
Experimental and Theoretical Study of the Synthesis of a Deep Eutectic Solvent Based on Protonated Caffeine, Ethylene Glycol, and ZnCl2

 , and
, and
Abstract
1. Introduction
2. Results and Discussion
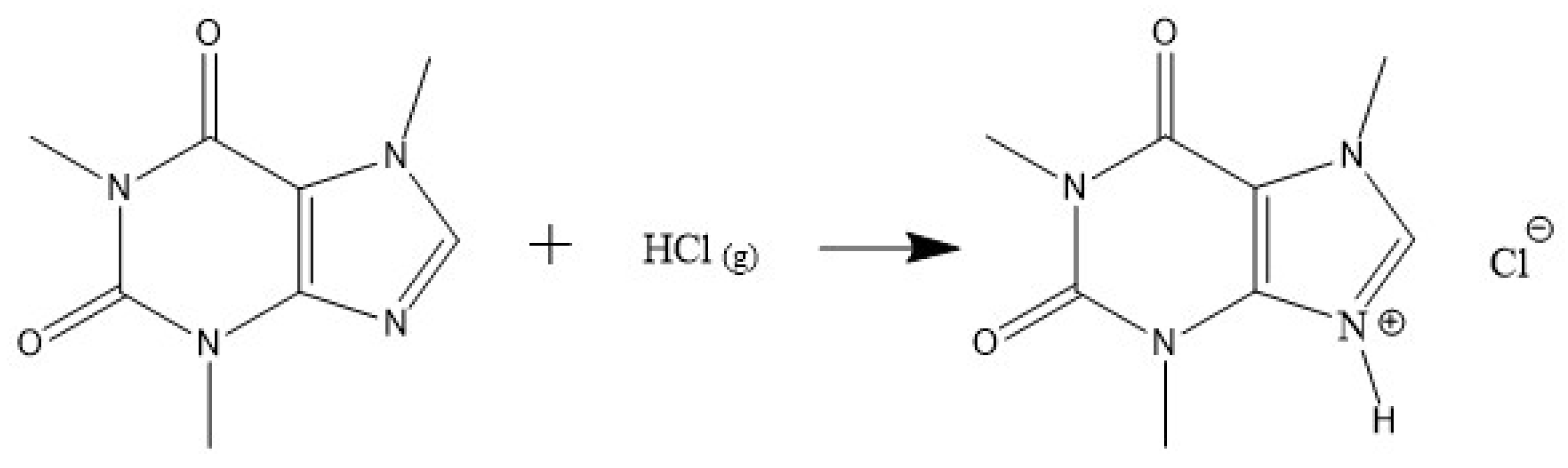
2.1. Activation of Caffeine’s Imidazole Ring
2.2. Synthesis of Protonated Caffeine-Based DES
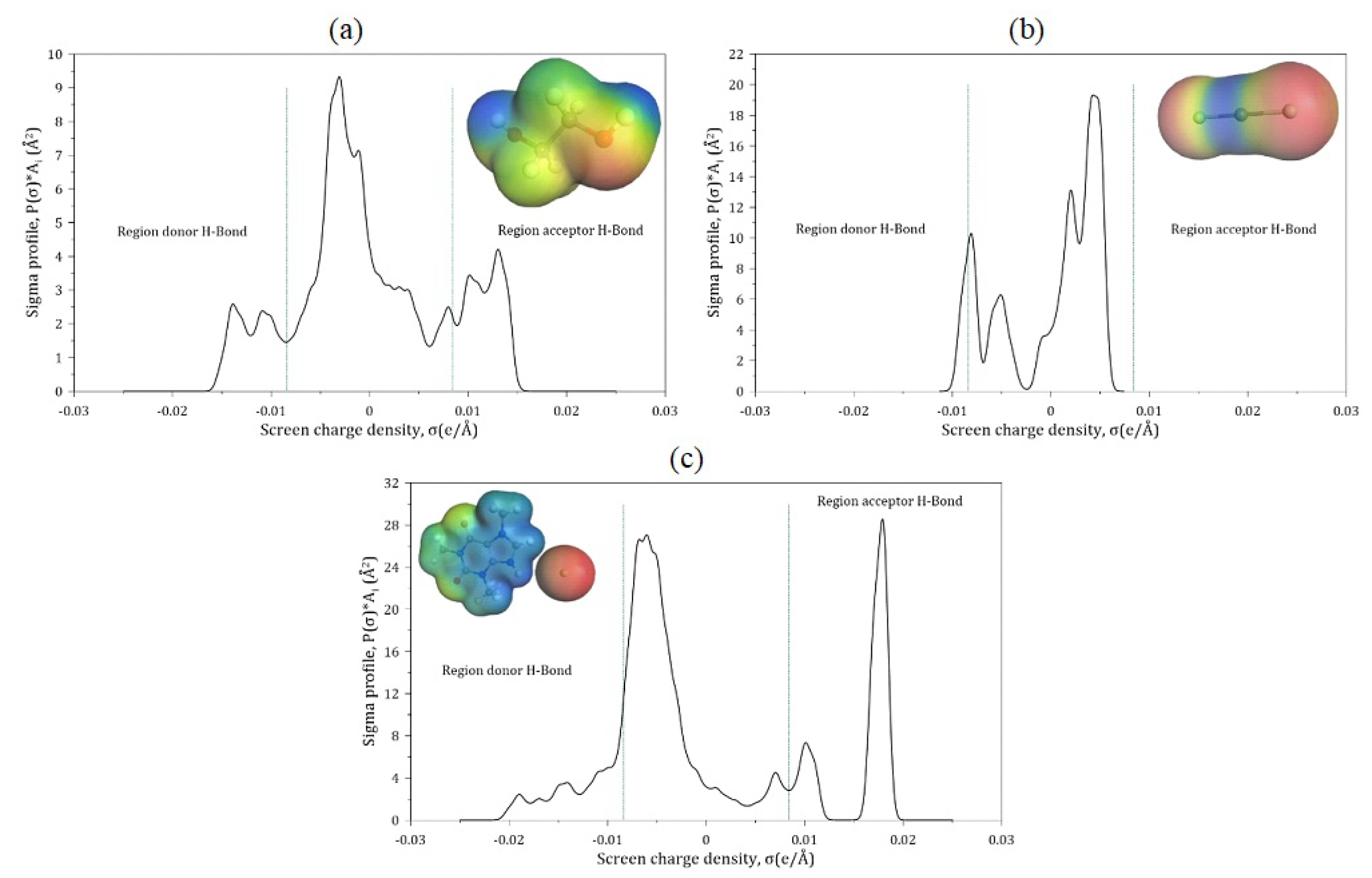
2.3. Sigma Profile
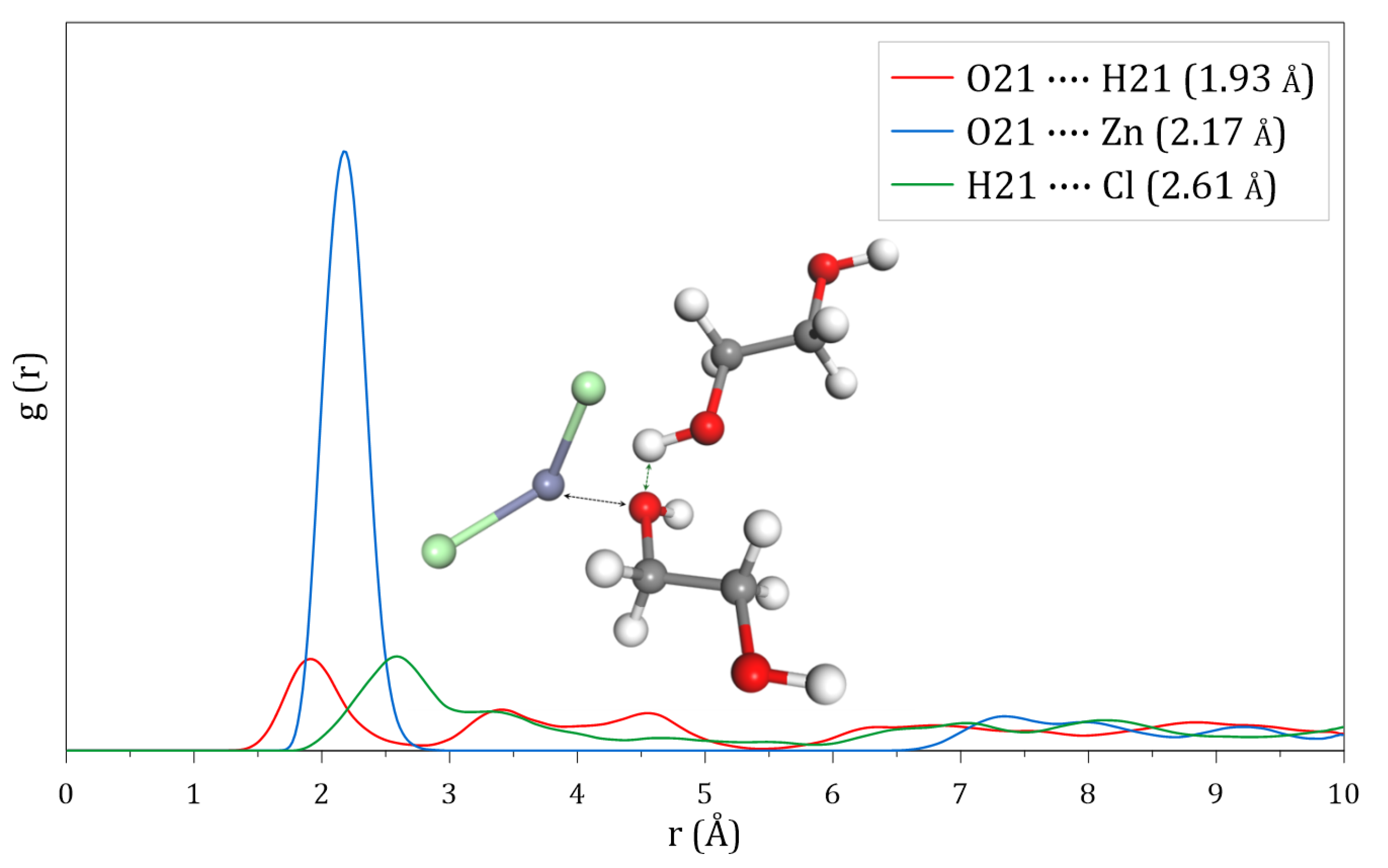
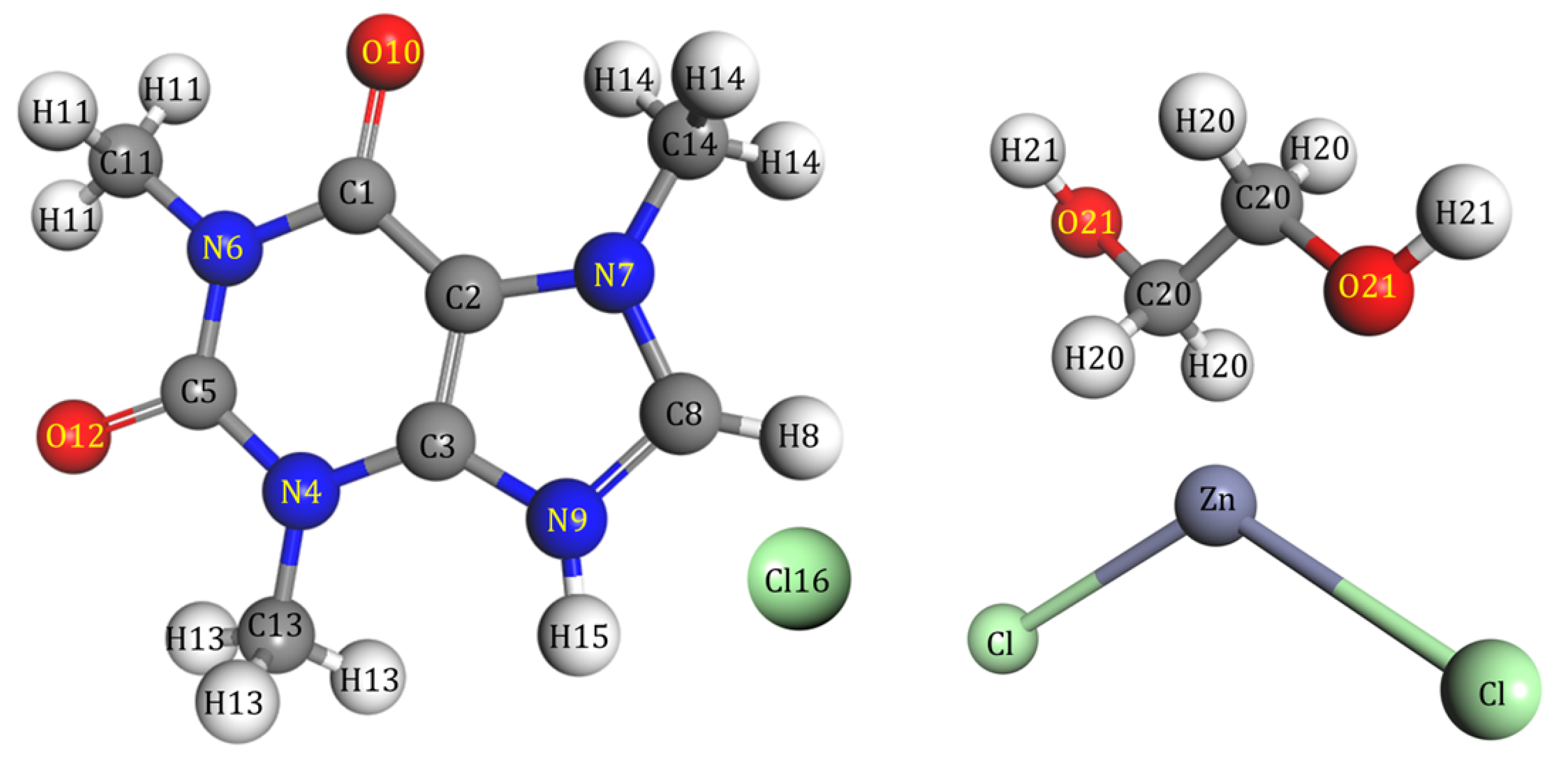
2.4. Radial Distribution Functions
3. Materials and Methods
3.1. Activation of Caffeine’s Imidazole Ring
3.1.1. Chemical Reagents
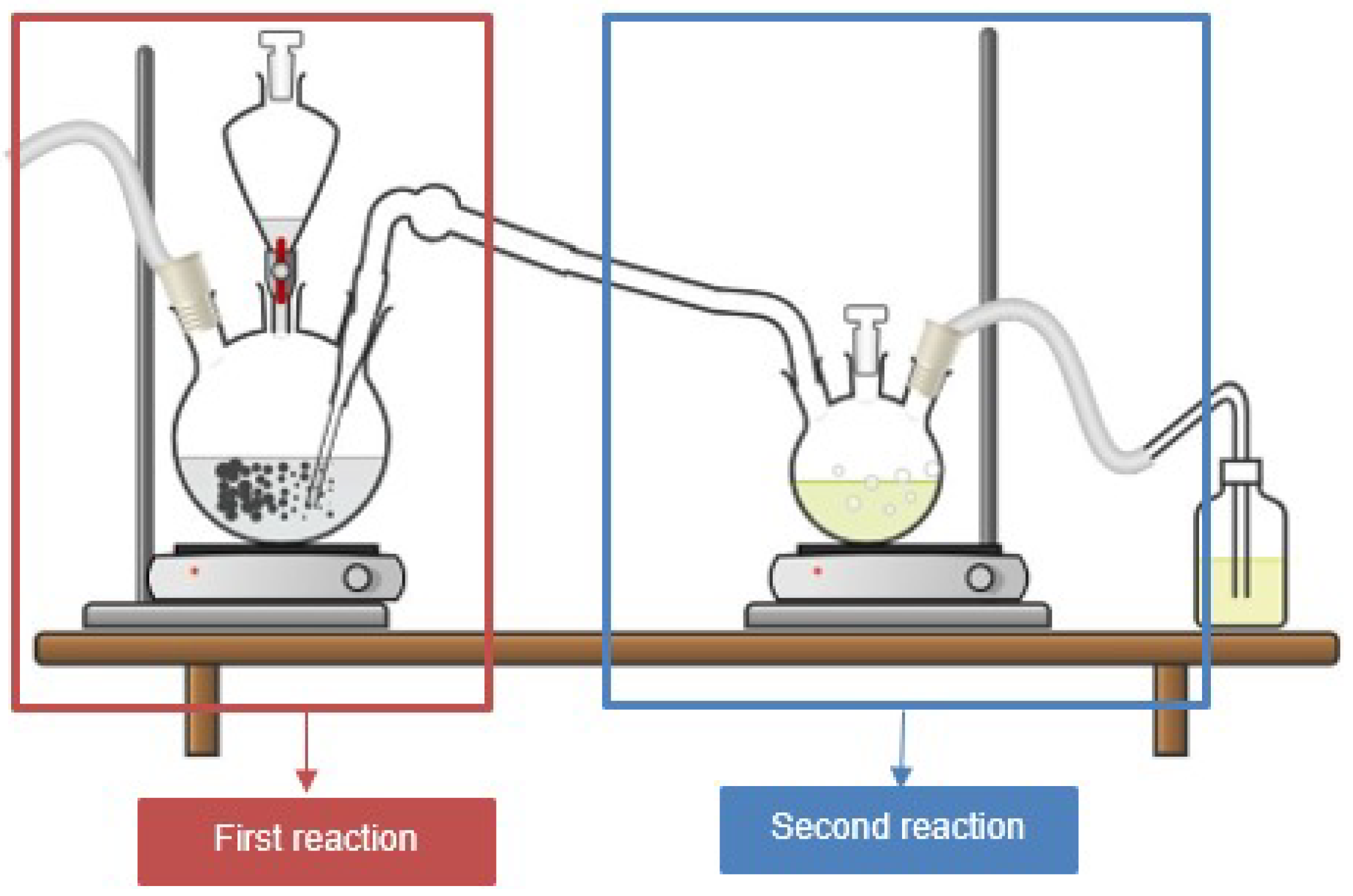
3.1.2. Reaction Setup and Procedure
- First stage, production of anhydrous hydrogen chloride: Anhydrous hydrogen chloride was generated by the reaction between sodium chloride and sulfuric acid, as shown in Figure 9. The system consisted of a round-bottomed flask containing sodium chloride, with sulfuric acid added dropwise, while the mixture was maintained at a constant temperature of C using a heating mantle, as reported by Groover [45]. An excess of hydrogen chloride (5 molar equivalents) was bubbled into the ethanol solution containing caffeine to drive the equilibrium of the protonation reaction toward product formation, ensuring complete protonation of the caffeine.
- Second stage, protonation reaction: The protonation reaction was carried out at C (Figure 10) with caffeine dissolved in anhydrous ethanol. A total of 110 mL of anhydrous ethanol was used to fully dissolve the caffeine at this temperature. The gaseous hydrogen chloride produced in the first stage was continuously bubbled into the ethanol solution, leading to the protonation of caffeine, which was indicated by the precipitation of a white solid after 1 h of reaction.
- Purification of caffeine hydrochloride: The solid product, caffeine hydrochloride (CafCl), was purified through rotary evaporation at C and 200 mbar to eliminate excess ethanol. The resulting solid was subsequently dried in a vacuum oven at C for 24 h to ensure complete removal of residual solvents. The final product yield was 88.4%.
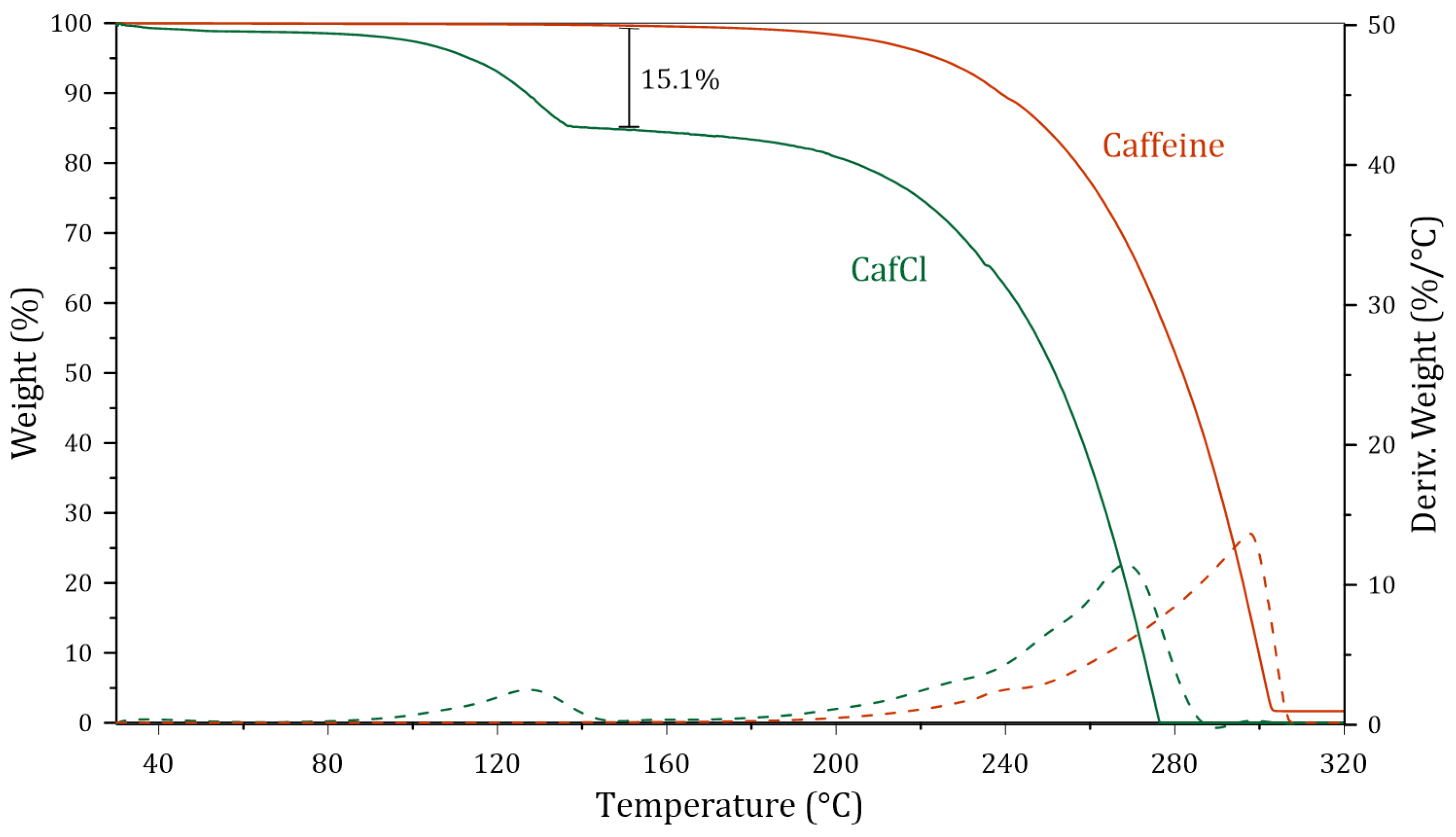
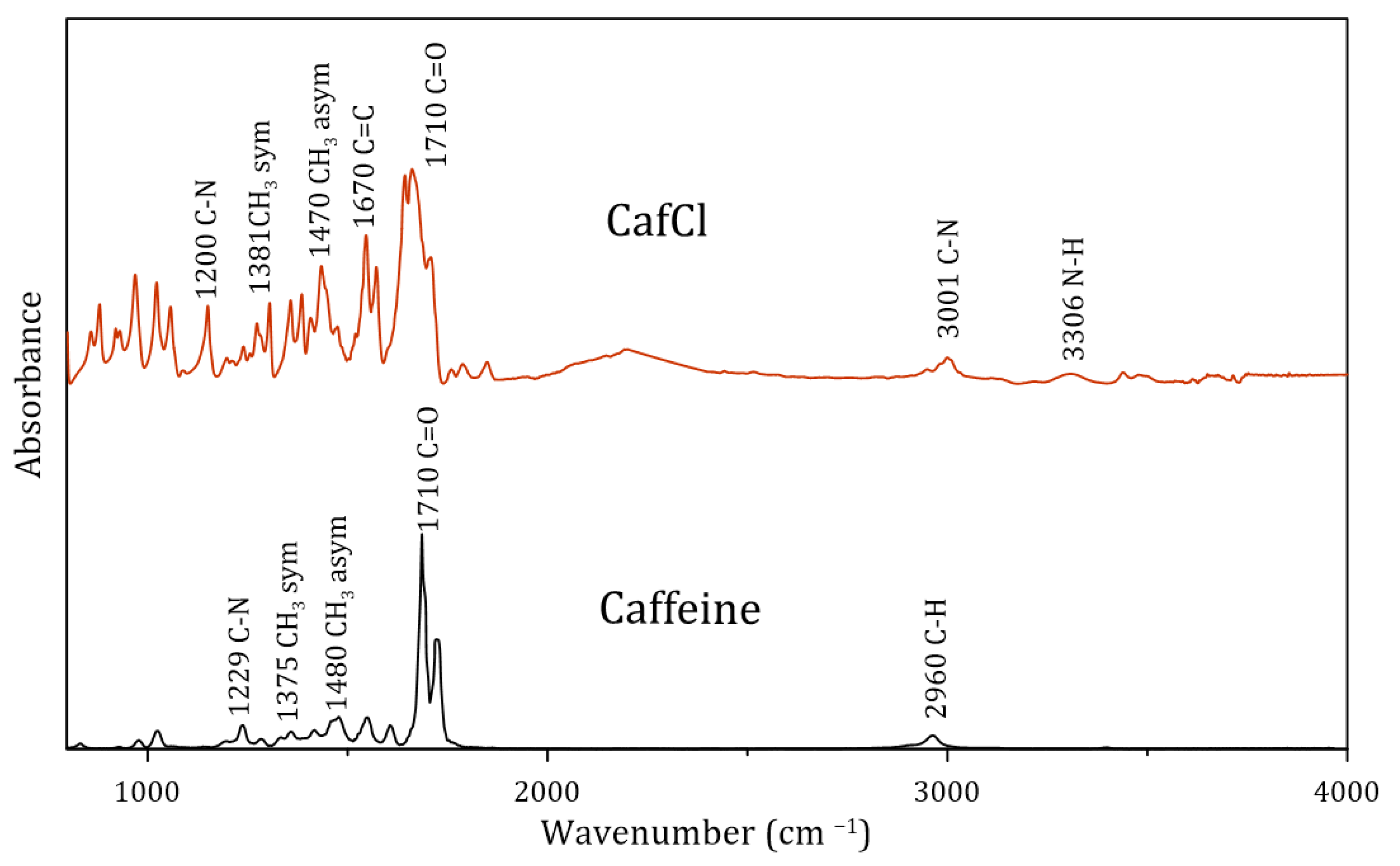
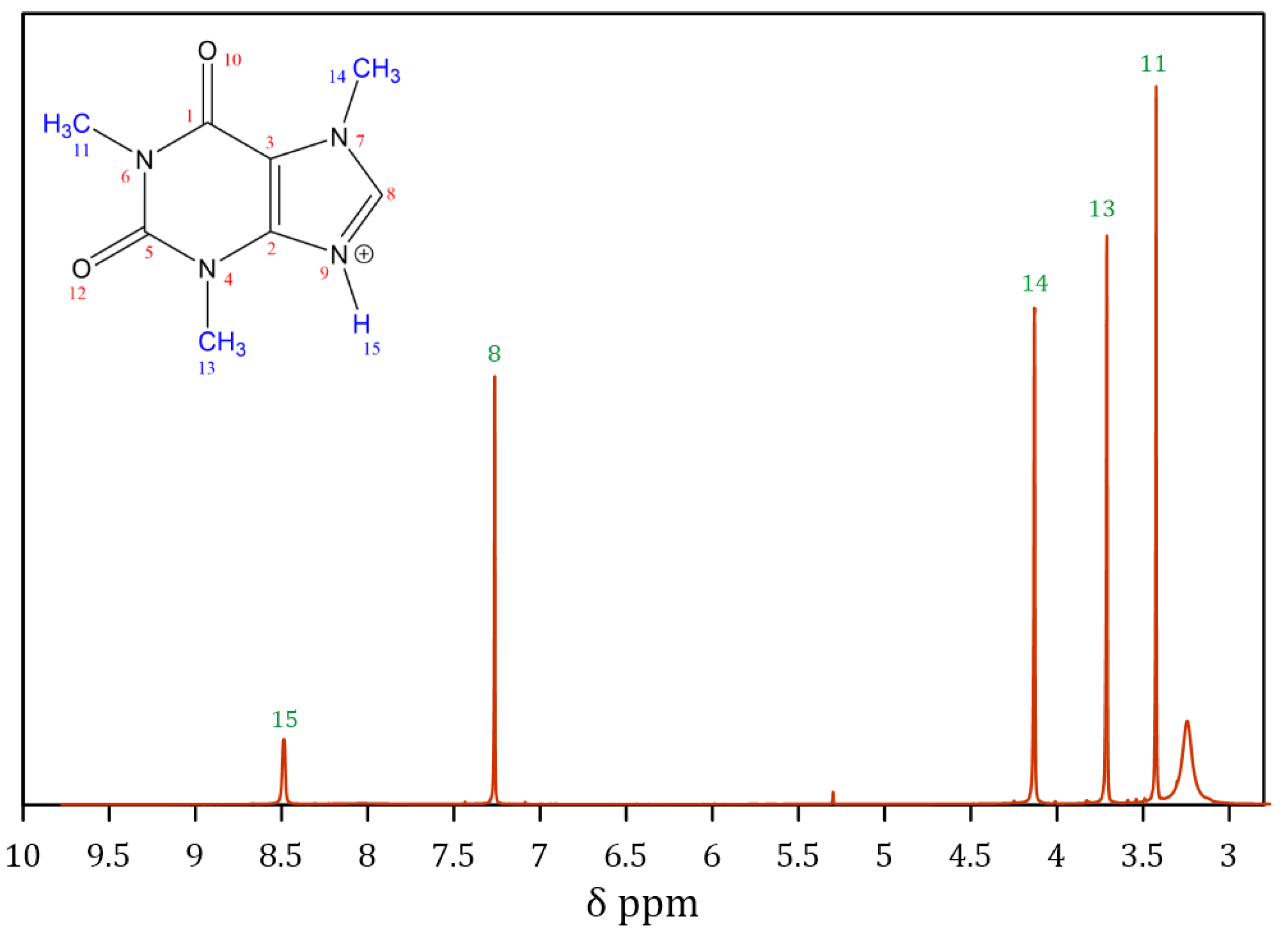
3.1.3. Characterization of Protonated Caffeine (CafCl)
3.2. Synthesis of Protonated Caffeine-Based DES
3.2.1. Chemical Reagents
3.2.2. Characterization of the Synthetized DES
3.3. Computational Detail
3.3.1. COSMO-RS Calculation
3.3.2. Ab Initio Molecular Dynamics (AIMD)
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Clarke, C.J.; Tu, W.C.; Levers, O.; Bröhl, A.; Hallett, J.P. Green and Sustainable Solvents in Chemical Processes. Chem. Rev. 2018, 118, 747–800. [Google Scholar] [CrossRef] [PubMed]
- Nanda, B.; Sailaja, M.; Mohapatra, P.; Pradhan, R.K.; Nanda, B.B. Green solvents: A suitable alternative for sustainable chemistry. Mater. Today: Proc. 2021, 47, 1234–1240. [Google Scholar] [CrossRef]
- Bains, W.; Petkowski, J.J.; Seager, S. Alternative Solvents for Life: Framework for Evaluation, Current Status and Future Research. 2022. Available online: https://arxiv.org/ftp/arxiv/papers/2401/2401.07296.pdf (accessed on 16 April 2024).
- Armenta, S.; Esteve-Turrillas, F.A.; Garrigues, S.; de la Guardia, M. Alternative green solvents in sample preparation. Green Anal. Chem. 2022, 1, 100007. [Google Scholar] [CrossRef]
- Lomba, L.; García, C.B.; Ribate, M.P.; Giner, B.; Zuriaga, E. Applications of deep eutectic solvents related to health, synthesis, and extraction of natural based chemicals. Appl. Sci. 2021, 11, 10156. [Google Scholar] [CrossRef]
- Gurkan, B.E.; Maginn, E.J.; Pentzer, E.B. Deep eutectic solvents: A new class of versatile liquids. Am. Chem. Soc. 2020, 124, 11313–11315. [Google Scholar] [CrossRef]
- Florindo, C.; Branco, L.C.; Marrucho, I.M. Quest for Green-Solvent Design: From Hydrophilic to Hydrophobic (Deep) Eutectic Solvents. ChemSusChem 2019, 12, 1549–1559. [Google Scholar] [CrossRef]
- Yang, Z. Natural Deep Eutectic Solvents and Their Applications in Biotechnology. In Advances in Biochemical Engineering/Biotechnology; Springer: Berlin/Heidelberg, Germany, 2019; Volume 168, pp. 31–59. [Google Scholar] [CrossRef]
- Francisco, M.; Van Den Bruinhorst, A.; Kroon, M.C. Low-transition-temperature mixtures (LTTMs): A new generation of designer solvents. Angew. Chem. Int. Ed. 2013, 52, 3074–3085. [Google Scholar] [CrossRef]
- Bösmann, A.; Datsevich, L.; Jess, A.; Lauter, A.; Schmitz, C.; Wasserscheid, P. Deep desulfurization of diesel fuel by extraction with ionic liquids. Chem. Commun. 2001, 23, 2494–2495. [Google Scholar] [CrossRef]
- Kumar, A.A.P.; Banerjee, T. Thiophene separation with ionic liquids for desulphurization: A quantum chemical approach. Fluid Phase Equilib 2009, 278, 1–8. [Google Scholar] [CrossRef]
- Nie, Y.; Li, C.; Meng, H.; Wang, Z. N,N-dialkylimidazolium dialkylphosphate ionic liquids: Their extractive performance for thiophene series compounds from fuel oils versus the length of alkyl group. Fuel Process. Technol. 2008, 89, 978–983. [Google Scholar] [CrossRef]
- Ghandi, K. A Review of Ionic Liquids, Their Limits and Applications. Green Sustain. Chem. 2014, 4, 44–53. [Google Scholar] [CrossRef]
- Lima, F.; Branco, L.C.; Silvestre, A.J.D.; Marrucho, I.M. Deep desulfurization of fuels: Are deep eutectic solvents the alternative for ionic liquids? Fuel 2021, 293, 120297. [Google Scholar] [CrossRef]
- United States Environmental Protection Agency. Control of Air Pollution from Motor Vehicles: Tier 3 Motor Vehicle Emission and Fuel Standards. 2014. Available online: http://www.epa.gov/oar/ (accessed on 16 April 2024).
- Gano, Z.S.; Mjalli, F.S.; Al-Wahaibi, T.; Al-Wahaibi, Y.; AlNashef, I.M. Extractive desulfurization of liquid fuel with FeCl3-based deep eutectic solvents: Experimental design and optimization by central-composite design. Chem. Eng. Process. Process Intensif. 2015, 93, 10–20. [Google Scholar] [CrossRef]
- Gano, Z.S.; Mjalli, F.S.; Al-Wahaibi, T.; Al-Wahaibi, Y.; Alnashef, I.M. Solubility of thiophene and dibenzothiophene in anhydrous FeCl3- and ZnCl2-based deep eutectic solvents. Ind. Eng. Chem. Res. 2014, 53, 6815–6823. [Google Scholar] [CrossRef]
- Shu, C.; Sun, T. Extractive desulfurisation of gasoline with tetrabutyl ammonium chloride-based deep eutectic solvents. Sep. Sci. Technol. 2016, 51, 1336–1343. [Google Scholar] [CrossRef]
- Warrag, S.E.; Adeyemi, I.; Rodriguez, N.R.; Nashef, I.M.; van Sint Annal, M.; Kroon, M.C.; Peters, C.J. Effect of the Type of Ammonium Salt on the Extractive Desulfurization of Fuels Using Deep Eutectic Solvents. J. Chem. Eng. Data 2018, 63, 1088–1095. [Google Scholar] [CrossRef]
- Thermo Fisher Scientific. Fisher Scientific. Available online: https://www.fishersci.es/shop/products/caffeine-99-thermo-scientific/11438643#?keyword=caffeine (accessed on 8 January 2025).
- Li, C.; Li, D.; Zou, S.; Li, Z.; Yin, J.; Wang, A.; Cui, Y.; Yao, Z.; Zhao, Q. Extraction desulfurization process of fuels with ammonium based deep eutectic solvents. Green Chem. 2013, 15, 2793–2799. [Google Scholar] [CrossRef]
- Verma, P.; Srivastava, A.; Srivastava, K.; Tandon, P.; Shimpi, M. Molecular Structure, Spectral Investigations, Hydrogen Bonding Interactions and Reactivity-Property Relationship of Caffeine-Citric Acid Cocrystal by Experimental and DFT Approach. Front. Chem. 2021, 9, 708538. [Google Scholar] [CrossRef]
- Jones, R.R.; Hooper, D.C.; Zhang, L.; Wolverson, D.; Valev, V.K. Raman Techniques: Fundamentals and Frontiers; Springer: New York, NY, USA, 2019. [Google Scholar] [CrossRef]
- Abiya, A.; Ilakiya, A.; Hemalakshmi, S.; Ramesh, S. Obtaining and Investigate of Caffeine from Contrasting Tea Specimen Using Liquid-Liquid Extraction. In Proceedings of the 2022 Conference on Chemical Analysis, Angkor Wat, Cambodia, 6–10 November 2022. [Google Scholar]
- Salami, M.; Ezabadi, A. Synthesis of the nano-magnetic ionic liquid based on caffeine and its catalytic application in the synthesis of xanthenes. In Research on Chemical Intermediates; Springer: Dordrecht, The Netherlands, 2020; Volume 46, pp. 4611–4626. [Google Scholar] [CrossRef]
- Hitachi High-Tech. Thermal Analysis of Caffeine. Available online: https://hha.hitachi-hightech.com/assets/uploads/assets/uploads/documents/Thermal_Analysis_of_Caffeine.pdf (accessed on 20 January 2025).
- Rajam, K.; Rajendran, S.; Banu, N.N. Effect of caffeine-Zn2+ system in preventing corrosion of carbon steel in well water. J. Chem. 2013, 2013, 521951. [Google Scholar] [CrossRef]
- Butt, S.; Hasan, S.M.F.; Hassan, M.M.; Alkharfy, K.M.; Neau, S.H. Directly compressed rosuvastatin calcium tablets that offer hydrotropic and micellar solubilization for improved dissolution rate and extent of drug release. Saudi Pharm. J. 2019, 27, 619–628. [Google Scholar] [CrossRef]
- Sitkowski, J.; Stefaniak, L.; Nicol, I.L.; Martin, M.L.; Martin, G.J.; Webb, G.A. Complete assignments of the 1H, 13C and 15N NMR spectra of caffeine. J. Magn. Reson. 1995, 51, 839–841. [Google Scholar] [CrossRef]
- Colherinhas, G.; Ludwig, V.; da Costa Ludwig, Z.M. GIAO-NMR spectroscopy of the xanthine’s structures in water solution using S-MC/QM methodology: An evaluation of the DFT-functionals’ efficiency. J. Mol. Liq. 2022, 347, 117955. [Google Scholar] [CrossRef]
- Li, J.G.; Hu, Y.F.; Peng, X.M.; Zhang, X.M. Study of physicochemical properties of FeCl3/[C4mim][Cl] ionic liquids. J. Chem. Thermodyn. 2016, 97, 277–281. [Google Scholar] [CrossRef]
- Sharma, S. Raman study of ferric chloride hexahydrate and ferric chloride hexadeutrate in crystalline, molten, and glassy states. J. Non-Cryst. Solids 1974, 15, 83–95. [Google Scholar] [CrossRef]
- Li, Q.; Dong, Y.; Hammond, K.D.; Wan, C. Revealing the role of hydrogen bonding interactions and supramolecular complexes in lignin dissolution by deep eutectic solvents. J. Mol. Liq. 2021, 344, 117779. [Google Scholar] [CrossRef]
- Krasil’nikov, V.N.; Tyutyunnik, A.P.; Zhukov, V.P.; Baklanova, I.V.; Gyrdasova, O.I.; Chulkov, E.V. Zinc glycolate Zn(OCH2CH2O): Synthesis and structure, spectral and optical properties, electronic structure and chemical bonding. J. Alloy. Compd. 2022, 924, 166320. [Google Scholar] [CrossRef]
- Calado, M.S.; Branco, A.S.H.; Najdanovic-Visak, V.; Visak, Z.P. Solubility of high-value compounds in environmentally friendly solvents-liquid poly(ethylene glycol) and ionic liquids: Experimental study and thermodynamic analysis. J. Chem. Thermodyn. 2014, 70, 154–159. [Google Scholar] [CrossRef]
- Andrejević, T.P.; Warżajtis, B.; Glišić, B.Đ.; Vojnovic, S.; Mojicevic, M.; Stevanović, N.L.; Nikodinovic-Runic, J.; Rychlewska, U.; Djuran, M.I. Zinc(II) complexes with aromatic nitrogen-containing heterocycles as antifungal agents: Synergistic activity with clinically used drug nystatin. J. Inorg. Biochem. 2020, 208, 111089. [Google Scholar] [CrossRef]
- Tfaili, S.; Gobinet, C.; Josse, G.; Angiboust, J.F.; Baillet, A.; Manfait, M.; Piot, O. Vibrational spectroscopies for the analysis of cutaneous permeation: Experimental limiting factors identified in the case of caffeine penetration. Anal. Bioanal. Chem. 2013, 405, 1325–1332. [Google Scholar] [CrossRef]
- Zareef, M.; Hassan, M.M.; Arslan, M.; Ahmad, W.; Ali, S.; Ouyang, Q.; Li, H.; Wu, X.; Chen, Q. Rapid prediction of caffeine in tea based on surface-enhanced Raman spectroscopy coupled multivariate calibration. Microchem. J. 2020, 159, 105431. [Google Scholar] [CrossRef]
- Baranska, M.; Proniewicz, L.M. Raman mapping of caffeine alkaloid. Vib. Spectrosc. 2008, 48, 153–157. [Google Scholar] [CrossRef]
- Yang, Q.; Wu, L.; Shi, C.; Wu, X.; Chen, X.; Wu, W.; Yang, H.; Wang, Z.; Zeng, L.; Peng, Y. Qualitative and Quantitative Analysis of Caffeine in Medicines by Terahertz Spectroscopy Using Machine Learning Method. IEEE Access 2021, 9, 140008–140021. [Google Scholar] [CrossRef]
- Kupenko, I.; Dubrovinsky, L.; Dmitriev, V.; Dubrovinskaia, N. In situ Raman spectroscopic study of the pressure induced structural changes in ammonia borane. J. Chem. Phys. 2012, 137, 074506. [Google Scholar] [CrossRef]
- Jahanbakhsh-Bonab, P.; Khoshnazar, Z.; Jahanbin Sardroodi, J.; Heidaryan, E. A computational probe into the physicochemical properties of cyclodextrin-based deep eutectic solvents for extraction processes. Carbohydr. Polym. Technol. Appl. 2024, 8, 100596. [Google Scholar] [CrossRef]
- Ghareh Bagh, F.S.; Shahbaz, K.; Mjalli, F.S.; Hashim, M.A.; Alnashef, I.M. Zinc (II) Chloride-Based Deep Eutectic Solvents for Application as Electrolytes: Preparation and Characterization. J. Mol. Liq. 2015, 204, 76–83. [Google Scholar] [CrossRef]
- Abbott, A.P.; Barron, J.C.; Ryder, K.S.; Wilson, D. Eutectic-Based Ionic Liquids with Metal-Containing Anions and Cations. Chem.–Eur. J. 2007, 13, 6495–6501. [Google Scholar] [CrossRef]
- Jha, D.; Maheshwari, P.; Singh, Y.; Haider, M.B.; Kumar, R.; Balathanigaimani, M.S. A comparative review of extractive desulfurization using designer solvents: Ionic liquids & deep eutectic solvents. J. Oil Gas Energy 2023, 110, 101313. [Google Scholar] [CrossRef]
- Pye, C.C.; Ziegler, T.; van Lenthe, E.; Louwen, J.N. An implementation of the conductor-like screening model of solvation within the Amsterdam density functional package—Part II. COSMO for real solvents. Can. J. Chem. 2009, 87, 790–797. [Google Scholar] [CrossRef]
- Echeverry-Vargas, L.; Estrada, D.; Gutierrez, L. Molecular Dynamics Simulations of the Interactions between a Hydrolyzed Polyacrylamide with the Face and Edge Surfaces of Molybdenite. Polymers 2022, 14, 3680. [Google Scholar] [CrossRef]
- Lemaoui, T.; Darwish, A.; Hammoudi, N.; Hatab, F.; Attoui, A.; Alnashef, I.; Benguerba, Y. Prediction of Electrical Conductivity of Deep Eutectic Solvents Using COSMO-RS Sigma Profiles as Molecular Descriptors: A Quantitative Structure-Property Relationship Study. Ind. Eng. Chem. Res. 2020, 59, 13343–13354. [Google Scholar] [CrossRef]
- Delley, B. An All-Electron Numerical Method for Solving the Local Density Functional for Polyatomic Molecules. J. Chem. Phys. 1990, 92, 508–517. [Google Scholar] [CrossRef]
- Delley, B. From molecules to solids with the DMol3 approach. J. Chem. Phys. 2000, 113, 7756–7764. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 1993, 98, 5648–5652. [Google Scholar] [CrossRef]
- Vosko, S.H.; Wilk, L.; Nusair, M. Accurate spin-dependent electron liquid correlation energies for local spin density calculations: A critical analysis. Can. J. Phys. 1980, 58, 1200–1211. [Google Scholar] [CrossRef]
- Mullins, E.; Oldland, R.; Liu, Y.A.; Wang, S.; Sandler, A.A.; Chen, C.C.; Zwolak, M.; Seavey, K. Sigma-profile database for using COSMO-based thermodynamic methods. Ind. Eng. Chem. Res. 2006, 45, 4389–4415. [Google Scholar] [CrossRef]
- Toikka, A.M.; Petrov, A.V. Comparative analysis of molecular interactions in quaternary fluid system performed by classical and ab initio molecular dynamics. Mendeleev Commun. 2023, 33, 413–415. [Google Scholar] [CrossRef]
- Becke, A.D. Density-functional exchange-energy approximation with correct asymptotic behavior. Phys. Rev. 1988, 38, 3098–3100. [Google Scholar] [CrossRef]
- Nosé, S. A molecular dynamics method for simulations in the canonical ensemble. Mol. Phys. 1984, 52, 255–268. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Proton | Theoretical Caffeine | Literature Caffeine | Experimental CafCl | Literature CafCl | Experimental Peak Integration |
|---|---|---|---|---|---|
| 8 | 7.5 | 7.58 | 7.27 | 7.99 | 1.0 |
| 11 | 3.48 | 3.37 | 3.27 | 3.13 | 3.10 |
| 13 | 3.51 | 3.55 | 3.71 | 3.33 | 3.10 |
| 14 | 4.0 | 4.01 | 4.13 | 3.82 | 3.04 |
| 15 | 8.5 | 11.27 | 0.99 |
| HBD | HBA | Molar Ratio | Observations About CafCl Incorporation |
|---|---|---|---|
| Ethylene glycol (ETG) | Choline chloride (ChCl) | 2:1 | Partial incorporation with high viscosity |
| Benzoic acid | Choline chloride | 1:1 | Crystal formation, partial incorporation with high viscosity |
| Benzamide | Choline chloride | 2:1 | Partial incorporation with high viscosity |
| Ethylene glicol | Tetraethylammonium bromide | 2:1 | Complete incorporation with medium viscosity |
| Ethylene glicol | Hydrated iron chloride | 3:1 | Partial incorporation with high viscosity |
| Ethylene glicol | Zinc chloride | 2:1 | Complete incorporation with manageable viscosity |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Benavides-Maya, L.S.; Torres-Perdomo, M.F.; Ocampo-Carmona, L.M.; Echeverry-Vargas, L. Experimental and Theoretical Study of the Synthesis of a Deep Eutectic Solvent Based on Protonated Caffeine, Ethylene Glycol, and ZnCl2. Molecules 2025, 30, 1557. https://doi.org/10.3390/molecules30071557
Benavides-Maya LS, Torres-Perdomo MF, Ocampo-Carmona LM, Echeverry-Vargas L. Experimental and Theoretical Study of the Synthesis of a Deep Eutectic Solvent Based on Protonated Caffeine, Ethylene Glycol, and ZnCl2. Molecules. 2025; 30(7):1557. https://doi.org/10.3390/molecules30071557
Chicago/Turabian StyleBenavides-Maya, Laura Sofía, Manuel Felipe Torres-Perdomo, Luz M. Ocampo-Carmona, and Luver Echeverry-Vargas. 2025. "Experimental and Theoretical Study of the Synthesis of a Deep Eutectic Solvent Based on Protonated Caffeine, Ethylene Glycol, and ZnCl2" Molecules 30, no. 7: 1557. https://doi.org/10.3390/molecules30071557
APA StyleBenavides-Maya, L. S., Torres-Perdomo, M. F., Ocampo-Carmona, L. M., & Echeverry-Vargas, L. (2025). Experimental and Theoretical Study of the Synthesis of a Deep Eutectic Solvent Based on Protonated Caffeine, Ethylene Glycol, and ZnCl2. Molecules, 30(7), 1557. https://doi.org/10.3390/molecules30071557