Insights into the Antimicrobial Mechanisms of a Scorpion Defensin on Staphylococcus aureus Using Transcriptomic and Proteomic Analyses

 , ,
, , 
Abstract
1. Introduction
2. Results
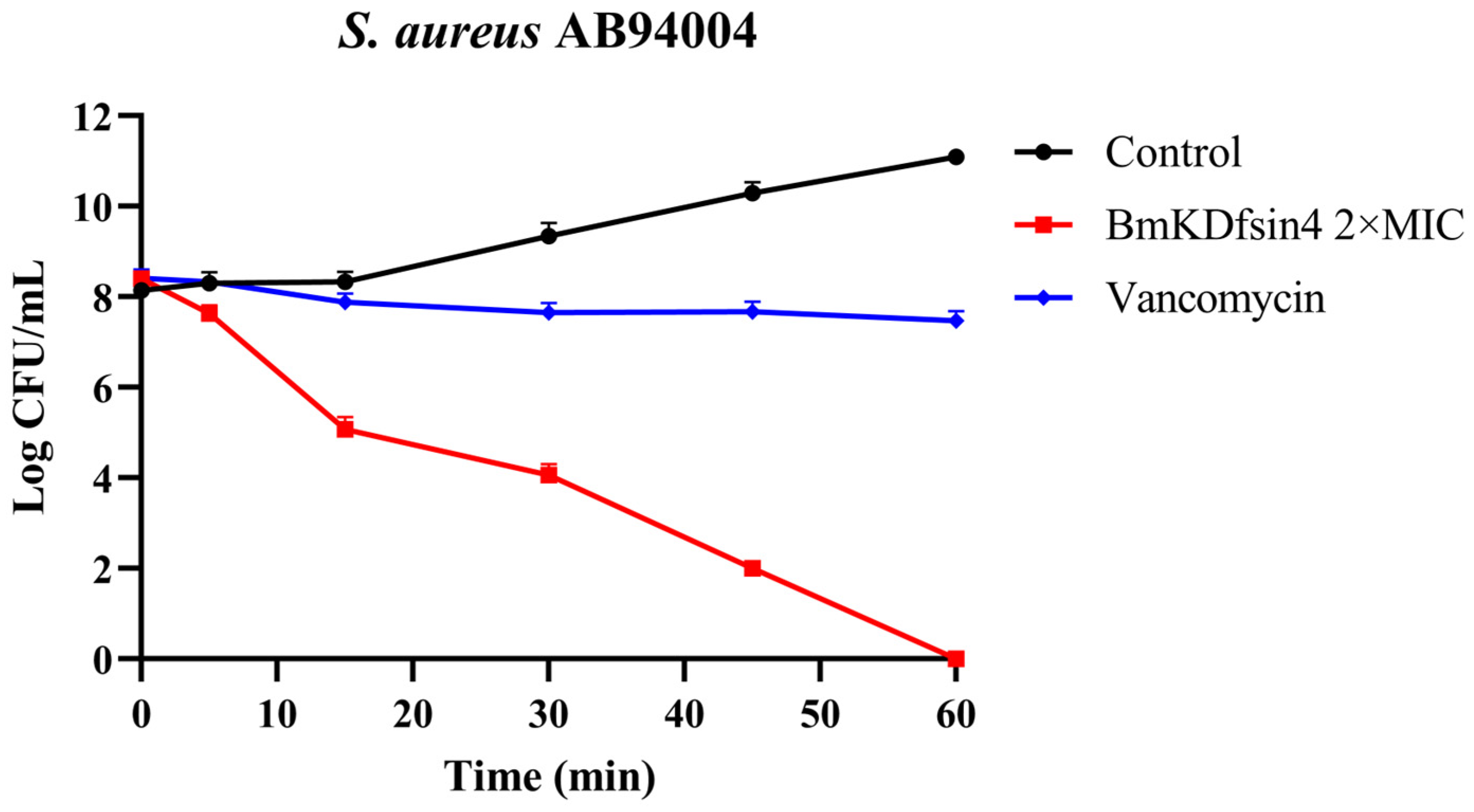
2.1. Bactericidal Effect of BmKDfsin4 on S. aureus AB94004
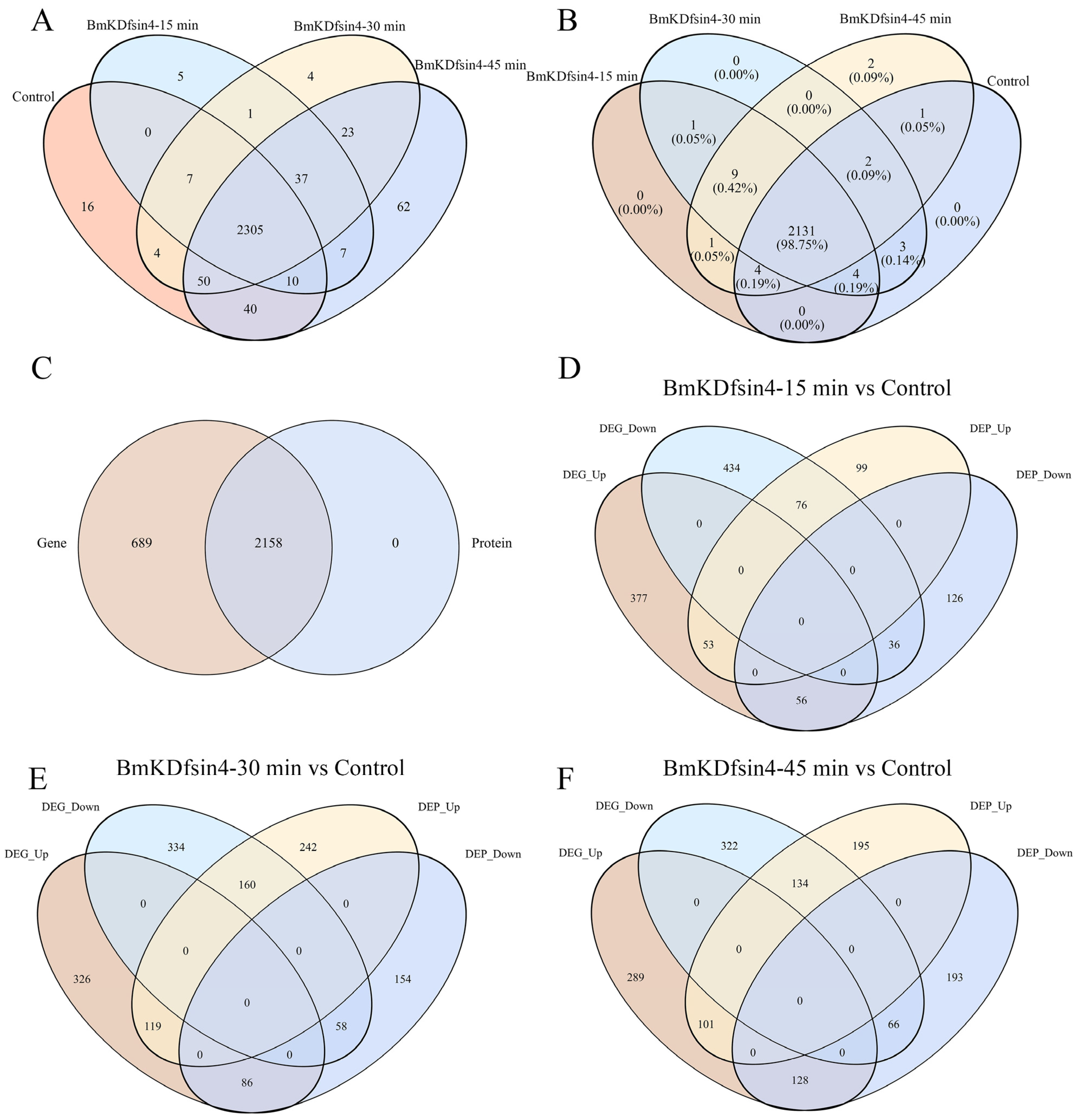
2.2. Global Transcriptomic and Proteomic Analyses of BmKDfsin4-Treated S. aureus AB94004
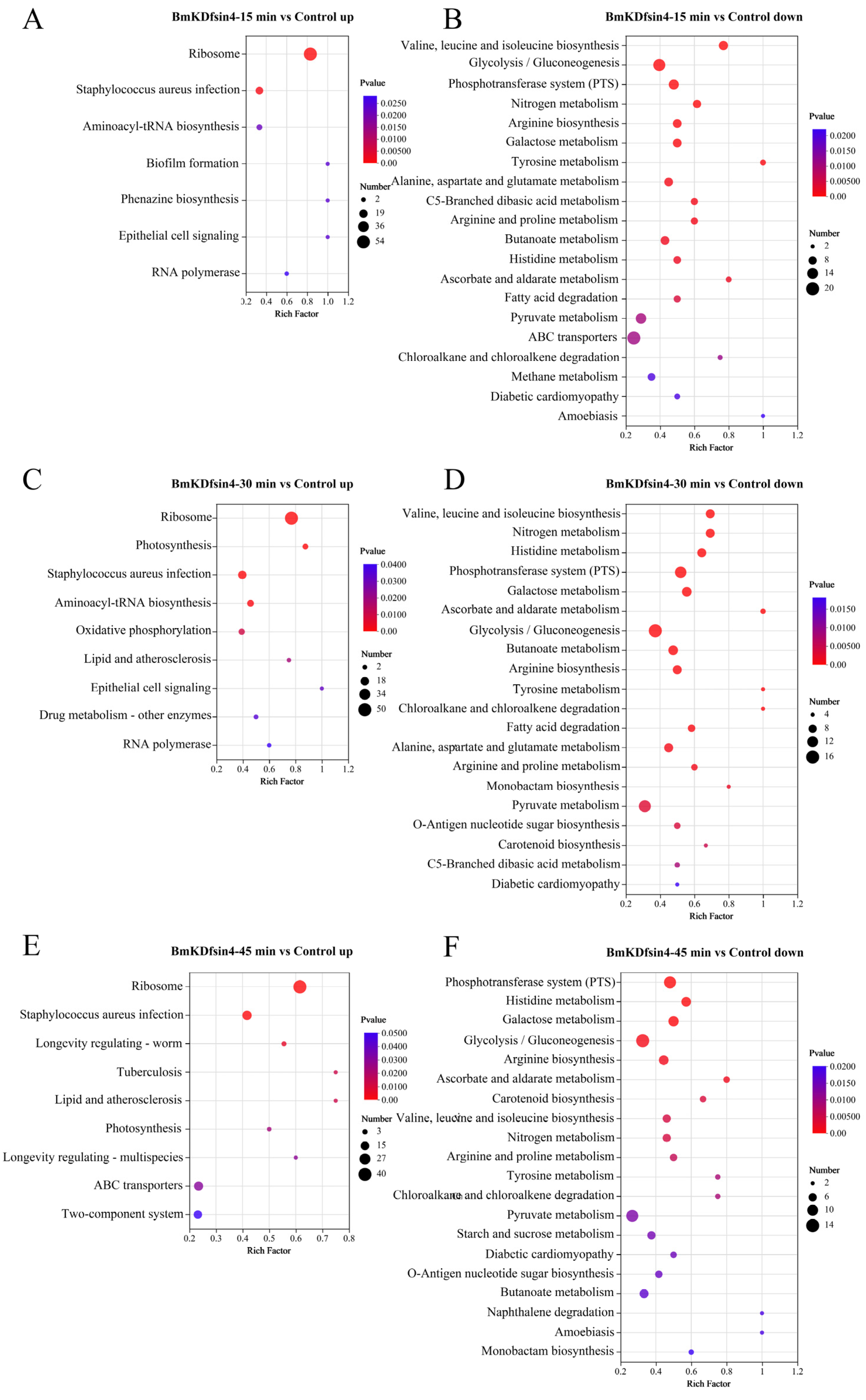
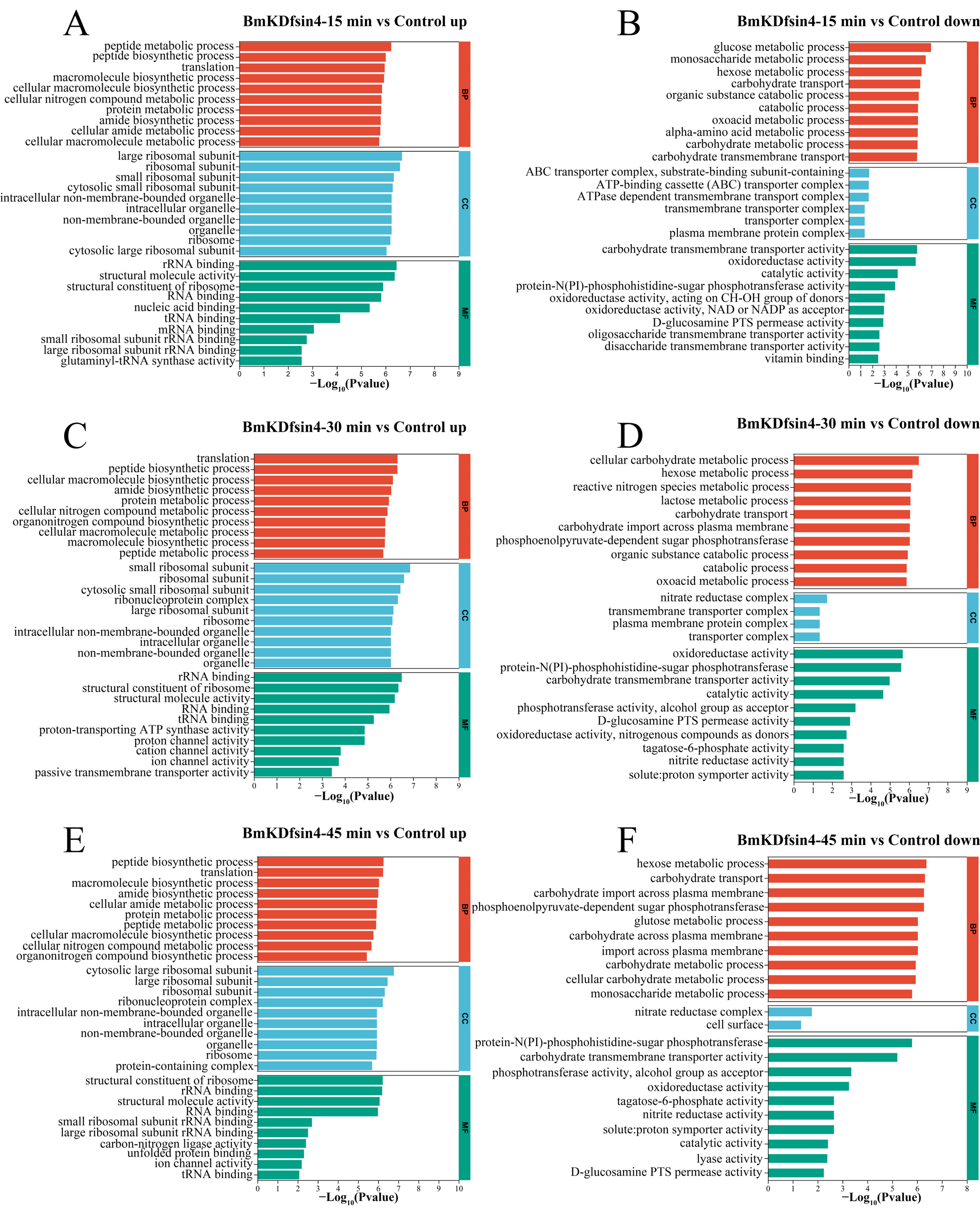
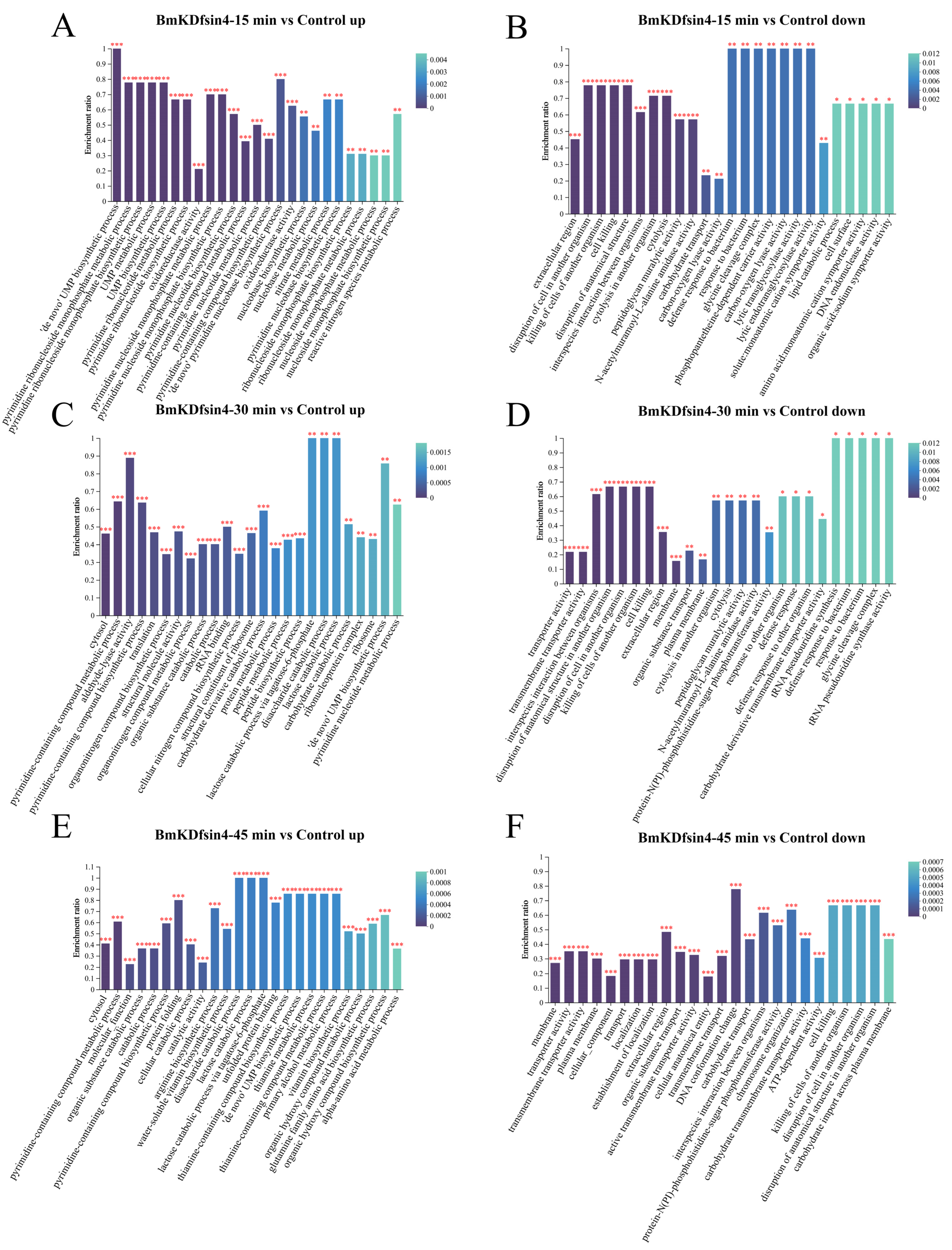
2.3. Functional Enrichment Analysis of DEGs and DEPs
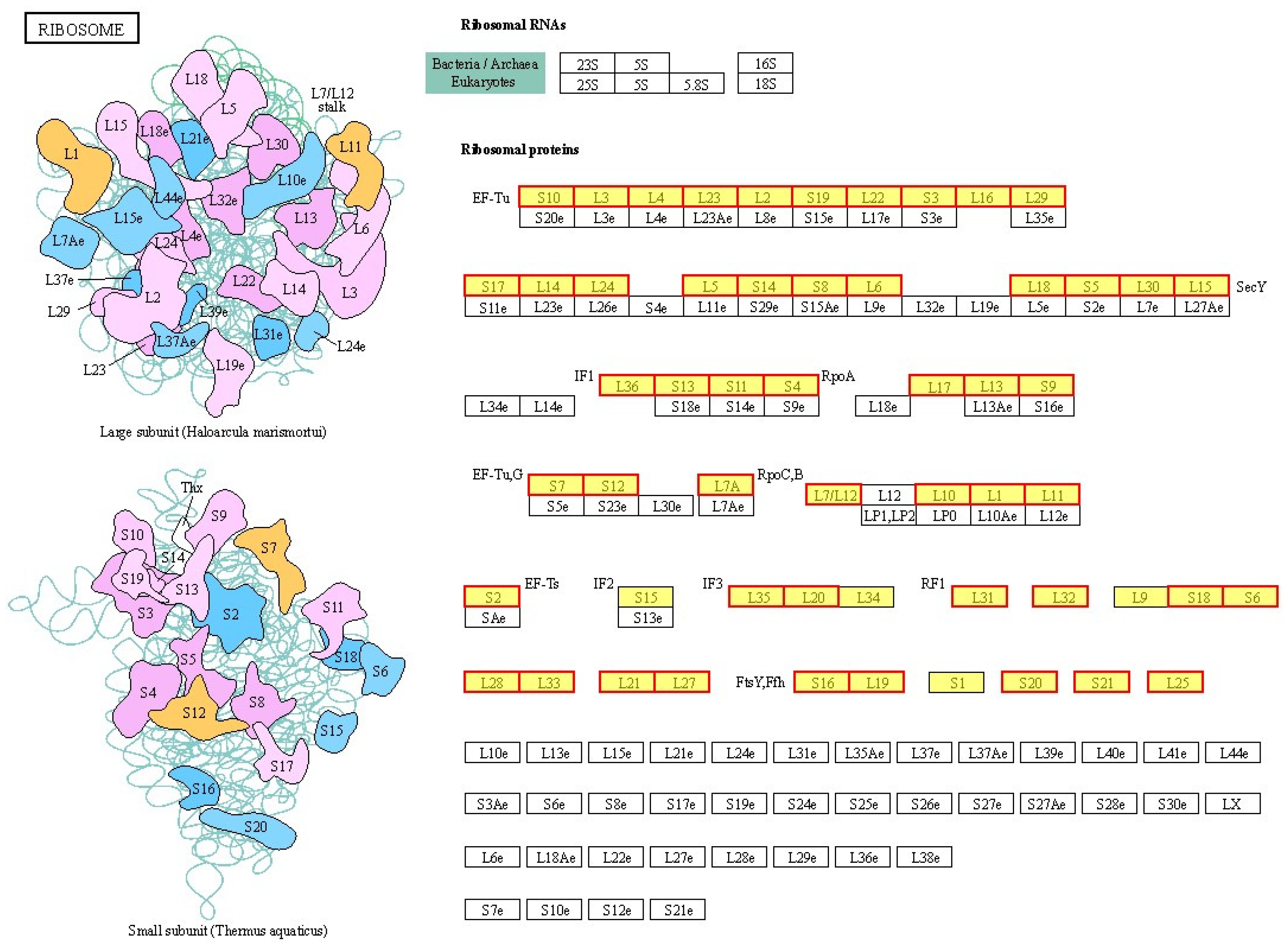
2.4. Effects of BmKDfsin4 on the Ribosome Assembly and Protein Synthesis of S. aureus
2.5. Reduction in Amino Acid Biosynthesis and Metabolism in S. aureus Due to BmKDfsin4 Treatment
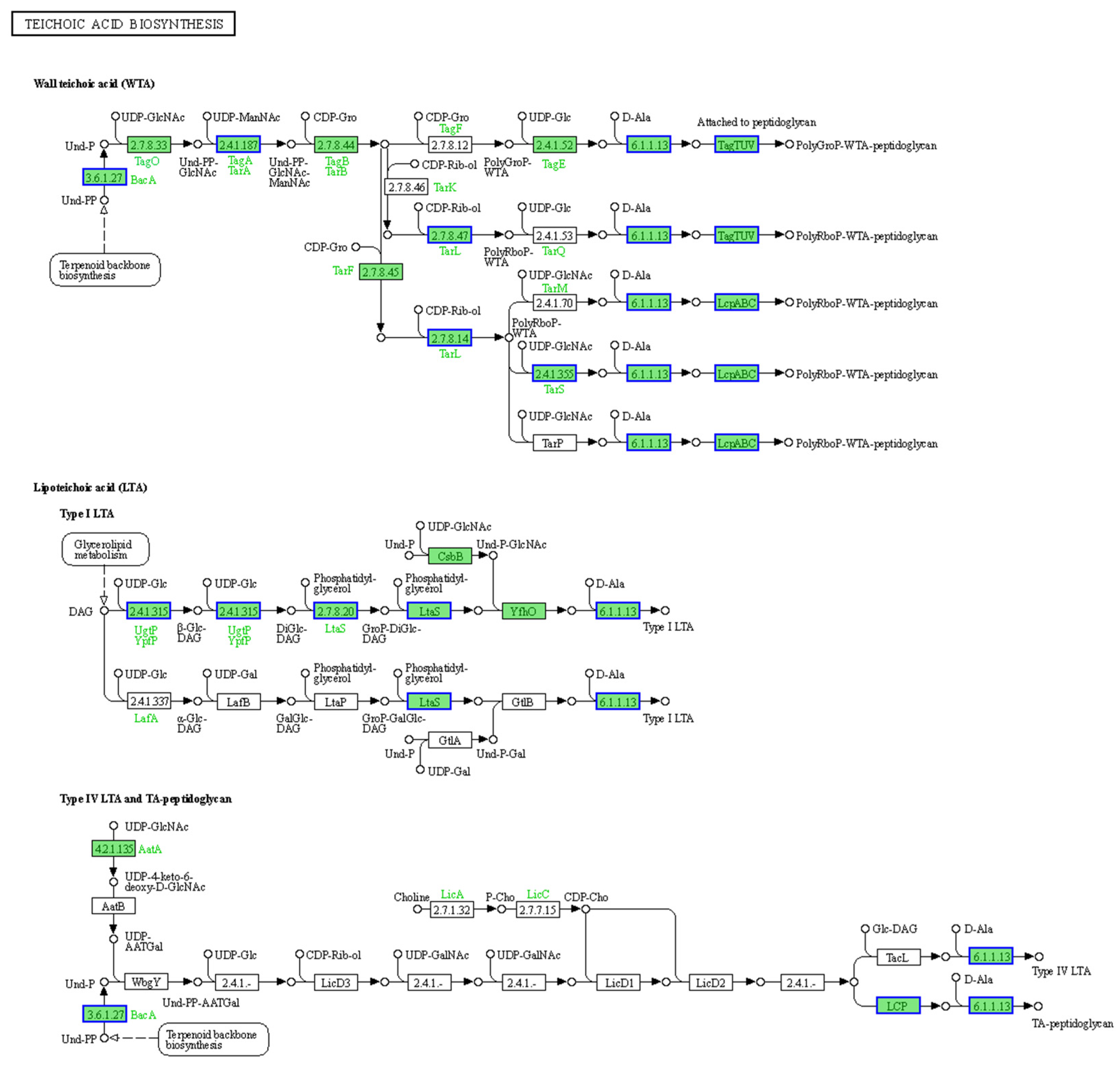
2.6. Inhibition of Cell Wall Biosynthesis in S. aureus AB94004 Due to BmKDfsin4 Treatment
2.7. Inhibitory Effect of BmKDfsin4 on the Metabolism of S. aureus AB9004
3. Discussion
4. Materials and Methods
4.1. Bactericidal Kinetics
4.2. RNA Extraction
4.3. Transcriptomic Analysis
4.4. Protein Extraction and Proteomic Analysis
4.5. Bioinformatics and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Tong, S.Y.C.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Bayer, A.; Cosgrove, S.E.; Daum, R.S.; Fridkin, S.K.; Gorwitz, R.J.; Kaplan, S.L.; Karchmer, A.W.; Levine, D.P.; Murray, B.E.; et al. Clinical practice guidelines by the infectious diseases society of america for the treatment of methicillin-resistant Staphylococcus aureus infections in adults and children. Clin. Infect. Dis. 2011, 52, e18–e55. [Google Scholar] [CrossRef]
- Sadaka, A.; Durand, M.L.; Sisk, R.; Gilmore, M.S. Staphylococcus aureus and its Bearing on Ophthalmic Disease. Ocul. Immunol. Inflamm. 2017, 25, 111–121. [Google Scholar] [CrossRef] [PubMed]
- Gechev, T.S.; Hille, J.; Woerdenbag, H.J.; Benina, M.; Mehterov, N.; Toneva, V.; Fernie, A.R.; Mueller-Roeber, B. Natural products from resurrection plants: Potential for medical applications. Biotechnol. Adv. 2014, 32, 1091–1101. [Google Scholar] [CrossRef]
- Kumar, P.; Kizhakkedathu, J.N.; Straus, S.K. Antimicrobial Peptides: Diversity, Mechanism of Action and Strategies to Improve the Activity and Biocompatibility In Vivo. Biomolecules 2018, 8, 4. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.; Ding, J.; Liao, C.; Xu, J.; Liu, X.; Lu, W. Defensins: The natural peptide antibiotic. Adv. Drug Deliv. Rev. 2021, 179, 114008. [Google Scholar] [CrossRef]
- Tavares, L.S.; Silva, C.d.; Souza, V.C.; Silva, V.L.; Diniz, C.G.; Santos, M.D. Strategies and molecular tools to fight antimicrobial resistance: Resistome, transcriptome, and antimicrobial peptides. Front. Microbiol. 2013, 4, 412. [Google Scholar] [CrossRef]
- Fu, J.; Zong, X.; Jin, M.; Min, J.; Wang, F.; Wang, Y. Mechanisms and regulation of defensins in host defense. Signal Transduct. Target. Ther. 2023, 8, 300. [Google Scholar] [CrossRef]
- Freiberg, C.; Brötz-Oesterhelt, H.; Labischinski, H. The impact of transcriptome and proteome analyses on antibiotic drug discovery. Curr. Opin. Microbiol. 2004, 7, 451–459. [Google Scholar] [CrossRef]
- Freiberg, C.; Brötz-Oesterhelt, H. Functional genomics in antibacterial drug discovery. Drug Discov. Today 2005, 10, 927–935. [Google Scholar] [CrossRef]
- Hesketh, A.; Deery, M.J.; Hong, H.-J. High-Resolution Mass Spectrometry Based Proteomic Analysis of the Response to Vancomycin-Induced Cell Wall Stress in Streptomyces coelicolor A3(2). J. Proteome Res. 2015, 14, 2915–2928. [Google Scholar] [CrossRef] [PubMed]
- Ran, W.; Yue, Y.; Long, F.; Zhong, K.; Bai, J.; Xiao, Y.; Bu, Q.; Huang, Y.; Wu, Y.; Gao, H. Antibacterial Mechanism of 2R,3R-Dihydromyricetin Against Staphylococcus aureus: Deciphering Inhibitory Effect on Biofilm and Virulence Based on Transcriptomic and Proteomic Analyses. Foodborne Pathog. Dis. 2023, 20, 90–99. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Liu, X.; Song, L.; Wang, Y.; Zhang, J.; Song, Y.; Zhuang, H.; Shen, J.; Yang, J.; Peng, C.; et al. The Antibacterial Activities and Effects of Baicalin on Ampicillin Resistance of MRSA and Stenotrophomonas maltophilia. Foodborne Pathog. Dis. 2024. [Google Scholar] [CrossRef]
- Aghamiri, S.; Zandsalimi, F.; Raee, P.; Abdollahifar, M.-A.; Tan, S.C.; Low, T.Y.; Najafi, S.; Ashrafizadeh, M.; Zarrabi, A.; Ghanbarian, H.; et al. Antimicrobial peptides as potential therapeutics for breast cancer. Pharmacol. Res. 2021, 171, 105777. [Google Scholar] [CrossRef]
- Hancock, R.E.W.; Sahl, H.-G. Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat. Biotechnol. 2006, 24, 1551–1557. [Google Scholar]
- Zasloff, M. Magainins, a class of antimicrobial peptides from Xenopus skin: Isolation, characterization of two active forms, and partial cDNA sequence of a precursor. Proc. Natl. Acad. Sci. USA 1987, 84, 5449–5453. [Google Scholar] [CrossRef]
- Kalfa, V.C.; Jia, H.P.; Kunkle, R.A.; McCray, P.B.; Tack, B.F.; Brogden, K.A. Congeners of SMAP29 Kill Ovine Pathogens and Induce Ultrastructural Damage in Bacterial Cells. Antimicrob. Agents Chemother. 2001, 45, 3256–3261. [Google Scholar] [CrossRef]
- Boman, H.G.; Agerberth, B.; Boman, A. Mechanisms of action on Escherichia coli of cecropin P1 and PR-39, two antibacterial peptides from pig intestine. Infect. Immun. 1993, 61, 2978–2984. [Google Scholar] [CrossRef]
- Piscopo, M.; Tenore, G.C.; Notariale, R.; Maresca, V.; Maisto, M.; de Ruberto, F.; Heydari, M.; Sorbo, S.; Basile, A. Antimicrobial and antioxidant activity of proteins from Feijoa sellowiana Berg. fruit before and after in vitro gastrointestinal digestion. Nat. Prod. Res. 2020, 34, 2607–2611. [Google Scholar] [CrossRef]
- Marinaro, C.; Lettieri, G.; Verrillo, M.; Morelli, M.; Carraturo, F.; Guida, M.; Piscopo, M. Possible Molecular Mechanisms Underlying the Decrease in the Antibacterial Activity of Protamine-like Proteins after Exposure of Mytilus galloprovincialis to Chromium and Mercury. Int. J. Mol. Sci. 2023, 24, 9345. [Google Scholar] [CrossRef]
- Brogden, K.A. Antimicrobial peptides: Pore formers or metabolic inhibitors in bacteria? Nat. Rev. Microbiol. 2005, 3, 238–250. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Deng, S.; Wang, X.; Thunders, M.; Qiu, J.; Li, Y. Discovery and Mechanism of Action of a Novel Antimicrobial Peptide from an Earthworm. Microbiol. Spectr. 2023, 11, e03206-22. [Google Scholar] [CrossRef]
- Selsted, M.E.; Ouellette, A.J. Mammalian defensins in the antimicrobial immune response. Nat. Immunol. 2005, 6, 551–557. [Google Scholar] [CrossRef]
- Jin, J.Y.; Zhou, L.; Wang, Y.; Li, Z.; Zhao, J.G.; Zhang, Q.Y.; Gui, J.F. Antibacterial and antiviral roles of a fish β-defensin expressed both in pituitary and testis. PLoS ONE 2010, 5, e12883. [Google Scholar] [CrossRef]
- Lv, C.; Han, Y.; Yang, D.; Zhao, J.; Wang, C.; Mu, C. Antibacterial activities and mechanisms of action of a defensin from manila clam Ruditapes philippinarum. Fish. Shellfish. Immunol. 2020, 103, 266–276. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Han, L.; Ni, Y.; Yu, Z.; Wang, D.; Zhou, J.; Li, B.; Zhang, W.; He, K. In vitro and In vivo Antibacterial Effects of Nisin Against Streptococcus suis. Probiotics Antimicrob. Proteins 2021, 13, 598–610. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, H.; Zuo, Z.; Qin, C.; Liu, Y.; Cao, Z.; Wu, Y. Novel structural determinants and bacterial death-related regulatory effects of the scorpion defensin BmKDfsin4 against gram-positive bacteria. Int. J. Biol. Macromol. 2024, 282, 137151. [Google Scholar] [CrossRef]
- Oldham, M.L.; Chen, J. Crystal structure of the maltose transporter in a pretranslocation intermediate state. Science 2011, 332, 1202–1205. [Google Scholar] [CrossRef]
- Oldham, M.L.; Khare, D.; Quiocho, F.A.; Davidson, A.L.; Chen, J. Crystal structure of a catalytic intermediate of the maltose transporter. Nature 2007, 450, 515–521. [Google Scholar] [CrossRef]
- Ho, H.; Miu, A.; Alexander, M.K.; Garcia, N.K.; Oh, A.; Zilberleyb, I.; Reichelt, M.; Austin, C.D.; Tam, C.; Shriver, S.; et al. Structural basis for dual-mode inhibition of the ABC transporter MsbA. Nature 2018, 557, 196–201. [Google Scholar] [CrossRef]
- Liu, M.; Chu, B.; Sun, R.; Ding, J.; Ye, H.; Yang, Y.; Wu, Y.; Shi, H.; Song, B.; He, Y.; et al. Antisense Oligonucleotides Selectively Enter Human-Derived Antibiotic-Resistant Bacteria through Bacterial-Specific ATP-Binding Cassette Sugar Transporter. Adv. Mater. 2023, 35, e2300477. [Google Scholar] [CrossRef] [PubMed]
- Jin, S.; Sun, D.; Song, D.; Wang, N.; Fu, H.; Ji, F.; Zhang, Y. iTRAQ-based quantitative proteomic analysis of embryonic developmental stages in Amur sturgeon, Acipenser schrenckii. Sci. Rep. 2018, 8, 6255. [Google Scholar] [CrossRef]
- Zhang, Z.; Fan, Z.; Yi, M.; Liu, Z.; Ke, X.; Gao, F.; Cao, J.; Wang, M.; Chen, G.; Lu, M. Characterization of the core gut microbiota of Nile tilapia (Oreochromis niloticus): Indication of a putative novel Cetobacterium species and analysis of its potential function on nutrition. Arch. Microbiol. 2022, 204, 690. [Google Scholar] [CrossRef]
- File, T.M., Jr. Overview of resistance in the 1990s. Chest 1999, 115, 3s–8s. [Google Scholar] [CrossRef] [PubMed]
- Neu, H.C. The crisis in antibiotic resistance. Science 1992, 257, 1064–1073. [Google Scholar] [CrossRef]
- Schneider, J.J.; Unholzer, A.; Schaller, M.; Schäfer-Korting, M.; Korting, H.C. Human defensins. J. Mol. Med. 2005, 83, 587–595. [Google Scholar] [CrossRef]
- Zeng, Z.; Zhang, Q.; Hong, W.; Xie, Y.; Liu, Y.; Li, W.; Wu, Y.; Cao, Z. A Scorpion Defensin BmKDfsin4 Inhibits Hepatitis B Virus Replication in Vitro. Toxins 2016, 8, 124. [Google Scholar] [CrossRef]
- Wang, J.; Sheng, Z.; Liu, Y.; Chen, X.; Wang, S.; Yang, H. Combined proteomic and transcriptomic analysis of the antimicrobial mechanism of tannic acid against Staphylococcus aureus. Front. Pharmacol. 2023, 14, 1178177. [Google Scholar] [CrossRef]
- Gao, Z.; Jiang, S.; Zhong, W.; Liu, T.; Guo, J. Linalool controls the viability of Escherichia coli by regulating the synthesis and modification of lipopolysaccharide, the assembly of ribosome, and the expression of substrate transporting proteins. Food Res. Int. 2023, 164, 112337. [Google Scholar] [CrossRef]
- Koenigsknecht, M.J.; Ramos, I.; Downs, D.M. Glutamine Phosphoribosylpyrophosphate Amidotransferase-independent Phosphoribosyl Amine Synthesis from Ribose 5-Phosphate and Glutamine or Asparagine. J. Biol. Chem. 2007, 282, 28379–28384. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pathway ID | KEGG Description | p Value | Rich Factor | Regulation |
|---|---|---|---|---|
| map00740 | Riboflavin metabolism | 5.42 × 10−6 | 0.90 | up |
| map00240 | Pyrimidine metabolism | 2.39 × 10−5 | 0.42 | up |
| map03010 | Ribosome | 3.20 × 10−5 | 0.48 | up |
| map00030 | Pentose phosphate pathway | 1.69 × 10−4 | 0.62 | up |
| map00270 | Cysteine and methionine metabolism | 5.95 × 10−4 | 0.57 | up |
| map01240 | Biosynthesis of cofactor | 7.59 × 10−4 | 0.37 | up |
| map00910 | Nitrogen metabolism | 1.14 × 10−3 | 0.46 | up |
| map00230 | Purine metabolism | 2.65 × 10−3 | 0.43 | up |
| map01232 | Nucleotide metabolism | 2.85 × 10−3 | 0.47 | up |
| map01240 | Biosynthesis of cofactor | 5.21 × 10−3 | 0.31 | up |
| map00730 | Thiamine metabolism | 7.60 × 10−3 | 0.60 | up |
| map00360 | Phenylalanine metabolism | 8.93 × 10−3 | 1.00 | up |
| map01502 | Vancomycin resistance | 1.93 × 10−2 | 0.67 | up |
| map00983 | Drug metabolism—other enzyme | 2.06 × 10−2 | 0.63 | up |
| map00350 | Tyrosine metabolism | 3.02 × 10−2 | 0.75 | up |
| map00750 | Vitamin B6 metabolism | 3.02 × 10−2 | 0.75 | up |
| map00362 | Benzoate degradation | 1.20 × 10−2 | 0.50 | down |
| map03020 | RNA polymerase | 2.90 × 10−2 | 0.60 | down |
| map00552 | Teichoic acid biosynthesis | 3.40 × 10−2 | 0.41 | down |
| map02060 | Phosphotransferase system (PTS) | 3.40 × 10−2 | 0.41 | down |
| Accession | Protein Name | Log2 FC | p Value | Regulation | GO Term |
|---|---|---|---|---|---|
| PR417_000186 | Ribosomal_S14 | 0.68 | 0.004935 | up | BP |
| PR417_000260 | Ribosomal_S15 | 3.86 | 0.01018 | up | BP |
| PR417_000310 | Ribosomal_L28 | 0.98 | 0.001746 | up | BP |
| PR417_000560 | Ribosomal_S20p | 1.21 | 0.0006025 | up | BP |
| PR417_000570 | Ribosomal_S21 | 2.01 | 0.004936 | up | BP |
| PR417_001047 | Ribosomal_S10 | 0.88 | 0.0007706 | up | BP |
| PR417_001051 | Ribosomal_L2 | 1.53 | 0.0000644 | up | BP |
| PR417_001052 | Ribosomal_S19 | 2.92 | 0.0007064 | up | BP |
| PR417_001054 | Ribosomal_S3_C | 0.70 | 0.0002152 | up | BP |
| PR417_001055 | Ribosomal_L16 | 1.29 | 0.002452 | up | BP |
| PR417_001057 | Ribosomal_S17 | 1.51 | 0.001027 | up | BP |
| PR417_001059 | Ribosomal_L24 | 2.01 | 0.001125 | up | BP |
| PR417_001064 | Ribosomal_L18p | 1.59 | 0.0003322 | up | BP |
| PR417_001066 | Ribosomal_L30 | 1.65 | 0.0000517 | up | BP |
| PR417_001067 | Ribosomal_L27A | 0.77 | 0.000431 | up | BP |
| PR417_001073 | Ribosomal_S11 | 0.84 | 0.0000539 | up | BP |
| PR417_001217 | Ribosomal_S4 | 0.75 | 0.001019 | up | BP |
| PR417_001258 | Ribosomal_L35p | 3.92 | 0.0004422 | up | BP |
| PR417_001259 | Ribosomal_L20 | 0.77 | 0.002422 | up | BP |
| PR417_001303 | Ribosomal_S1 | 0.72 | 0.001404 | up | CC |
| PR417_001445 | Ribosomal_L31 | 1.21 | 0.006626 | up | BP |
| PR417_001541 | Ribosomal_S18 | 1.93 | 0.0001338 | up | BP |
| PR417_001543 | Ribosomal_S6 | 2.54 | 0.0000811 | up | BP |
| PR417_001894 | Ribosomal_L9 | 0.70 | 0.006551 | up | BP |
| PR417_002081 | Ribosomal_L10 | 0.67 | 0.0001408 | up | BP |
| PR417_002082 | Ribosomal_L12 | 1.68 | 0.0007194 | up | BP |
| PR417_002088 | Ribosomal_S7 | 0.94 | 0.003338 | up | BP |
| Gene ID | Gene Name | Gene Description | Log2 FC | p Value | Regulation |
|---|---|---|---|---|---|
| PR417_000270 | proS | proline–tRNA ligase | 1.05 | 1.01 × 10−34 | up |
| PR417_000515 | hisS | histidine–tRNA ligase | 2.00 | 9.38 × 10−57 | up |
| PR417_000530 | alaS | alanine–tRNA ligase | 0.95 | 4.72 × 10−16 | up |
| PR417_000580 | glyS1 | glycine–tRNA ligase | 1.22 | 3.33 × 10−46 | up |
| PR417_001207 | tyrS | tyrosine–tRNA ligase | 0.94 | 1.14 × 10−6 | up |
| PR417_001255 | thrS | threonine–tRNA ligase | 1.03 | 1.54 × 10−9 | up |
| PR417_001275 | valS | valine–tRNA ligase | 0.81 | 2.02 × 10−17 | up |
| PR417_001324 | asnS | asparagine–tRNA ligase | 0.87 | 3.27 × 10−14 | up |
| PR417_001745 | leuS | leucine–tRNA ligase | 1.18 | 5.81 × 10−26 | up |
| PR417_001888 | serS | serine–tRNA ligase | 0.60 | 4.58 × 10−8 | up |
| PR417_002019 | lysS | lysine–tRNA ligase | 0.63 | 4.96 × 10−11 | up |
| PR417_002406 | ileS | isoleucine–tRNA ligase | 1.85 | 9.62 × 10−77 | up |
| PR417_001117 | pheT | phenylalanine–tRNA ligase subunit beta | 1.39 | 2.01 × 10−26 | up |
| PR417_001118 | pheS | phenylalanine–tRNA ligase subunit alpha | 1.05 | 9.27 × 10−17 | up |
| Accession | Gene Name | Protein ID | Description | Log2 FC | |
|---|---|---|---|---|---|
| Gene | Protein | ||||
| PR417_001228 | ald | HDK3247889.1 | Alanine dehydrogenase | −0.55 | −0.72 |
| PR417_000076 | puuE | HDK3246759.1 | Aspartate aminotransferase family protein | −1.91 | −0.67 |
| PR417_001891 | metX | HDK3248527.1 | Homoserine O-acetyltransferase | −0.07 | −0.60 |
| PR417_001358 | argJ | HDK3248015.1 | Bifunctional glutamate N-acetyltransferase/amino-acid acetyltransferase ArgJ | 0.86 | −16.61 |
| PR417_000873 | argH | HDK3247540.1 | Argininosuccinate lyase | −3.66 | |
| PR417_000874 | argG | HDK3247541.1 | Argininosuccinate synthase | −2.77 | |
| PR417_001186 | purF | HDK3247847.1 | Amidophosphoribosyl transferase | −0.53 | - |
| PR417_001184 | purN | HDK3247845.1 | Phosphoribosyl glycinamide formyl transferase | −0.41 | - |
| PR417_000028 | pruA | HDK3246711.1 | L-glutamate gamma-semialdehyde dehydrogenase | −0.90 | - |
| PR417_000076 | - | HDK3246759.1 | Aspartate aminotransferase family protein | −1.91 | −0.668 |
| PR417_002017 | gltB | HDK3248649.1 | Glutamate synthase large subunit | −1.45 | |
| PR417_002018 | gltD | HDK3248650.1 | Glutamate synthase subunit beta | −1.23 | |
| PR417_000871 | - | HDK3247538.1 | Glu/Leu/Phe/Val dehydrogenase | −1.08 | |
| PR417_000225 | glnA | HDK3246901.1 | Type I glutamate–ammonia ligase | −0.96 | - |
| Accession | Protein Name | Protein ID | Description | Log2 FC |
|---|---|---|---|---|
| PR417_000847 | DltC | HDK3247514.1 | D-alanine-activating enzyme/D-alanine-D-alanyl | −0.84 |
| PR417_000449 | LtaS | HDK3247125.1 | Polyglycerol-phosphate lipoteichoic acid synthase LtaS | −0.95 |
| PR417_002396 | MurD | HDK3249008.1 | UDP-N-acetylmuramoyl-L-alanyl-D-glutamate synthetase | −0.59 |
| PR417_000273 | UppS | HDK3246949.1 | UDP pyrophosphate synthase UppS | −0.85 |
| PR417_001798 | - | HDK3248439.1 | LCP protein family | −1.00 |
| PR417_000593 | PBP | HDK3247268.1 | Penicillin-binding protein | −0.60 |
| PR417_002630 | - | HDK3249238.1 | Polyisoprenyl-teichoic acid–peptidoglycan teichoic acid transferase | −0.62 |
| PR417_000365 | TagA | HDK3247041.1 | Teichoic acid biosynthesis protein TagA | −0.74 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2025 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yang, X.; Zhang, H.; Lu, S.; Guo, Y.; Li, Y.; Qin, C.; Zuo, Z.; Wu, Y.; Cao, Z. Insights into the Antimicrobial Mechanisms of a Scorpion Defensin on Staphylococcus aureus Using Transcriptomic and Proteomic Analyses. Molecules 2025, 30, 1542. https://doi.org/10.3390/molecules30071542
Yang X, Zhang H, Lu S, Guo Y, Li Y, Qin C, Zuo Z, Wu Y, Cao Z. Insights into the Antimicrobial Mechanisms of a Scorpion Defensin on Staphylococcus aureus Using Transcriptomic and Proteomic Analyses. Molecules. 2025; 30(7):1542. https://doi.org/10.3390/molecules30071542
Chicago/Turabian StyleYang, Xuhua, Haozhen Zhang, Sijia Lu, Yiyuan Guo, Yitong Li, Chenhu Qin, Zheng Zuo, Yingliang Wu, and Zhijian Cao. 2025. "Insights into the Antimicrobial Mechanisms of a Scorpion Defensin on Staphylococcus aureus Using Transcriptomic and Proteomic Analyses" Molecules 30, no. 7: 1542. https://doi.org/10.3390/molecules30071542
APA StyleYang, X., Zhang, H., Lu, S., Guo, Y., Li, Y., Qin, C., Zuo, Z., Wu, Y., & Cao, Z. (2025). Insights into the Antimicrobial Mechanisms of a Scorpion Defensin on Staphylococcus aureus Using Transcriptomic and Proteomic Analyses. Molecules, 30(7), 1542. https://doi.org/10.3390/molecules30071542