.5 (C-6), 120.5 (C-3′, C-5′), 119.8 (C-3), 116.1 (C-5″), 115.5 (C-1), 35.6 (CH2), 23.3 (4″-CH3, 6″-CH3), 15.3 (CH3). IR (ATR) ν῀ [cm−1] = 3446, 3241, 2914, 1663, 1613, 1583, 1526, 1505, 1458, 1398, 1338, 1320, 1264, 1249, 1228, 1190, 1170, 1093, 984, 952, 901, 873, 832, 802, 741. HRMS (EI): m/z = [M]•+ calculated for C30H26N4O2S2•+: 538.1492; found: 538.1494. Purity (HPLC): >98%.
6-(3-Nitrophenyl)benzo[d]thiazol-2-amine (13). 2-Amino-6-bromobenzothiazole (12, 1.86 g, 8.14 mmol, 1.00 eq) and Pd(PPh3)4 (0.941 g, 0.814 mmol, 0.100 eq) were weighed out into a flask, which was put under nitrogen afterwards. Degassed 1,4-dioxane (40 mL) was added via syringe, and the mixture was stirred for 10 min at room temperature before 3-nitrophenyl boronic acid (1.63 g, 9.77 mmol, 1.20 eq), Cs2CO3 (13.3 g, 40.7 mmol, 5.00 eq), and degassed water (17.5 mL) were added under nitrogen atmosphere. The reaction mixture was stirred for 21 h at 80 °C, cooled to room temperature, diluted with water (200 mL), and extracted with EtOAc (4 × 100 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 7:3) to yield the title compound as an orange solid (1.35 g, 4.97 mmol, 61%). m.p.: 218–219 °C (decomposition), Rf: 0.05 (hexanes/EtOAc 7:3). 1H NMR (500 MHz, CDCl3) δ [ppm] = 8.46 (t, J = 2.1 Hz, 1H, 2′-H), 8.18 (ddd, J = 8.2, 2.3, 1.0 Hz, 1H, 4′-H), 7.92 (ddd, J = 7.7, 1.9, 1.0 Hz, 1H, 6′-H), 7.87 (d, J = 2.1 Hz, 1H, 7-H), 7.67–7.56 (m, 3H, 4-H, 5-H, 5′-H), 5.30 (s, 2H, NH2). 13C NMR (126 MHz, CDCl3) δ [ppm] = 166.5 (C-2), 152.6 (C-3a), 148.9 (C-3′), 142.8 (C-1′), 133.3 (C-6), 133.1 (C-7a), 133.0 (C-6′), 129.9 (C-5′), 125.5 (C-5), 121.9 (C-2′), 121.9 (C-4′), 119.9 (C-4), 119.7 (C-7). IR (ATR) ν῀ [cm−1] = 3422, 3290, 3066, 1638, 1599, 1530, 1512, 1486, 1456, 1345, 1304, 1275, 1094, 1064, 906, 876, 862, 801, 742, 728, 714, 685, 674. HRMS (EI): m/z = [M]•+ calculated for C13H9N3O2S•+: 271.0410; found: 271.0411.
tert-Butyl (4-(2-aminobenzo[d]thiazol-6-yl)phenyl)carbamate (14). 2-Amino-6-bromobenzothiazole (12, 1.21 g, 5.28 mmol, 1.00 eq) and Pd(PPh3)4 (610 mg, 0.528 mmol, 0.100 eq) were weighed out into a flask, which was put under nitrogen afterwards. Degassed 1,4-dioxane (30 mL) was added via syringe, and the mixture was stirred for 10 min at room temperature before 4-(N-boc-amino)phenylboronic acid pinacol ester (2.02 g, 6.34 mmol, 1.20 eq), Cs2CO3 (8.60 g, 26.4 mmol, 5.00 eq), and degassed water (10 mL) were added under nitrogen atmosphere. The reaction mixture was stirred for 20 h at 80 °C, cooled to room temperature, diluted with water (250 mL), and extracted with EtOAc (4 × 100 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 1:1) to yield the title compound as a light-yellow solid (498 mg, 1.46 mmol, 28%). m.p.: 320 °C (decomposition), Rf: 0.16 (hexanes/EtOAc 1:1). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 9.40 (s, 1H, NHCO), 7.92 (d, J = 1.9 Hz, 1H, 7-H), 7.57–7.54 (m, 2H, 2′-H, 6′-H), 7.53–7.48 (m, 4H, 3′-H, 5′-H, NH2), 7.47 (dd, J = 8.4, 1.9 Hz, 1H, 5-H), 7.36 (d, J = 8.3 Hz, 1H, 4-H), 1.49 (s, 9H, C(CH3)3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 166.6 (NHCO), 152.8 (C-2), 151.8 (C-3a), 138.4 (C-4′), 133.9 (C-1′), 132.9 (C-6), 131.8 (C-7a), 126.5 (C-2′, C-6′), 123.8 (C-5), 118.43 (C-7), 118.38 (C-3′, C-5′), 117.8 (C-4), 79.1 (C(CH3)3), 28.1 (C(CH3)3). IR (ATR) ν῀ [cm−1] = 3351, 2981, 2932, 1699, 1635, 1588, 1522, 1504, 1459, 1417, 1389, 1367, 1314, 1301, 1233, 1160, 1110, 1053, 1023, 835, 812, 772, 762. HRMS (EI): m/z = [M]•+ calculated for C18H19N3O2S•+: 341.1192; found: 341.1193.
2-Bromo-N-(6-(3-nitrophenyl)benzo[d]thiazol-2-yl)acetamide (15). At 0 °C, bromoacetyl bromide (851 µL, 9.77 mmol, 1.20 eq) was added dropwise to a stirred suspension of amine 13 (2.21 g, 8.14 mmol, 1.00 eq) and NEt3 (1.36 mL, 9.77 mmol, 1.20 eq) in anhydrous DCM/DMF (6:1, 35 mL). After heating to reflux for 3 h, water (150 mL) and brine (50 mL) were added, and the reaction mixture was extracted with EtOAc (4 × 100 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 8:2) to yield the title compound as a light-yellow solid (827 mg, 2.11 mmol, 26%). m.p.: 186 °C, Rf: 0.07 (hexanes/EtOAc 8:2). 1H NMR (500 MHz, CDCl3) δ [ppm] = 9.74 (s, 1H, NHCO), 8.51 (t, J = 2.0 Hz, 1H, 2′-H), 8.23 (ddd, J = 8.2, 2.2, 1.0 Hz, 1H, 4′-H), 8.09 (dd, J = 1.9, 0.6 Hz, 1H, 7-H), 7.97 (ddd, J = 7.7, 1.8, 1.0 Hz, 1H, 6′-H), 7.92 (dd, J = 8.4, 0.6 Hz, 1H, 4-H), 7.73 (dd, J = 8.5, 1.9 Hz, 1H, 5-H), 7.65 (t, J = 8.0 Hz, 1H, 5′-H), 4.15 (s, 2H, CH2). 13C NMR (126 MHz, CDCl3 δ [ppm] = 164.2 (NHCO), 157.9 (C-2), 149.0 (C-3′), 148.7 (C-3a), 142.5 (C-1′), 135.4 (C-6), 133.6 (C-7a), 133.3 (C-6′), 130.1 (C-5′), 126.1 (C-5), 122.3 (C-4′), 122.3 (C-2′), 122.1 (C-4), 120.3 (C-7), 27.9 (CH2). IR (ATR) ν῀ [cm−1] = 2968, 1653, 1610, 1574, 1548, 1530, 1452, 1342, 1320, 1281, 1111, 1000, 874, 863, 825, 799, 770, 746, 730, 718, 679. HRMS (EI): m/z = [M]•+ calculated for C15H1079BrN3O3S•+: 390.9621; found: 390.9615.
tert-Butyl (4-(2-(2-bromoacetamido)benzo[d]thiazol-6-yl)phenyl)carbamate (16). At 0 °C, bromoacetyl bromide (206 µL, 2.37 mmol, 1.00 eq) was added dropwise to a stirred suspension of amine 14 (809 mg, 2.37 mmol, 1.00 eq) and NEt3 (661 µL, 4.74 mmol, 2.00 eq) in anhydrous EtOAc (20 mL). After heating to reflux for 3 h, water (100 mL) and brine (50 mL) were added, and the reaction mixture was extracted with EtOAc (4 × 100 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 7:3) to yield the title compound as a light-yellow solid (495 mg, 1.07 mmol, 45%). m.p.: 300 °C (decomposition), Rf: 0.28 (hexanes/EtOAc 7:3). 1H NMR (500 MHz, CDCl3) δ [ppm] = 8.00 (d, J = 1.8 Hz, 1H, 7-H), 7.85 (d, J = 8.4 Hz, 1H, 4-H), 7.68 (dd, J = 8.5, 1.8 Hz, 1H, 5-H), 7.60–7.55 (m, 2H, 2′-H, 6′-H), 7.50–7.43 (m, 2H, 3′-H, 5′-H), 7.26 (s, 1H, 4-NHCO), 6.59 (bs, 1H, 4′-NHCO), 4.14 (s, 2H, CH2), 1.55 (s, 9H, C(CH3)3). 13C NMR (126 MHz, CDCl3) δ [ppm] = 164.2 (2-NHCO), 157.5 (4′-NHCO), 152.9 (C-2), 146.7 (C-3a), 138.1 (C-4′), 137.8 (C-6), 135.3 (C-1′), 132.9 (C-7a), 128.0 (C-2′, C-6′), 126.1 (C-5), 121.3 (C-4), 119.5 (C-7), 119.1 (C-3′, C-5′), 80.9 (C(CH3)3), 28.5 (C(CH3)3), 27.9 (CH2). IR (ATR) ν῀ [cm−1] = 3370, 3215, 1687, 1602, 1543, 1524, 1502, 1454, 1393, 1368, 1329, 1299, 1272, 1234, 1158, 1058, 982, 834, 812, 754, 720. HRMS (EI): m/z = [M-H]− calculated for C20H1979BrN3O3S−: 460.0338; found: 460.0336.
2-((4,6-Dimethylpyrimidin-2-yl)thio)-N-(6-(3-nitrophenyl)benzo[d]thiazol-2-yl)acetamide (17). At room temperature, potassium tert-butoxide (236 mg, 2.10 mmol, 2.00 eq) was added to a stirred solution of 2-bromoacetamide 15 (413 mg, 1.05 mmol, 1.00 eq) and 4,6-dimethylpyrimidine-2-thiol (177 mg, 1.26 mmol, 1.20 eq) in anhydrous DMF (5 mL). After 3 h, water (50 mL) and brine (100 mL) were added, and the reaction mixture was extracted with EtOAc (3 × 100 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 7:3) to yield the title compound as a yellow solid (319 mg, 0.707 mmol, 67%). m.p.: 205 °C, Rf: 0.09 (hexanes/EtOAc 7:3). 1H NMR (400 MHz, DMSO-d6) δ [ppm] = 12.75 (s, 1H, NHCO), 8.51 (t, J = 2.1 Hz, 1H, 2′-H), 8.49–8.44 (m, 1H, 7-H), 8.24–8.19 (m, 2H, 4′-H, 6′-H), 7.90–7.85 (m, 2H, 4-H, 5-H), 7.78 (t, J = 8.0 Hz, 1H, 5′-H), 6.97 (s, 1H, 5″-H), 4.20 (s, 2H, CH2), 2.29 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (101 MHz, DMSO-d6) δ [ppm] = 168.8 (NHCO), 168.3 (C-2″), 167.1 (C-4″, C-6″), 159.1 (C-2), 148.9 (C-3a), 148.5 (C-3′), 141.6 (C-1′), 133.3 (C-6′), 133.2 (C-6), 132.6 (C-7a), 130.5 (C-5′), 125.4 (C-5), 121.9 (C-4′), 121.1 (C-2′), 121.0 (C-4), 120.5 (C-7), 116.2 (C-5″), 34.5 (CH2), 23.2 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 2985, 1694, 1666, 1608, 1564, 1526, 1452, 1342, 1304, 1262, 1156, 893, 864, 800, 745, 730, 716, 676. HRMS (EI): m/z = [M]•+ calculated for C21H17N5O3S2•+: 451.0767; found: 451.0762.
tert-Butyl (4-(2-(2-((4,6-dimethylpyrimidin-2-yl)thio)acetamido)benzo[d]thiazol-6 yl)phenyl)carbamate (18). At room temperature, potassium tert-butoxide (274 mg, 2.44 mmol, 2.00 eq) was added to a stirred solution of 2-bromoacetamide 16 (564 mg, 1.22 mmol, 1.00 eq) and 4,6-dimethylpyrimidine-2-thiol (205 mg, 1.46 mmol, 1.20 eq) in anhydrous DMF (5 mL). After 3 h, water (50 mL) and brine (100 mL) were added, and the reaction mixture was extracted with EtOAc (4 × 100 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the title compound was yielded as a yellow solid (499 mg, 0.956 mmol, 78%). m.p.: 220 °C, Rf: 0.10 (hexanes/EtOAc 7:3). 1H NMR (400 MHz, DMSO-d6) δ [ppm] = 12.64 (s, 1H, 2-NHCO), 9.44 (s, 1H, 4′-NHCO), 8.23 (d, J = 1.8 Hz, 1H, 7-H), 7.78 (d, J = 8.5 Hz, 1H, 4-H), 7.70 (dd, J = 8.5, 1.9 Hz, 1H, 5-H), 7.65–7.60 (m, 2H, 2′-H, 6′-H), 7.59–7.51 (m, 2H, 3′-H, 5′-H), 6.96 (s, 1H, 5″-H), 4.19 (s, 2H, CH2), 2.29 (s, 6H, 4″-CH3, 6″-CH3), 1.49 (s, 9H, C(CH3)3). 13C NMR (101 MHz, DMSO-d6) δ [ppm] = 168.8 (C-2″), 168.1 (2-NHCO), 167.0 (C-4″, C-6″), 158.0 (C-2), 152.7 (4′-NHCO), 147.7 (C-3a), 138.9 (C-4′), 135.5 (C-6), 133.5 (C-1′), 132.4 (C-7a), 126.9 (C-2′, C-6′), 124.7 (C-5), 120.7 (C-4), 119.0 (C-7), 118.4 (C-3′, C-5′), 116.2 (C-5″), 79.1 (C(CH3)3), 34.4 (CH2), 28.13 (C(CH3)3), 23.2 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 2976, 1720, 1700, 1583, 1536, 1458, 1367, 1326, 1264, 1233, 1154, 1119, 1053, 1024, 846, 819, 779, 478. HRMS (EI): m/z = [M]•+ calculated for C26H27N5O3S2•+: 521.1550; found: 521.1550.
N-(6-(3-Aminophenyl)benzo[d]thiazol-2-yl)-2-((4,6-dimethylpyrimidin-2-yl)thio)acetamide (19). Under nitrogen atmosphere, nitro compound 17 (317 mg, 0.808 mmol, 1.00 eq) was suspended in MeOH/water (10:1, 2.2 mL), and iron powder (266 mg, 4.04 mmol, 5.00 eq) and ammonium chloride (216 mg, 4.04 mmol, 5.00 eq) were added. The reaction mixture was heated to reflux for 3 h, and, after cooling, it was diluted with water (100 mL). Subsequently, the mixture was extracted with EtOAc (4 × 50 mL), and the combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 1:1) to yield the title compound as a light-yellow solid (251 mg, 0.596 mmol, 74%). m.p.: 118–122 °C, Rf: 0.12 (hexanes/EtOAc 1:1). 1H NMR (400 MHz, DMSO-d6) δ [ppm] = 12.65 (s, 1H, NHCO), 8.14 (d, J = 1.8 Hz, 1H, 7-H), 7.79 (d, J = 8.4 Hz, 1H, 4-H), 7.62 (dd, J = 8.5, 1.9 Hz, 1H, 5-H), 7.10 (t, J = 7.8 Hz, 1H, 5′-H), 6.96 (s, 1H, 5″-H), 6.88 (t, J = 2.0 Hz, 1H, 2′-H), 6.82 (ddd, J = 7.6, 1.8, 1.0 Hz, 1H, 6′-H), 6.56 (ddd, J = 7.9, 2.2, 1.0 Hz, 1H, 4′-H), 5.15 (s, 2H, NH2), 4.19 (s, 2H, CH2), 2.29 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (101 MHz, DMSO-d6) δ [ppm] = 168.8 (NHCO), 168.1 (C-2″), 167.0 (C-4″, C-6″), 158.1 (C-2), 149.1 (C-3′), 147.9 (C-3a), 140.7 (C-1′), 136.8 (C-6), 132.3 (C-7a), 129.4 (C-5′), 125.0 (C-5), 120.6 (C-4), 119.4 (C-7), 116.1 (C-5″), 114.5 (C-6′), 113.0 (C-4′), 112.3 (C-2′), 34.5 (CH2), 23.3 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 2921, 1693, 1602, 1583, 1560, 1532, 1453, 1265, 1135, 864, 824, 782, 733, 694. HRMS (EI): m/z = [M]•+ calculated for C21H19N5OS2•+: 421.1026; found: 421.1011.
N-(6-(4-Aminophenyl)benzo[d]thiazol-2-yl)-2-((4,6-dimethylpyrimidin-2-yl)thio)acetamide (20). At room temperature, Boc-protected amine 18 (1.12 g, 2.15 mmol, 1.00 eq) was suspended in chloroform (10 mL), and TFA (3.22 mL, 42.9 mmol, 25.0 eq) was added. After the reaction mixture was stirred for 24 h, it was alkalized with saturated NaHCO3 solution and extracted with DCM (4 × 50 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 1:1) to yield the title compound as a beige solid (755 mg, 1.79 mmol, 83%). m.p.: 146 °C (decomposition), Rf: 0.12 (hexanes/EtOAc 1:1). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 12.58 (s, 1H, NHCO), 8.11 (d, J = 1.8 Hz, 1H, 7-H), 7.72 (d, J = 8.4 Hz, 1H, 4-H), 7.61 (dd, J = 8.5, 1.9 Hz, 1H, 5-H), 7.44–7.37 (m, 2H, 2′-H, 6′-H), 6.97 (s, 1H, 5″-H), 6.68–6.61 (m, 2H, 3′-H, 5′-H), 5.22 (s, 2H, NH2), 4.18 (s, 2H, CH2), 2.30 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (101 MHz, DMSO-d6) δ [ppm] = 168.9 (C-2″), 168.0 (NHCO), 167.1 (C-4″, C-6″), 157.4 (C-2), 148.3 (C-4′), 146.8 (C-3a), 136.6 (C-6), 132.4 (C-7a), 127.7 (C-2′, C-6′), 127.3 (C-1′), 124.9 (C-5), 124.1 (C-4), 120.6 (C-7), 117.9 (C-5″), 116.2 (C-3′, C-5′), 34.4 (CH2), 23.3 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 2922, 1692, 1603, 1583, 1536, 1455, 1265, 1184, 1136, 818. HRMS (EI): m/z = [M]•+ calculated for C21H19N5OS2•+: 421.1026; found: 421.1024.
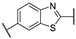
N-(6-(3-Acetamidophenyl)benzo[d]thiazol-2-yl)-2-((4,6-dimethylpyrimidin-2-yl)thio)acetamide (FM94). At room temperature, acetic anhydride (22.3 µL, 0.237 mmol, 2.00 eq) was added dropwise to a stirred solution of amine 19 (50.0 mg, 0.119 mmol, 1.00 eq) and NEt3 (33.1 µL, 0.237 mmol, 2.00 eq) in anhydrous DCM (2 mL). After 3 h, water (50 mL) was added, and the reaction mixture was extracted with DCM (4 × 50 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (DCM/MeOH 100:1) to yield the title compound as a white solid (53.0 mg, 0.114 mmol, 96%). m.p.: 162 °C (decomposition), Rf: 0.03 (DCM/MeOH 100:1). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 12.68 (s, 1H, 2-NHCO), 10.03 (s, 1H, 3′-NHCO), 8.21 (d, J = 1.9 Hz, 1H, 7-H), 7.93 (t, J = 1.9 Hz, 1H, 2′-H), 7.83 (d, J = 8.4 Hz, 1H, 4-H), 7.66 (dd, J = 8.5, 1.9 Hz, 1H, 5-H), 7.56 (dt, J = 7.2, 2.1 Hz, 1H, 4′-H), 7.44–7.32 (m, 2H, 5′-H, 6′-H), 6.96 (s, 1H, 5″-H), 4.20 (s, 2H, CH2), 2.29 (s, 6H, 4″-CH3, 6″-CH3), 2.07 (s, 3H, CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 168.8 (C-2″), 168.4 (3′-NHCO), 168.2 (2-NHCO), 167.1 (C-4″, C-6″), 162.3 (C-2′), 158.4 (C-2), 148.2 (C-3a), 140.5 (C-1′), 139.9 (C-3′), 135.9 (C-6), 132.4 (C-7a), 129.3 (C-5′), 125.1 (C-5), 121.6 (C-6′), 120.8 (C-4), 119.7 (C-7), 117.9 (C-4′), 117.4 (C-2′), 116.2 (C-5″), 34.5 (CH2), 24.1 (CH3), 23.3 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 2922, 1694, 1605, 1583, 1537, 1456, 1428, 1314, 1263, 1146, 1033, 874, 824, 790, 696. HRMS (EI): m/z = [M]•+ calculated for C23H21N5O2S2•+: 463.1131; found: 463.1128. Purity (HPLC): >99%.
N-(3-(2-(2-((4,6-Dimethylpyrimidin-2-yl)thio)acetamido)benzo[d]thiazol-6-yl)phenyl)benzamide (FM95). At room temperature, benzoyl chloride (26.6 µL, 0.229 mmol, 2.00 eq) was added dropwise to a stirred solution of amine 19 (48.3 mg, 0.115 mmol, 1.00 eq) and NEt3 (31.9 µL, 0.229 mmol, 2.00 eq) in anhydrous DCM (2 mL). After 3 h, water (50 mL) was added, and the reaction mixture was extracted with DCM (4 × 50 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (DCM/MeOH 120:1) to yield the title compound as a white solid (39.7 mg, 0.0755 mmol, 66%). m.p.: 262 °C, Rf: 0.17 (DCM/MeOH 120:1). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 12.70 (s, 1H, 2-NHCO), 10.34 (s, 1H, 3′-NHCO), 8.27 (d, J = 1.8 Hz, 1H, 7-H), 8.17 (t, J = 1.5 Hz, 1H, 2′-H), 8.02–7.97 (m, 2H, 2‴-H, 6‴-H), 7.86 (d, J = 8.4 Hz, 1H, 4-H), 7.85–7.79 (m, 1H, 4′-H), 7.73 (dd, J = 8.5, 1.9 Hz, 1H, 5-H), 7.63–7.59 (m, 1H, 4‴-H), 7.58–7.53 (m, 2H, 3‴-H, 5‴-H), 7.49–7.44 (m, 2H, 5′-H, 6′-H), 6.96 (s, 1H, 5″-H), 4.20 (s, 2H, CH2), 2.30 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 168.8 (C-2″), 168.2 (2-NHCO), 167.1 (C-4″, C-6″), 165.6 (3′-NHCO), 158.4 (C-2), 148.2 (C-3a), 140.4 (C-1′), 139.8 (C-3′), 135.8 (C-6), 134.9 (C-1‴), 132.5 (C-7a), 131.6 (C-4‴), 129.3 (C-5′), 128.4 (C-3‴, C-5‴), 127.7 (C-2‴, C-6‴), 125.1 (C-5), 122.1 (C-6′), 120.9 (C-4), 119.7 (C-7), 119.1 (C-4′), 118.7 (C-2′), 116.2 (C-5′), 34.5 (CH2), 23.3 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 3295, 2956, 1718, 1647, 1582, 1526, 1464, 1432, 1389, 1368, 1307, 1264, 1152, 884, 876, 884, 892, 762, 706, 694. HRMS (EI): m/z = [M]•+ calculated for C28H23N5O2S2•+: 525.1288; found: 525.1294. Purity (HPLC): >99%.
N-(3-(2-(2-((4,6-Dimethylpyrimidin-2-yl)thio)acetamido)benzo[d]thiazol-6-yl)phenyl)thiophene-2-carboxamide (FM96). At room temperature, 2-thiophenecarbonyl chloride (15.6 µL, 0.145 mmol, 1.10 eq) was added dropwise to a stirred solution of amine 19 (55.7 mg, 0.132 mmol, 1.00 eq) and NEt3 (20.3 µL, 0.145 mmol, 1.10 eq) in anhydrous DCM (2 mL). After 1 h, water (50 mL) was added, and the reaction mixture was extracted with DCM (4 × 50 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 6:4) to yield the title compound as a white solid (57.0 mg, 0.107 mmol, 81%). m.p.: 226 °C, Rf: 0.25 (hexanes/EtOAc 6:4). 1H NMR (400 MHz, DMSO-d6) δ [ppm] = 12.70 (s, 1H, 2-NHCO), 10.32 (s, 1H, 3′-NHCO), 8.27 (d, J = 1.9 Hz, 1H, 7-H), 8.10 (s, 1H, 2′-H), 7.93–7.81 (m, 2H, 4-H, 5‴-H), 7.81–7.68 (m, 2H, 5-H, 6′-H), 7.51–7.42 (m, 2H, 4′-H, 5′-H), 7.25 (dd, J = 5.0, 3.7 Hz, 1H, 4‴-H), 6.97 (s, 1H, 5″-H), 4.20 (s, 2H, CH2), 2.30 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 168.8 (2-NHCO), 168.2 (C-2‴), 167.1 (C-4″, C-6″), 156.0 (3′-NHCO), 158.5 (C-2), 148.3 (C-3a), 140.4 (C-1′), 140.0 (C-2‴), 139.3 (C-3′), 135.7 (C-6), 132.5 (C-7a), 132.0 (C-5‴), 129.4 (C-5′), 129.2 (C-3‴), 128.1 (C-4‴), 125.1 (C-5), 122.2 (C-4′), 120.9 (C-4), 119.7 (C-7), 119.1 (C-6′), 118.8 (C-2′), 116.2 (C-5″), 34.5 (CH2), 23.3 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 2964, 1704, 1657, 1605, 1583, 1544, 1491, 1454, 1313, 1266, 1212, 1152, 875, 837, 792, 732, 696. HRMS (EI): m/z = [M]•+ calculated for C26H21N5O2S3•+: 531.0852; found: 531.0843. Purity (HPLC): >99%.
2-((4,6-Dimethylpyrimidin-2-yl)thio)-N-(6-(3-(phenylsulfonamido)phenyl)benzo[d]thiazol-2-yl)acetamide (FM104). At room temperature, benzenesulfonyl chloride (23.4 µL, 0.184 mmol, 0.95 eq) was added dropwise to a stirred solution of amine 19 (81.5 mg, 0.193 mmol, 1.00 eq) and NEt3 (25.6 µL, 0.184 mmol, 0.95 eq) in anhydrous DCM (5 mL). After 1 h, water (25 mL) and brine (25 mL) were added, and the reaction mixture was extracted with DCM (3 × 50 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (DCM/MeOH 100:1) to yield the title compound as a white solid (16.3 mg, 0.0290 mmol, 15%). m.p.: 204–209 °C, Rf: 0.06 (DCM/MeOH 100:1). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 12.69 (s, 1H, NHCO), 10.40 (s, 1H, NHSO2), 8.13 (d, J = 1.9 Hz, 1H, 7-H), 7.85–7.78 (m, 3H, 4-H, 2‴-H, 6‴-H), 7.63–7.59 (m, 1H, 4‴-H), 7.58–7.53 (m, 3H, 5-H, 3‴-H, 5‴-H), 7.41–7.35 (m, 2H, 2′-H, 6′-H), 7.32 (t, J = 7.8 Hz, 1H, 5′-H), 7.07 (ddd, J = 7.8, 2.2, 1.1 Hz, 1H, 4′-H), 6.96 (s, 1H, 5″-H), 4.19 (s, 2H, CH2), 2.29 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 168.8 (C-2″), 168.2 (NHCO), 167.1 (C-4″, C-6″), 158.6 (C-2), 148.3 (C-3a), 141.0 (C-1′), 139.4 (C-1‴), 138.3 (C-3′), 135.2 (C-6), 133.0 (C-4‴), 132.5 (C-7a), 129.8 (C-5′), 129.3 (C-3‴, C-5‴), 126.7 (C-2‴, C-6‴), 125.0 (C-5), 122.7 (C-6′), 120.9 (C-4), 119.7 (C-7), 118.8 (C-4′), 118.4 (C-2′), 116.2 (C-5″), 34.5 (CH2), 23.3 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 3186, 2965, 1720, 1602, 1580, 1454, 1331, 1309, 1265, 1154, 1088, 955, 882, 836, 794, 751, 714, 685. HRMS (EI): m/z = [M]•+ calculated for C27H23N5O3S3•+: 561.0958; found: 561.0956. Purity (HPLC): >99%.
N-(3-(2-(2-((4,6-Dimethylpyrimidin-2-yl)thio)acetamido)benzo[d]thiazol-6-yl)phenyl)-5-methylthiophene-2-carboxamide (FM108). At room temperature, previously prepared 5-methylthiophene-2-carbonyl chloride (26.8 µL, 0.220 mmol, 2.00 eq) was added dropwise to a stirred solution of amine 19 (46.4 mg, 0.110 mmol, 1.00 eq) and NEt3 (30.7 µL, 0.220 mmol, 2.00 eq) in anhydrous DCM (5 mL). After 2 h, water (50 mL) was added, and the reaction mixture was extracted with DCM (3 × 50 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (DCM/MeOH 100:2) to yield the title compound as a white solid (27.6 mg, 0.0506 mmol, 46%). m.p.: 122 °C (decomposition), Rf: 0.04 (DCM/MeOH 100:2). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 12.69 (s, 1H, 2-NHCO), 10.19 (s, 1H, 3′-NHCO), 8.26 (d, J = 1.9 Hz, 1H, 7-H), 8.09 (t, J = 1.3 Hz, 1H, 2′-H), 7.89–7.83 (m, 2H, 4-H, 3‴-H), 7.75–7.70 (m, 2H, 5-H, 4′-H), 7.48–7.40 (m, 2H, 5′-H, 6′-H), 6.97 (s, 1H, 5″-H), 6.94 (dd, J = 3.7, 1.1 Hz, 1H, 4‴-H), 4.20 (s, 2H, CH2), 2.51 (s, 3H, CH3), 2.30 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 168.8 (C-2″), 168.2 (2-NHCO), 167.1 (C-4″, C-6″), 159.9 (3′-NHCO), 158.4 (C-2), 148.2 (C-3a), 146.0 (C-5‴), 140.4 (C-1′), 139.5 (C-3′), 137.4 (C-2‴), 135.7 (C-6), 132.5 (C-7a), 129.4 (C-3‴), 129.3 (C-5′), 126.7 (C-4‴), 125.1 (C-5), 122.1 (C-6′), 120.9 (C-4), 119.7 (C-7), 119.0 (C-4′), 118.6 (C-2′), 116.2 (C-5″), 34.5 (CH2), 23.3 (4″-CH3, 6″-CH3), 15.3 (CH3). IR (ATR) ν῀ [cm−1] = 3268, 2956, 1720, 1626, 1581, 1544, 1525, 1486, 1455, 1422, 1307, 1264, 1212, 1152, 1095, 978, 881, 852, 834, 816, 790, 756, 742, 698, 664. HRMS (EI): m/z = [M]•+ calculated for C30H26N4O2S2•+: 545.1008; found: 545.1005. Purity (HPLC): >99%.
N-(4-(2-(2-((4,6-Dimethylpyrimidin-2-yl)thio)acetamido)benzo[d]thiazol-6-yl)phenyl)benzamide (FM127). At room temperature, benzoyl chloride (43.0 µL, 0.371 mmol, 2.00 eq) was added dropwise to a stirred solution of amine 20 (78.1 mg, 0.185 mmol, 1.00 eq) and NEt3 (51.6 µL, 0.371 mmol, 2.00 eq) in anhydrous DCM (5 mL). After 3 h, water (50 mL) was added, and the reaction mixture was extracted with DCM (4 × 50 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 1:1) to yield the title compound as a white solid (44.3 mg, 0.0843 mmol, 46%). m.p.: 262 °C, Rf: 0.14 (hexanes/EtOAc 1:1). 1H NMR (400 MHz, DMSO-d6) δ [ppm] = 12.67 (s, 1H, 2-NHCO), 10.35 (s, 1H, 4′-NHCO), 8.30 (d, J = 1.8 Hz, 1H, 7-H), 8.02–7.94 (m, 2H, 2‴-H, 6‴-H), 7.94–7.88 (m, 2H, 3′-H, 5′-H), 7.82 (d, J = 8.5 Hz, 1H, 4-H), 7.78–7.71 (m, 3H, 5-H, 2′-H, 6′-H), 7.64–7.52 (m, 3H, 3‴-H, 4‴-H, 5‴-H), 6.97 (s, 1H, 5″-H), 4.20 (s, 2H, CH2), 2.30 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (101 MHz, DMSO-d6) δ [ppm] = 168.9 (C-2″), 168.2 (2-NHCO), 167.1 (C-4″, C-6″), 165.6 (4′-NHCO), 158.2 (C-2), 147.9 (C-3a), 138.6 (C-4′), 135.4 (C-6), 135.0 (C-1‴), 134.9 (C-1′), 132.5 (C-7a), 131.6 (C-4‴), 128.4 (C-3‴, C-5‴), 127.7 (C-2‴, C-6‴), 126.9 (C-5), 124.8 (C-4), 120.7 (C-3′, C-5′), 119.2 (C-7), 116.2 (C-5″), 34.5 (CH2), 23.3 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 3367, 1684, 1673, 1604, 1566, 1550, 1527, 1493, 1455, 1394, 1342, 1324, 1298, 1267, 1245, 1227, 1189, 1129, 1031, 897, 870, 834, 808, 750, 702, 690, 676. HRMS (EI): m/z = [M]•+ calculated for C28H23N5O2S2•+: 525.1288; found: 525.1281. Purity (HPLC): >99%.
N-(6-(4-Acetamidophenyl)benzo[d]thiazol-2-yl)-2-((4,6-dimethylpyrimidin-2-yl)thio)acetamide (FM128). At room temperature, acetic anhydride (23.7 µL, 0.253 mmol, 2.00 eq) was added dropwise to a stirred solution of amine 20 (53.3 mg, 0.126 mmol, 1.00 eq) and NEt3 (35.2 µL, 0.253 mmol, 2.00 eq) in anhydrous DCM (2 mL). After 3 h, water (50 mL) was added, and the reaction mixture was extracted with DCM (4 × 50 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 4:6) to yield the title compound as a white solid (35.1 mg, 0.0757 mmol, 60%). m.p.: 255 °C, Rf: 0.06 (hexanes/EtOAc 4:6). 1H NMR (500 MHz, DMSO-d6) δ [ppm] = 12.65 (s, 1H, 2-NHCO), 10.02 (s, 1H, 4′-NHCO), 8.24 (d, J = 1.8 Hz, 1H, 7-H), 7.79 (d, J = 8.4 Hz, 1H, 4-H), 7.71 (dd, J = 8.5, 1.9 Hz, 1H, 5-H), 7.69–7.64 (m, 4H, 2′-H, 3′-H, 5′-H, 6′-H), 6.96 (s, 1H, 5″-H), 4.19 (s, 2H, CH2), 2.29 (s, 6H, 3″-CH3, 4″-CH3), 2.07 (s, 3H, CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 168.8 (C-2″), 168.3 (4′-NHCO), 168.1 (2-NHCO), 167.1 (C-4″, C-6″), 158.1 (C-2), 147.8 (C-3a), 138.7 (C-4′), 135.5 (C-6), 134.4 (C-1′), 132.4 (C-7a), 127.0 (C-2′, C-6′), 124.8 (C-5), 120.8 (C-4), 119.3 (C-3′, C-5′), 119.1 (C-7), 116.2 (C-5″), 34.4 (CH2), 24.1 (CH3), 23.3 (3″-CH3, 4″-CH3). IR (ATR) ν῀ [cm−1] = 3361, 1679, 1658, 1600, 1551, 1531, 1460, 1394, 1340, 1330, 1292, 1275, 1264, 1225, 1192, 1128, 1092, 830, 807, 799, 748, 734, 692. HRMS (EI): m/z = [M]•+ calculated for C23H21N5O2S2•+: 463.1131; found: 463.1132. Purity (HPLC): >99%.
N-(4-(2-(2-((4,6-Dimethylpyrimidin-2-yl)thio)acetamido)benzo[d]thiazol-6-yl)phenyl)thiophene-2-carboxamide (FM129). At room temperature, 2-thiophenecarbonyl chloride (23.8 µL, 0.222 mmol, 1.20 eq) was added dropwise to a stirred solution of amine 20 (78.1 mg, 0.185 mmol, 1.00 eq) and NEt3 (23.8 µL, 0.222 mmol, 1.20 eq) in anhydrous DCM (5 mL). After 1 h, water (50 mL) was added, and the reaction mixture was extracted with DCM (4 × 50 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (DCM/MeOH 100:1) to yield the title compound as a white solid (75.7 mg, 0.142 mmol, 77%). m.p.: 280–284 °C, Rf: 0.29 (DCM/MeOH 100:1). 1H NMR (400 MHz, DMSO-d6) δ [ppm] = 12.68 (s, 1H, 2-NHCO), 10.33 (s, 1H, 4′-NHCO), 8.30 (d, J = 1.8 Hz, 1H, 7-H), 8.05 (dd, J = 3.8, 1.2 Hz, 1H, 3‴-H), 7.88 (dd, J = 5.0, 1.1 Hz, 1H, 5‴-H), 7.86–7.79 (m, 3H, 4-H, 3′-H, 5′-H), 7.79–7.72 (m, 3H, 5-H, 2′-H, 6′-H), 7.24 (dd, J = 5.0, 3.8 Hz, 1H, 4‴-H), 6.97 (s, 1H, 5″-H), 4.20 (s, 2H, CH2), 2.29 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (101 MHz, DMSO-d6) δ [ppm] = 168.8 (C-2″), 168.2 (2-NHCO), 167.0 (C-4″, C-6″), 159.9 (4′-NHCO), 158.2 (C-2), 147.9 (C-3a), 140.0 (C-2‴), 138.1 (C-4′), 135.3 (C-6), 135.1 (C-1′), 132.5 (C-7a), 132.0 (C-5‴), 129.2 (C-3‴), 128.1 (C-4‴), 127.0 (C-2′, C-6′), 124.8 (C-5), 120.8 (C-4), 120.7 (C-3′, C-5′), 119.2 (C-7), 116.2 (C-5″), 34.5 (CH2), 23.3 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 3371, 1682, 1659, 1600, 1531, 1457, 1421, 1326, 1263, 1228, 1188, 1091, 864, 832, 805, 750, 722. HRMS (EI): m/z = [M]•+ calculated for C26H21N5O2S3•+: 531.0852; found: 531.0860. Purity (HPLC): >99%.
2-((4,6-Dimethylpyrimidin-2-yl)thio)-N-(6-(3-(phenylsulfonamido)phenyl)benzo[d]thiazol-2-yl)acetamide (FM130). At room temperature, benzenesulfonyl chloride (26.1 µL, 0.204 mmol, 0.95 eq) was added dropwise to a stirred solution of amine 20 (90.6 mg, 0.215 mmol, 1.00 eq) and NEt3 (28.5 µL, 0.204 mmol, 0.95 eq) in anhydrous DCM (5 mL). After heating to reflux for 8 h, water (25 mL) and brine (25 mL) were added, and the reaction mixture was extracted with DCM (3 × 50 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (DCM/MeOH/AcOH 100:0.5:0.25) to yield the title compound as a white solid (68.0 mg, 0.121 mmol, 59%). m.p.: 233 °C, Rf: 0.07 (DCM/MeOH/AcOH 100:0.5:0.25). 1H NMR (500 MHz, CDCl3) δ [ppm] = 11.86 (s, 1H, NHCO), 7.91 (d, J = 1.8 Hz, 1H, 7-H), 7.84–7.78 (m, 2H, 2‴-H, 6‴-H), 7.74 (d, J = 8.4 Hz, 1H, 4-H), 7.57–7.52 (m, 2H, 5-H, 4‴-H), 7.52–7.48 (m, 2H, 2′-H, 6′-H), 7.48–7.43 (m, 2H, 3‴-H, 5‴-H), 7.18–7.12 (m, 2H, 3′-H, 5′-H), 6.88 (s, 1H, 5″-H), 6.78 (s, 1H, NHSO2), 3.98 (s, 2H, CH2), 2.57 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (126 MHz, CDCl3) δ [ppm] = 169.9 (C-2″), 168.4 (NHCO), 168.3 (C-4″, C-6″), 158.0 (C-2), 148.4 (C-3a), 139.2 (C-1‴), 138.2 (C-1′), 136.2 (C-6), 135.6 (C-4′), 133.4 (C-7a), 133.2 (C-4‴), 129.2 (C-3‴, C-5‴), 128.2 (C-2′, C-6′), 127.4 (C-2‴, C-6‴), 125.5 (C-5), 122.4 (C-3′, C-5′), 121.3 (C-4), 119.6 (C-7), 117.3 (C-5″), 34.8 (CH2), 24.0 (4″-CH3, 6″-CH3). IR (ATR) ν῀ [cm−1] = 3260, 2918, 1695, 1604, 1583, 1560, 1540, 1517, 1464, 1407, 1332, 1312, 1295, 1274, 1227, 1151, 1090, 1031, 929, 847, 824, 754, 718, 687. HRMS (EI): m/z = [M]•+ calculated for C27H23N5O3S3•+: 561.0958; found: 561.0954. Purity (HPLC): >99%.
N-(4-(2-(2-((4,6-Dimethylpyrimidin-2-yl)thio)acetamido)benzo[d]thiazol-6-yl)phenyl)-5-methylthiophene-2-carboxamide (FM131). At room temperature, previously prepared 5-methylthiophene-2-carbonyl chloride (25.2 µL, 0.202 mmol, 1.10 eq) was added dropwise to a stirred solution of amine 20 (77.4 mg, 0.184 mmol, 1.10 eq) and NEt3 (28.2 µL, 0.202 mmol, 1.10 eq) in anhydrous DCM (5 mL). After 2 h, water (50 mL) was added, and the reaction mixture was extracted with DCM (3 × 50 mL). The combined organic layers were dried over sodium sulfate. After evaporating the solvent in vacuo, the crude product was purified via flash column chromatography (hexanes/EtOAc 6:4) to yield the title compound as a white solid (45.3 mg, 0.083 mmol, 45%). m.p.: 294 °C, Rf: 0.10 (hexanes/EtOAc 6:4). 1H NMR (400 MHz, DMSO-d6) δ [ppm] = 12.66 (s, 1H, 2-NHCO), 10.19 (s, 1H, 4′-NHCO), 8.29 (d, J = 1.8 Hz, 1H, 7-H), 7.87–7.78 (m, 4H, 4-H, 3′-H, 5′-H, 3‴-H), 7.78–7.70 (m, 3H, 2′-H, 6′-H, 5-H), 6.97 (s, 1H, 5″-H), 6.94 (dd, J = 3.7, 1.2 Hz, 1H, 4‴-H), 4.20 (s, 2H, CH2), 2.30 (s, 6H, 4″-CH3, 6″-CH3). 13C NMR (126 MHz, DMSO-d6) δ [ppm] = 168.8 (C-2″), 168.1 (2-NHCO), 167.1 (C-4″, C-6″), 159.8 (4′-NHCO), 158.1 (C-2), 147.9 (C-3a), 145.9 (C-5‴), 138.2 (C-4′), 137.4 (C-2‴), 135.4 (C-6), 134.9 (C-1′), 132.5 (C-7a), 129.4 (C-3‴), 126.9 (C-2′, C-6′), 126.7 (C-4‴), 124.8 (C-5), 120.8 (C-4), 120.5 (C-3′, C-5′), 119.2 (C-7), 116.2 (C-5″), 34.4 (CH2), 23.3 (4″-CH3, 6″-CH3), 15.3 (CH3). IR (ATR) ν῀ [cm−1] = 3361, 1679, 1658, 1600, 1551, 1531, 1460, 1394, 1340, 1330, 1292, 1275, 1264, 1255, 1192, 1128, 1092, 899, 863, 830, 807, 799, 748, 734, 692. HRMS (EI): m/z = [M]•+ calculated for C30H26N4O2S2•+: 545.1008; found: 545.1008. Purity (HPLC): >99%.





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}