

Hydroperoxyl Radical Scavenging Activity of Bromophenols from Marine Red Alga Polysiphonia urceolata: Mechanistic Insights, Kinetic Analysis, and Influence of Physiological Media


Abstract
1. Introduction
2. Results and Discussion
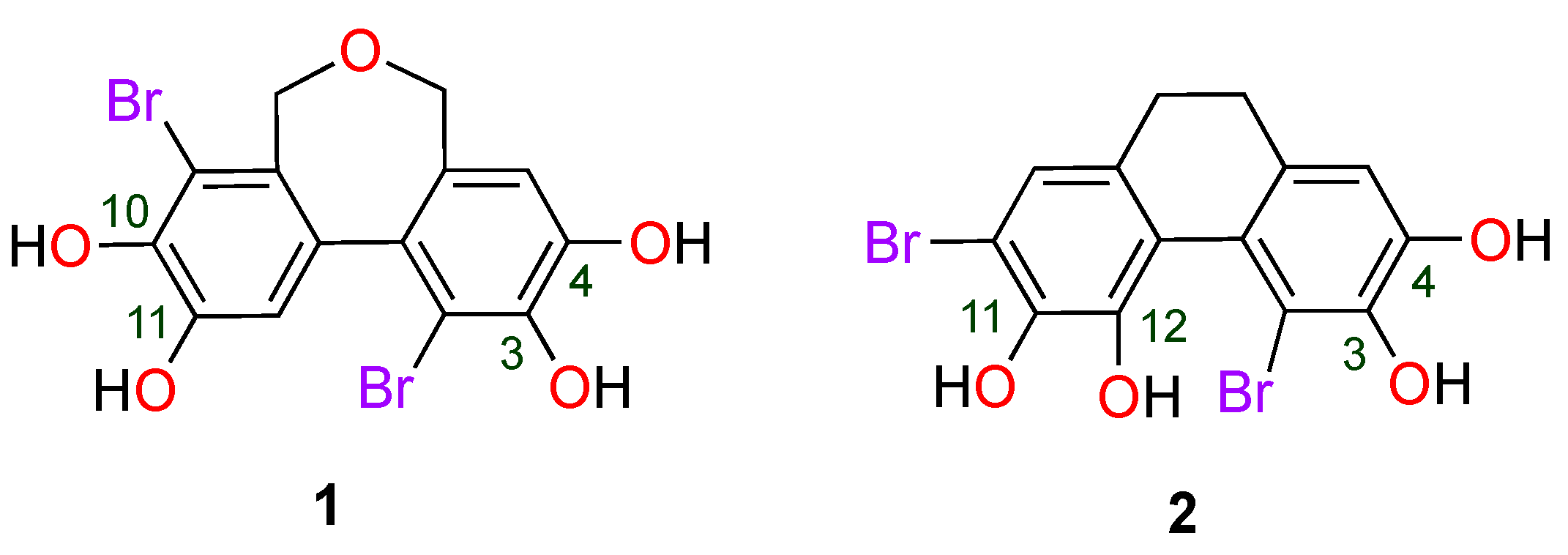
2.1. Molecular Geometry and Electronic Properties
2.2. Radical Scavenging Mechanism at Physiological Conditions
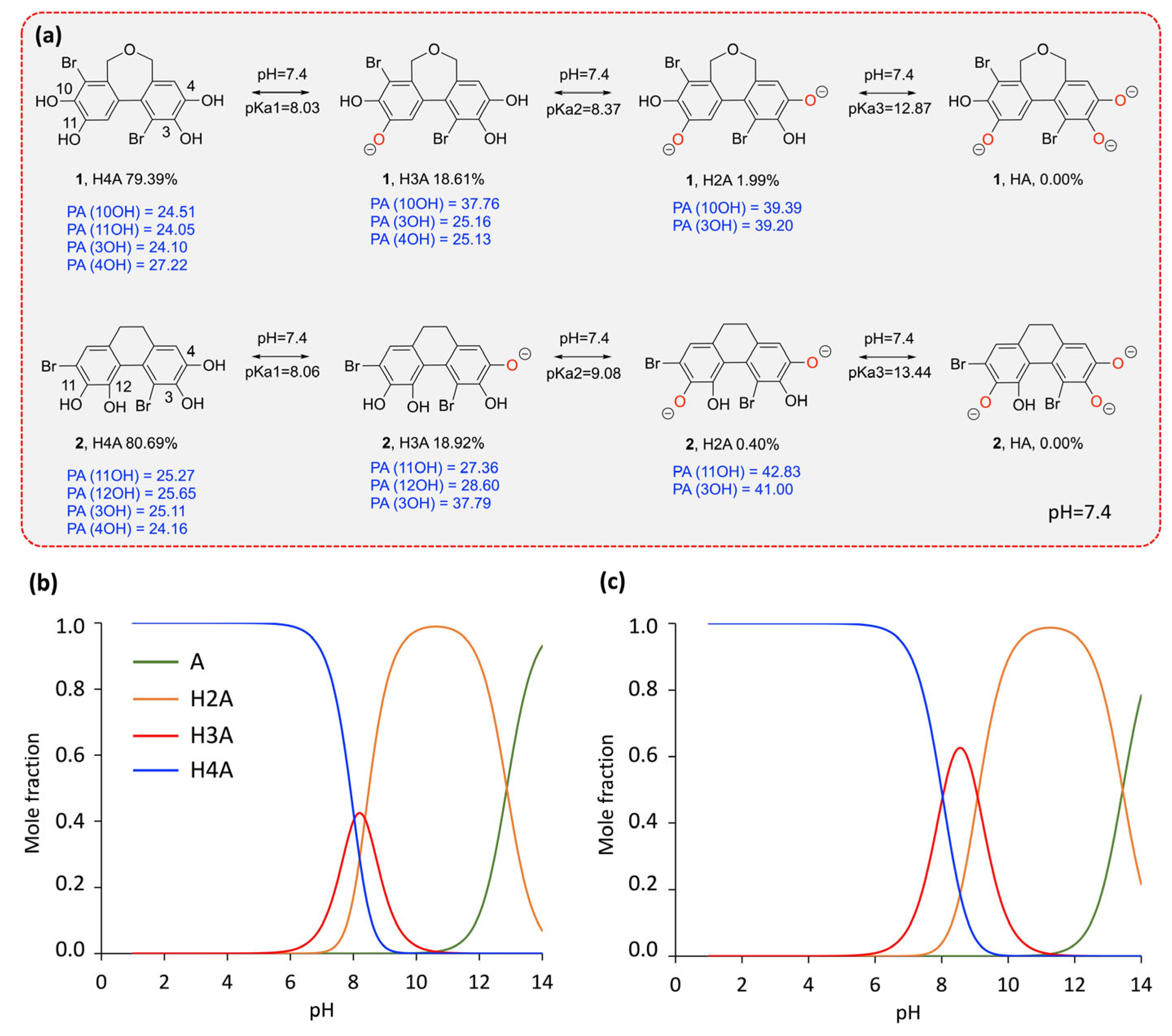
2.2.1. Acid–Base Equilibrium
2.2.2. Thermodynamic Assessment in Physiological Media
2.2.3. Kinetic Investigations in Physiological Media
3. Materials and Methods
4. Conclusions
Supplementary Materials
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Dong, H.; Hansen, P.E.; Dong, S.; Stagos, D.; Lin, X.; Liu, M. 3—Marine natural bromophenols: Sources, structures, main bioactivities, and toxicity. In Marine Phenolic Compounds; Pérez-Correa, J.R., Mateos, R., Domínguez, H., Eds.; Elsevier: Amsterdam, The Netherlands, 2023; pp. 87–112. [Google Scholar]
- Dong, H.; Dong, S.; Erik Hansen, P.; Stagos, D.; Lin, X.; Liu, M. Progress of Bromophenols in Marine Algae from 2011 to 2020: Structure, Bioactivities, and Applications. Mar. Drugs 2020, 18, 411. [Google Scholar] [CrossRef] [PubMed]
- Matulja, D.; Vranješević, F.; Kolympadi Markovic, M.; Pavelić, S.K.; Marković, D. Anticancer Activities of Marine-Derived Phenolic Compounds and Their Derivatives. Molecules 2022, 27, 1449. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; Yin, L.; Wang, Y.; Wang, S.; Song, F. A new bromobenzyl methyl sulphoxide from marine red alga Symphyocladia latiuscula. Nat. Prod. Res. 2013, 27, 723–726. [Google Scholar] [CrossRef] [PubMed]
- Cherian, C.; Jannet Vennila, J.; Sharan, L. Marine bromophenols as an effective inhibitor of virulent proteins (peptidyl arginine deiminase, gingipain R and hemagglutinin A) in Porphyromas gingivalis. Arch. Oral Biol. 2019, 100, 119–128. [Google Scholar] [CrossRef]
- Liu, M.; Wang, G.; Xiao, L.; Xu, X.; Liu, X.; Xu, P.; Lin, X. Bis(2,3-dibromo-4,5-dihydroxybenzyl) Ether, a Marine Algae Derived Bromophenol, Inhibits the Growth of Botrytis cinerea and Interacts with DNA Molecules. Mar. Drugs 2014, 12, 3838–3851. [Google Scholar] [CrossRef]
- Kim, S.-Y.; Kim, S.R.; Oh, M.-J.; Jung, S.-J.; Kang, S.Y. In Vitro antiviral activity of red alga, Polysiphonia morrowii extract and its bromophenols against fish pathogenic infectious hematopoietic necrosis virus and infectious pancreatic necrosis virus. J. Microbiol. 2011, 49, 102–106. [Google Scholar] [CrossRef]
- Kang, N.-J.; Han, S.-C.; Kang, H.-J.; Ko, G.; Yoon, W.-J.; Kang, H.-K.; Yoo, E.-S. Anti-Inflammatory Effect of 3-Bromo-4,5-Dihydroxybenzaldehyde, a Component of Polysiphonia morrowii, In Vivo and In Vitro. Toxicol. Res. 2017, 33, 325–332. [Google Scholar] [CrossRef]
- Paudel, P.; Park, S.E.; Seong, S.H.; Jung, H.A.; Choi, J.S. Bromophenols from Symphyocladia latiuscula Target Human Monoamine Oxidase and Dopaminergic Receptors for the Management of Neurodegenerative Diseases. J. Agric. Food Chem. 2020, 68, 2426–2436. [Google Scholar] [CrossRef]
- Paudel, P.; Seong, S.H.; Zhou, Y.; Park, H.J.; Jung, H.A.; Choi, J.S. Anti-Alzheimer’s Disease Activity of Bromophenols from a Red Alga, Symphyocladia latiuscula (Harvey) Yamada. ACS Omega 2019, 4, 12259–12270. [Google Scholar] [CrossRef]
- Tziveleka, L.-A.; Tammam, M.A.; Tzakou, O.; Roussis, V.; Ioannou, E. Metabolites with Antioxidant Activity from Marine Macroalgae. Antioxidants 2021, 10, 1431. [Google Scholar] [CrossRef]
- Boulebd, H. Mechanistic Insights into the Antioxidant and Pro-oxidant Activities of Bromophenols from Marine Algae: A DFT Investigation. J. Org. Chem. 2024, 89, 8168–8177. [Google Scholar] [CrossRef] [PubMed]
- Li, K.; Li, X.-M.; Ji, N.-Y.; Wang, B.-G. Bromophenols from the Marine Red Alga Polysiphonia urceolata with DPPH Radical Scavenging Activity. J. Nat. Prod. 2008, 71, 28–30. [Google Scholar] [CrossRef] [PubMed]
- Boulebd, H.; Amine Khodja, I.; Benarous, K.; Mą̨czyński, M.; Spiegel, M. A Comprehensive Experimental and Theoretical Investigation of the Antioxidant Properties of Hispidin and Isohispidin. J. Org. Chem. 2025, 90, 3257–3268. [Google Scholar] [CrossRef]
- Boulebd, H.; Pereira, D.M. Examination of the Antioxidant Activity of Psoralidin: Computational Mechanistic Study and Impact on the ROS Level in Human Keratinocytes. J. Org. Chem. 2023, 88, 5745–5751. [Google Scholar] [CrossRef]
- Amorati, R.; Pedulli, G.F.; Cabrini, L.; Zambonin, L.; Landi, L. Solvent and pH Effects on the Antioxidant Activity of Caffeic and Other Phenolic Acids. J. Agric. Food Chem. 2006, 54, 2932–2937. [Google Scholar] [CrossRef]
- Lemańska, K.; Szymusiak, H.; Tyrakowska, B.; Zieliński, R.; Soffers, A.E.M.F.; Rietjens, I.M.C.M. The influence of pH on antioxidant properties and the mechanism of antioxidant action of hydroxyflavones. Free Radic. Biol. Med. 2001, 31, 869–881. [Google Scholar] [CrossRef]
- Boulebd, H.; Carmena-Bargueño, M.; Pérez-Sánchez, H. Exploring the Antioxidant Properties of Caffeoylquinic and Feruloylquinic Acids: A Computational Study on Hydroperoxyl Radical Scavenging and Xanthine Oxidase Inhibition. Antioxidants 2023, 12, 1669. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R. A computational methodology for accurate predictions of rate constants in solution: Application to the assessment of primary antioxidant activity. J. Comput. Chem. 2013, 34, 2430–2445. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R. Computational strategies for predicting free radical scavengers’ protection against oxidative stress: Where are we and what might follow? Int. J. Quantum Chem. 2019, 119, e25665. [Google Scholar] [CrossRef]
- Alberto, M.E.; Russo, N.; Grand, A.; Galano, A. A physicochemical examination of the free radical scavenging activity of Trolox: Mechanism, kinetics and influence of the environment. Phys. Chem. Chem. Phys. 2013, 15, 4642–4650. [Google Scholar] [CrossRef]
- Boulebd, H. Radical scavenging behavior of butylated hydroxytoluene against oxygenated free radicals in physiological environments: Insights from DFT calculations. Int. J. Chem. Kinet. 2022, 54, 50–57. [Google Scholar] [CrossRef]
- Boulebd, H. Is cannabidiolic acid an overlooked natural antioxidant? Insights from quantum chemistry calculations. New J. Chem. 2022, 46, 162–168. [Google Scholar] [CrossRef]
- Frisch, M.J.; Trucks, G.W.; Schlegel, H.B.; Scuseria, G.E.; Robb, M.A.; Cheeseman, J.R.; Scalmani, G.; Barone, V.; Mennucci, B.; Petersson, G.A.; et al. Gaussian 09; Gaussian, Inc.: Wallingford, CT, USA, 2009. [Google Scholar]
- Zhao, Y.; Truhlar, D.G. The M06 suite of density functionals for main group thermochemistry, thermochemical kinetics, noncovalent interactions, excited states, and transition elements: Two new functionals and systematic testing of four M06-class functionals and 12 other functionals. Theor. Chem. Acc. 2008, 120, 215–241. [Google Scholar] [CrossRef]
- Galano, A.; Alvarez-Idaboy, J.R. Kinetics of radical-molecule reactions in aqueous solution: A benchmark study of the performance of density functional methods. J. Comput. Chem. 2014, 35, 2019–2026. [Google Scholar] [CrossRef]
- Zhao, Y.; Truhlar, D.G. How Well Can New-Generation Density Functionals Describe the Energetics of Bond-Dissociation Reactions Producing Radicals? J. Phys. Chem. A 2008, 112, 1095–1099. [Google Scholar] [CrossRef]
- Marenich, A.V.; Cramer, C.J.; Truhlar, D.G. Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions. J. Phys. Chem. B 2009, 113, 6378–6396. [Google Scholar] [CrossRef]
- Lu, T.; Chen, F. Multiwfn: A multifunctional wavefunction analyzer. J. Comput. Chem. 2012, 33, 580–592. [Google Scholar] [CrossRef]
- Humphrey, W.; Dalke, A.; Schulten, K. VMD: Visual molecular dynamics. J. Mol. Graph. 1996, 14, 33–38. [Google Scholar] [CrossRef]
- Galano, A.; Pérez-González, A.; Castañeda-Arriaga, R.; Muñoz-Rugeles, L.; Mendoza-Sarmiento, G.; Romero-Silva, A.; Ibarra-Escutia, A.; Rebollar-Zepeda, A.M.; León-Carmona, J.R.; Hernández-Olivares, M.A.; et al. Empirically Fitted Parameters for Calculating pKa Values with Small Deviations from Experiments Using a Simple Computational Strategy. J. Chem. Inf. Model. 2016, 56, 1714–1724. [Google Scholar] [CrossRef]
- Evans, M.G.; Polanyi, M. Some applications of the transition state method to the calculation of reaction velocities, especially in solution. Trans. Faraday Soc. 1935, 31, 875–894. [Google Scholar] [CrossRef]
- Eyring, H. The Activated Complex in Chemical Reactions. J. Chem. Phys. 1935, 3, 107–115. [Google Scholar] [CrossRef]
- Truhlar, D.G.; Hase, W.L.; Hynes, J.T. Current Status of Transition-State Theory. J. Phys. Chem. A 1983, 87, 2664–2682. [Google Scholar] [CrossRef]
- Pollak, E.; Pechukas, P. Symmetry numbers, not statistical factors, should be used in absolute rate theory and in Broensted relations. J. Am. Chem. Soc. 1978, 100, 2984–2991. [Google Scholar] [CrossRef]
- Fernández-Ramos, A.; Ellingson, B.A.; Meana-Pañeda, R.; Marques, J.M.; Truhlar, D.G. Symmetry numbers and chemical reaction rates. Theor. Chem. Acc. 2007, 118, 813–826. [Google Scholar] [CrossRef]
- Eckart, C. The penetration of a potential barrier by electrons. Phys. Rev. 1930, 35, 1303. [Google Scholar] [CrossRef]
- Collins, F.C.; Kimball, G.E. Diffusion-controlled reaction rates. J. Colloid Interface Sci. 1949, 4, 425–437. [Google Scholar] [CrossRef]
- Corchado, J.C.; Coitino, E.L.; Chuang, Y.-Y.; Fast, P.L.; Truhlar, D.G. Interpolated variational transition-state theory by mapping. J. Phys. Chem. A 1998, 102, 2424–2438. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Compound | Position | IF a (cm−1) | ΔG≠ b (kcal/mol) | κ c | k d (M−1 s−1) | Γ e (%) | koverall (M−1 s−1) |
|---|---|---|---|---|---|---|---|
| 1 | 3OH | −2026.7 | 15.4 | 44.7 | 1.35 × 103 | 83 | 1.64 × 103 |
| 4OH | −2440.8 | 17.8 | 360.9 | 1.78 × 102 | 11 | ||
| 10OH | −2040.1 | 18.1 | 109.4 | 3.70 × 101 | 2 | ||
| 11OH | −2434.1 | 18.4 | 401.6 | 7.02 × 101 | 4 | ||
| 2 | 3OH | −1880.6 | 11.9 | 9.7 | 9.75 × 104 | 93 | 9.75 × 105 |
| 4OH | −2467.8 | 15.4 | 127.6 | 3.91 × 103 | 4 | ||
| 10OH | −1901.2 | 15.93 | 39.7 | 5.18 × 102 | 0 | ||
| 11OH | −2307.9 | 15.42 | 102.2 | 3.16 × 103 | 3 |
| Comp. | Mechanism | State | ΔG≠ a (kcal/mol) | κ b | kapp c (M−1 s−1) | f d | kf e (M−1 s−1) | Γ f (%) | koverall (M−1 s−1) | |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | f-HAT | 3OH | H4A | 17.33 | 252.31 | 3.09 × 102 | 0.794 | 2.45 × 102 | 0 | 3.46 × 108 |
| 4OH | 19.72 | 1184.32 | 2.57 × 101 | 2.04 × 101 | 0 | |||||
| 10OH | 17.97 | 650.68 | 2.70 × 102 | 2.14 × 102 | 0 | |||||
| 11OH | 20.29 | 1361.70 | 1.13 × 101 | 8.97 × 100 | 0 | |||||
| f-HAT | 3OH | H3A | 16.16 | 166.10 | 1.47 × 103 | 0.186 | 2.73 × 102 | 0 | ||
| 4OH | 17.99 | 545.21 | 2.20 × 102 | 4.09 × 101 | 0 | |||||
| 10OH | - | - | 1.50 × 109 g | 2.79 × 108 | 81 | |||||
| SET | 5.74 | 16.25 h | 4.73 × 106 | 8.80 × 105 | 0 | |||||
| f-HAT | 3OH | H2A | - | - | 1.50 × 109 g | 0.020 | 3.00 × 107 | 9 | ||
| 10OH | - | - | 1.50 × 109 g | 3.70 × 107 | 9 | |||||
| SET | 8.34 | 16.46 h | 3.30 × 108 | 6.60 × 106 | 2 | |||||
| 2 | f-HAT | 3OH | H4A | 12.24 | 24.48 | 1.62 × 105 | 0.807 | 1.31 × 105 | 0 | 9.67 × 108 |
| 4OH | 14.96 | 205.97 | 1.39 × 104 | 1.12 × 104 | 0 | |||||
| 11OH | 13.34 | 72.39 | 7.51 × 104 | 6.06 × 104 | 0 | |||||
| 12OH | 15.47 | 407.47 | 1.14 × 104 | 9.20 × 103 | 0 | |||||
| f-HAT | 3OH | H3A | - | - | 1.60 × 109 g | 0.189 | 3.02 × 108 | 31 | ||
| 11OH | - | - | 1.60 × 109 g | 3.02 × 108 | 31 | |||||
| 12OH | - | - | 1.60 × 109 g | 3.02 × 108 | 31 | |||||
| SET | 6.09 | 16.28 h | 1.97 × 108 | 3.72 × 107 | 4 | |||||
| f-HAT | 3OH | H2A | - | - | 1.60 × 109 g | 0.004 | 6.40 × 106 | 1 | ||
| 12OH | - | - | 1.60 × 109 g | 6.50 × 106 | 1 | |||||
| SET | 2.20 | 13.18 h | 2.35 × 109 | 9.40 × 106 | 1 | |||||
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boulebd, H. Hydroperoxyl Radical Scavenging Activity of Bromophenols from Marine Red Alga Polysiphonia urceolata: Mechanistic Insights, Kinetic Analysis, and Influence of Physiological Media. Molecules 2025, 30, 1697. https://doi.org/10.3390/molecules30081697
Boulebd H. Hydroperoxyl Radical Scavenging Activity of Bromophenols from Marine Red Alga Polysiphonia urceolata: Mechanistic Insights, Kinetic Analysis, and Influence of Physiological Media. Molecules. 2025; 30(8):1697. https://doi.org/10.3390/molecules30081697
Chicago/Turabian StyleBoulebd, Houssem. 2025. "Hydroperoxyl Radical Scavenging Activity of Bromophenols from Marine Red Alga Polysiphonia urceolata: Mechanistic Insights, Kinetic Analysis, and Influence of Physiological Media" Molecules 30, no. 8: 1697. https://doi.org/10.3390/molecules30081697
APA StyleBoulebd, H. (2025). Hydroperoxyl Radical Scavenging Activity of Bromophenols from Marine Red Alga Polysiphonia urceolata: Mechanistic Insights, Kinetic Analysis, and Influence of Physiological Media. Molecules, 30(8), 1697. https://doi.org/10.3390/molecules30081697